

Abstract
Background and Purpose
Systemic administration of Toll-like receptor 4 (TLR4) and TLR9 agonists prior to cerebral ischemia, have been shown to reduce ischemic injury by reprogramming the brain’s response to stroke. Our goal was to explore the mechanism of TLR induced neuroprotection by determining whether a TLR7 agonist also protects against stroke injury.
Methods
C57Bl/6, TNF−/−, interferon regulatory factor (IRF)7−/−, or type I interferon receptor (IFNAR)−/− mice were subcutaneously administered the TLR7 agonist Gardiquimod (GDQ) 72 hr prior to middle cerebral artery occlusion (MCAO). Infarct volume and functional outcome were determined following reperfusion. Plasma cytokine responses and induction of mRNA for IFN related genes in the brain were measured. IFNAR−/− mice were also treated with the TLR4 agonist (lipopolysaccharide) or the TLR9 agonist (CpG) prior to MCAO and infarct volumes measured.
Results
The results show that GDQ reduces infarct volume as well as functional deficits in mice. GDQ pretreatment provided robust neuroprotection in TNF−/− mice indicating that TNF was not essential. GDQ induced a significant increase in plasma IFNα levels and both IRF7−/− and IFNAR−/− mice failed to be protected, implicating a role for IFN signaling in TLR7 mediated protection.
Conclusion
Our studies provide the first evidence that TLR7 preconditioning can mediate neuroprotection against ischemic injury. Moreover, we show that the mechanism of protection is unique from other TLR preconditioning ligands in that it is independent of TNF and dependent on IFNAR.
Keywords: Ischemia, Neuroprotection
Introduction
Toll-like receptors (TLRs) are sentinels of the innate immune system, which have recently been shown to be involved in stroke injury. In a model of brain focal ischemia, mice deficient in TLR2 or TLR4 showed significantly less brain damage compared to their wildtype counterparts, highlighting a deleterious role for these receptors in ischemic injury1–5. In accordance with this, TLR2 and TLR4 expression on monocytes was associated with poor functional outcome in human ischemic stroke patients and correlated with higher serum levels of proinflammatory cytokines6. Thus, finding ways to modulate the TLR response to stroke could provide a potential therapeutic target to reduce ischemic injury.
An important aspect of the TLR family is their ability to auto- and cross-regulate the response to subsequent TLR signaling by priming initially with a small amount of TLR ligand. The priming event can lead to suppression and redirection of the subsequent response to stimulation with a secondary TLR ligand. For example, pretreating cultured murine macrophages with a small dose of a TLR4, TLR7 or TLR9 ligand reduces NFB activation and TNF, and enhances interferon (IFN) β in response to subsequent TLR4 activation7. Importantly NFκB and TNF have been shown to play damaging roles in brain ischemia8–10, while IFNβ is neuroprotective11–13. Taken together with the evidence that TLRs play a role in stroke injury, we postulated previously that TLR activation in the setting of ischemia could be redirected via prior stimulation with a TLR ligand. Indeed, we and others have shown that exogenous administration of small doses of ligands for at least three TLRs (TLR2, TLR4, and TLR9) prior to stroke provides protection14–18. In addition, preconditioning with the TLR4 or TLR9 ligands leads to a reprogrammed response to stroke. The reprogrammed response is characterized by enhanced interferon regulatory factor (IRF)-mediated transcription and increased production of IFN-associated genes following ischemia in LPS and CpG preconditioned animals11, 19.
The mechanism by which TLR preconditioning induces ischemic tolerance and provides protection remains incompletely understood. However, an important role for TNF has been shown for LPS and CpG preconditioning because TNF-deficient mice cannot be protected by either of these TLR ligands10, 18. An important role for an IFN response also exists because mice deficient in either IRF3 or IRF7 failed to be protected with LPS or CpG preconditioning11, 19. To further delineate mechanisms underlying TLR preconditioning, we investigated the potential for a TLR7 agonist to induce neuroprotection. As discussed above, TLR7 has been shown to provide cross-tolerance to a subsequent TLR4 stimulation and thus we postulated that preconditioning through TLR7 would also provide protection against ischemic injury. In addition, as TLR7 signaling induces a more substantial type I IFN response compared to TLR4 or TLR9, which show minimal to no induction of type I IFNs (IFNα and IFNβ20 we hypothesized that TLR7-preconditioning, through its increase in expression of type I IFNs may provide a route to neuroprotection that is unique from TLR4 and TLR9.
The results provided here are the first evidence that TLR7 preconditioning confers robust protection against focal ischemia. We show that the reduced damage is associated with upregulation of IFN-associated genes, which is similar to our previous findings with TLR4- and TLR9-preconditioning. Surprisingly, we find that TLR7-mediated preconditioning works through a TNF-independent mechanism, which contrasts with TLR4 and TLR9. We found that TLR7 preconditioning required IRF7 for the induction of IFNα to confer neuroprotection.
Furthermore, only TLR7 preconditioning required the presence of the cognate receptor for type I IFNs (IFNAR) – a feature not shared by TLR4 or TLR9 preconditioning. Collectively, these novel findings highlight a new mechanism of TLR preconditioning-induced protection that relies on the production and signaling of type I IFNs.
Methods
Mice
C57Bl/6 and B6.129S-Tnftm1Gkl/J (TNF−/−) mice were obtained from Jackson Laboratories (West Sacramento, CA). TLR7−/− mice were purchased from OrientalBioService (Osaka, Japan), IRF7−/− mice were provided by Dr. Ian Rifkin (Boston University School of Medicine, Boston, MA) and IFNAR−/− mice were provided by Dr. Anthony French (Washington University School of Medicine, St. Louis, MO). These strains were backcrossed ≥8 generations onto C57Bl/6. All studies were performed with male mice between 10–14 weeks of age.
All mice were given free access to food and water and were housed in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. Animal protocols were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee and met the guidelines set forth by the National Institutes of Health.
Drug treatments
Mice were given a subcutaneous (s.c.) injection of Gardiquimod (GDQ; 10–40 ug/mouse, Invivogen), ODN 1826 (CpG; 40 μg/mouse, Invivogen), lipopolysaccharide (LPS; 20 μg/mouse, Sigma) or saline. To determine the effective time window of protection, mice were injected from 1–14 days with GDQ, prior to middle cerebral artery occlusion (MCAO). For all other experiments, mice were treated 72hr prior to MCAO.
Ischemia-reperfusion model
Mice were subjected to focal cerebral ischemia by MCAO as described previously19. The number of animals per group and treatment are reported in the figure legends. Cerebral blood flow (CBF) was monitored throughout the procedure by laser Doppler flowmetry (Transonic System Inc.). Body temperature was maintained at 37°C during and after the surgery with a heating pad. Following 45–60 min of occlusion, the monofilament was removed and blood flow was restored (reperfusion). The duration of MCAO was optimized based on the surgeon per study to obtain consistent baseline infarct sizes across studies. Twenty-four hours following MCAO, mice were deeply anesthetized, brains removed and cut into 1mm coronal sections for measurement of infarct size as previously described19. A total of 179 C57BL/6 mice were used for experiments with 22 excluded due to early attrition or failure to maintain CBF reduction of <20% of baseline during study. For the genetically engineered mice: TLR7−/− 16 total, 4 excluded; TNF−/− 16 total, 2 excluded; IRF7−/− 19 total, 1 excluded; IFNAR−/− 48 total, 4 excluded. There was no effect of genotype or treatment on mortality rate associated with the model.
Analysis of serum cytokine levels
Mice were deeply anesthetized with isoflurane and blood was collected via cardiac puncture. ELISA kits were used to analyze serum levels of TNF (R&D Systems), IFNα and IFNβ (PBL InterferonSource). Samples were run in duplicate.
Neurological evaluation
Twenty-four hours following MCAO, mice were scored on body movement (focal) and physical appearance (general well being) using a scale designed specifically to assess neurological deficits in mice as has been previously described21. Sensorimotor deficits were evaluated using the corner test, which measures the extent to which the mouse favors (turns towards) the ipsilateral (right) side after approaching a confining corner. Each mouse was tested 10 times. Naïve mice turn to each side equally, whereas after a stroke, mice tend to turn preferentially to the side ipsilateral to the stroke (right). All analyses were performed by researchers blinded to treatment to prevent experimental bias.
Tissue processing and Quantitative real time PCR
Total RNA was isolated from the brain cortex using the Qiagen Rneasy Lipid Mini Kit (Qiagen). RNA was reverse transcribed using an Omniscript Reverse Transcription kit (Qiagen). Quantitative PCR (qtPCR) was performed using TaqMan Gene Expression Assays (Applied Biosystems) on an ABI-prism 7700. Results were normalized to β-Actin expression. The relative quantification was determined using the comparative CT method (2−DDCt).
Statistical Analyses
Data are presented as mean ± SEM and were analyzed using Student t-test, 1-way ANOVA or 2-way ANOVA with Bonferroni’s post-hoc test as indicated in figure legends. Differences were considered significant when p<0.05. Prism4 (Graphpad) was used for all statistical analyses.
Results
GDQ preconditioning reduces ischemic damage in an in vivo model of stroke
To determine whether GDQ could protect against ischemia, mice were pre-treated with various doses of GDQ (10 – 40 μg/mouse, s.c.) 72 hr prior to MCAO (60 min) and the infarct size determined 24 hr later. Results show that GDQ significantly reduced ischemic damage in a dose-dependent manner (Fig. 1A), with a maximal protective effect achieved at the dose of 40 μg/mouse (28 ± 3.6% compared with saline at 58 ± 0.94%). In addition we found that the neuroprotection induced by GDQ preconditioning was still evident 72 hr post-MCAO (24.78 ± 2.4% compared with saline treated 39.72 ± 2.2%; Fig. 1B), indicating that GDQ-induced neuroprotection is a sustained effect.
Figure 1. GDQ preconditioning reduces Ischemic injury.

(A) C57Bl/6 mice were preconditioned with escalating doses of GDQ (N = 8; 10, 20 or 40 μg per mouse; s.c.) or saline (N = 5) 72hr prior to 60 min MCAO. Infarct size was determined 24 hr following MCAO. (B) C57Bl/6 mice were pre-treated with GDQ (N = 8; 20 μg/mouse, s.c.) or saline (n = 6) 72hr prior to MCAO. Infarct size was determined 72 hr following MCAO. (C) C57Bl/6 mice were pre-treated with GDQ (N = 5 – 6; 20 μg/mouse, s.c.) or saline (n = 6) at various times prior to MCAO. Infarct size was determined 24 hr following MCAO. Two-way ANOVA, Bonferroni post hoc, *p<0.01, **p<0.01, ***p<0.001 versus saline controls. (D&E) C57BL/6 mice were treated with GDQ (N = 5; 40 μg/mouse, s.c.) or saline (N = 6) 72hr prior to MCAO (60 min). Mice were then examined using the (D) neurological score (focal and general) and (E) corner test to determine neurological and sensorimotor deficits 24hr following MCAO. Student’s t-test, *p<0.01, **p<0.01, ***p<0.001 versus saline controls.
To determine the effective time window of GDQ preconditioning, mice were treated with GDQ 1 – 14 days prior to MCAO. We found that GDQ preconditioning significantly decreased infarct size when administered 1 day prior to MCAO (35% reduction in infarct volume), and this effect was still evident when GDQ was administered 7 days prior to MCAO (20% reduction). However protection was lost when GDQ was given 14 days before MCAO (Fig. 1C), indicating that the neuroprotective time window of TLR7 preconditioning lasts for at least one week. This time window of neuroprotection is comparable to those we have reported previously for LPS and CpG preconditioning10, 18.
GDQ preconditioning reduces ischemia-induced neurological deficits
To determine whether neurological deficits associated with the stroke injury are attenuated by GDQ preconditioning, we examined mice using focal and general assessment scales21. Mice pre-treated with GDQ scored better in the focal and general categories compared to saline controls, providing evidence that GDQ attenuates neurological deficits as well as reducing infarct size (Fig. 1D). To assess sensorimotor deficits, mice were subjected to the corner test following MCAO. Results from this test have been shown to correlate with infarct volume and can reveal the extent of post-infarct recovery22, 23. Mice preconditioned with GDQ showed significantly fewer sensorimotor deficits, represented by a decreased tendency to turn to the right (62.50% ± 8.54%) compared to saline-treated animals (87.50% ± 4.79%; Fig. 1E).
TLR7 mediates GDQ-induced protection against ischemic injury
We tested whether the neuroprotective effects were specifically exerted through TLR7 since previous work by others showed that some TLR7 agonists were able to signal through adenosine receptors24. We preconditioned TLR7−/− mice with GDQ 72 hr before subjecting them to MCAO (45 min). Infarct size in GDQ-preconditioned TLR7−/− mice (44.38% ± 3.16%) did not differ significantly (p=0.4) from saline-treated controls (38.52% ± 6.75%), indicating that TLR7−/− mice are not protected by GDQ preconditioning (Fig. 2). Thus, GDQ preconditioning-induced neuroprotection is mediated via TLR7 signaling.
Figure 2. GDQ-induced neuroprotection is mediated through TLR7.
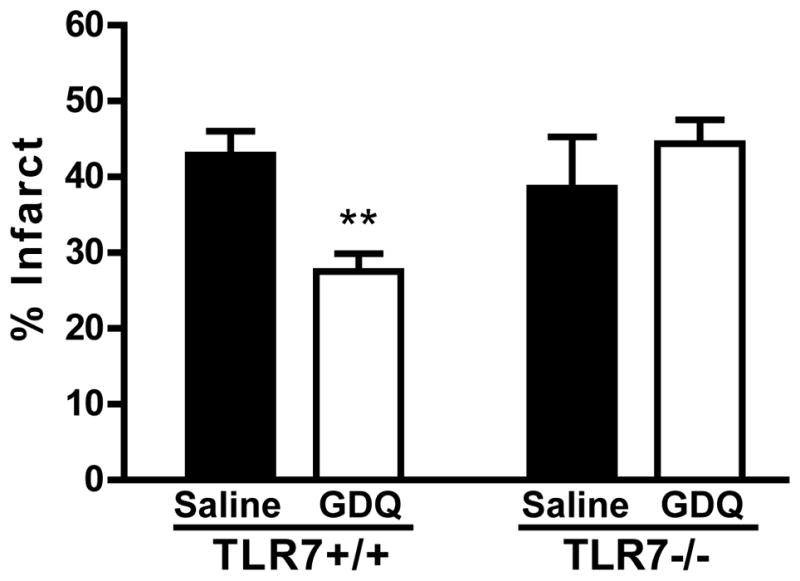
TLR7+/+ (N = 7) or TLR7−/− (N = 5 – 7) mice were preconditioned with GDQ (40 μg/mouse, s.c.) or saline 72hr prior to MCAO (45 min). Infarct size was determined 24hr following MCAO. Two-way ANOVA, Bonferroni post hoc, **p<0.01 versus saline control for respective genotype.
GDQ preconditioning results in an IFN-associated response to stroke in the brain
We have shown previously that CpG and LPS preconditioning reprogram the brain’s response to MCAO by upregulating expression of a network of IFN-associated genes after stroke, which may contribute to the neuroprotection observed in preconditioned animals11, 19. To determine whether GDQ preconditioning induces a similar reprogramming of the brain’s response to stroke, we examined the expression level of 5 of the IFN-associated genes (Usp18, Oasl2, Isg15, Trim30, Ifit1) following MCAO. Twenty-four hours following MCAO, GDQ-preconditioned animals showed significant increased levels of Usp18, Oasl2, Isg15 and Ifit1 (Table 1), when compared with non-preconditioned animals. Trim30 gene expression trended toward significant induction with a fold increase of 1.81 ± 0.3, p=0.06. These results indicate that similar to LPS and CpG preconditioning, GDQ preconditioning reprograms the genomic response to stroke to a predominantly type I IFN response that is not evident in the setting of stroke alone.
Table 1.
IFN-associated genes in the brains of GDQ-preconditioned animals following MCAO (24 hours).
| Gene | Fold change* | p-value# |
|---|---|---|
| Usp18 | 4.98 +/− 2.07 | 0.005 |
| Oasl2 | 3.75 +/− 0.8 | 0.005 |
| ISG15 | 3.54 +/− 0.9 | 0.01 |
| Ifit1 | 2.02 +/− 0.9 | 0.007 |
| Trim30 | 1.81 +/− 0.3 | 0.06 |
qt-PCR results showing fold change compared to MCAO (n=4–6/treatment).
based on Student t-test of GDQ+MCAO vs MCAO.
TNF is not required for GDQ-induced neuroprotection
We have shown that both LPS and CpG preconditioning require TNF as a critical mediator of neuroprotection10, 18. To determine whether TLR7-mediated protection depends on TNF we measured serum levels of TNF in GDQ treated mice at 1, 3 and 24 hr post injection. GDQ did not induce any measurable changes in TNF levels (Fig. 3A). It should be noted that while our low, protective dose of GDQ did not induce an increase in serum TNF levels, previous studies have shown that higher doses of other TLR7 ligands (e.g. Imiquimod) can induce serum TNF25. The unaltered TNF serum levels in mice treated with a protective dose of GDQ suggest that TNF may not be critical to GDQ-induced protection.
Figure 3. TNFα is not required for TLR7-induced neuroprotection.
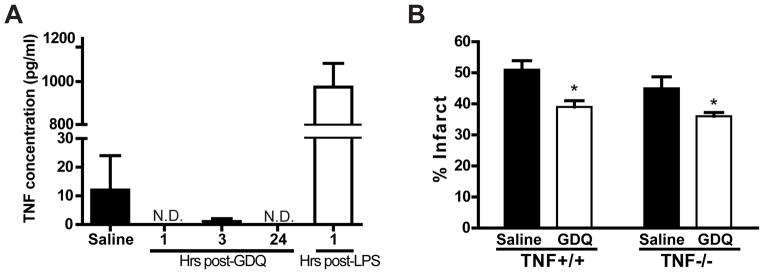
(A) C57Bl/6 mice were injected with either GDQ (40 ug/mouse, s.c.), LPS (20 μg/mouse, s.c.), or saline. Blood was collected at various time-points following injection and serum levels of TNFα determined via ELISA; N.D.= not detected (N = 4 – 6). (B) TNFα−/− (N = 6 – 8) and TNF+/+ mice (N = 7) were injected with GDQ (40 μg/mouse, s.c.) or saline 72hr prior to MCAO (50 min). Infarct size was determined 24hr following MCAO. Two-way ANOVA, Bonferroni post hoc, *p<0.05, ***p<0.001 versus saline controls.
To determine whether TNF is required for GDQ-induced neuroprotection we examined the effects of preconditioning TNF−/− mice with GDQ. TNF−/− and TNF+/+ mice were preconditioned with GDQ 72 hr prior to MCAO (50 min). GDQ-treated TNF+/+ mice had significantly reduced infarcts (39.43% ± 3.66%) compared to saline controls (50.8% ± 2.92%). Interestingly, TNF−/− mice preconditioned with GDQ were also protected (saline: 45.35% ± 2.94% versus GDQ: 36.02% ± 1.21%), indicating that TNF does not play a role in GDQ-induced neuroprotection (Fig. 3B). This contrasts sharply with LPS and CpG preconditioning and suggests a novel TNF-independent mechanism through which TLR7 mediates neuroprotection against ischemia.
Gardiquimod administration increases IFNα but not IFNβ
TLR7 signaling activates the transcription factor NFκB and proinflammatory cytokines as well as interferon regulatory factors (IRFs) and induction of type I Interferons (IFNα and IFNβ). We postulated that GDQ preconditioning relies on the interferon response as our results showed that TLR7-induced neuroprotection was independent of TNF. To determine the role of type I IFNs in TLR7 preconditioning, we measured the changes in serum levels of IFNα and IFNβ following treatment with GDQ as well as with the TLR4 and TLR9 ligands, LPS and CpG. GDQ induced a dramatic increase in IFNα levels at 1 hr and 2 hr (5-fold and 10-fold, respectively) but returned to baseline levels by 24 hr following injection. CpG induced only a modest increase in IFNα, whereas no increase in IFNα was observed with LPS (Fig. 4A). The same doses of GDQ, LPS and CpG failed to induce detectable levels of serum IFNβ (data not shown). As expected, TLR7−/− mice showed no increase in IFNα following GDQ preconditioning compared to saline treated mice (data not shown). Thus, GDQ preconditioning causes a robust increase in IFNα in the systemic circulation, while CpG and LPS preconditioning induces little to no expression of type I IFNs.
Figure 4. GDQ-preconditioning induces IFNα and requires IRF7 for neuroprotection.

(A) C57Bl/6 mice were injected with GDQ (40 ug/mouse, s.c.), CpG (40 μg/mouse, s.c.), LPS (20 μg/mouse, s.c.), or saline. Blood was collected at indicated times and serum IFNα levels were measured (N = 4–10). Two-way ANOVA, Bonferroni post hoc, *p<0.05, ***p<0.001 versus saline controls. (B) IRF7−/− and IRF7+/+ mice were injected with 40 μg GDQ or saline. Blood was collected at 2hr and serum IFNα measured (N = 3 – 8). One-way ANOVA, Bonferroni post hoc, *p<0.05 versus saline controls. (C) IRF7+/+ (N = 8 – 9) or IRF7−/− mice (N = 7 – 9) were preconditioned with GDQ (40 ug/mouse, s.c.) or saline 72hr prior to MCAO (45 min). Infarct size was determined 24hr following MCAO. Two-way ANOVA, Bonferroni post hoc, **p<0.01 versus saline controls.
IRF7 is a critical mediator for GDQ-induced neuroprotection
TLR7 mediates the induction of IFNα through the transcription factor IRF7. Thus mice deficient in IRF7 should not increase IFNα in response to GDQ and could be used to explore whether IFNα plays a critical role in GDQ preconditioning. We confirmed that GDQ stimulated IFNα induction required IRF7 activation (Fig. 4B), as the increase in IFNα observed in IRF7+/+ mice (6.5-fold over saline) was absent in IRF7−/− mice (no significant increase). To determine whether IRF7 is a critical effector of GDQ-mediated protection we treated IRF7−/− and IRF7+/+ mice with GDQ (40 μg/mouse) 72 hr prior to MCAO (45 min) and measured infarct size 24 hr later. GDQ-treated IRF7−/− mice were not protected by GDQ preconditioning (42.11% ± 2.87%) showing no significant difference in infarct size compared to saline controls (38.94% ± 2.43%; Fig. 4C). Hence, IRF7 is essential for the protective effects of GDQ preconditioning, an effect that may likely occur through IFNα.
IFNAR mediates GDQ-induced neuroprotection
To further determine whether IFNα plays a novel role in TLR7 mediated neuroprotection we used mice deficient in the interferon α/β receptor (IFNAR−/− mice). Mice were preconditioned with GDQ, CpG, or LPS 72 hr prior to MCAO (45 min). We found that IFNAR−/− mice preconditioned with GDQ displayed a significant reduction in protection compared to IFNAR+/+ mice (Fig. 5; #p<0.05), with no significant decrease in infarct size compared to saline treated mice (p>0.05; Fig. 5). In contrast, CpG and LPS preconditioning induced marked neuroprotection against ischemic injury in the IFNAR−/− mice, reducing infarct levels equivalent to that seen in IFNAR+/+ mice (Fig. 5). This result, along with the aforementioned IFNα data, suggests that IFNα, acting through its cognate receptor IFNAR, is a major mediator of TLR7-induced neuroprotection and that this mechanism of neuroprotection is not evident in preconditioning via TLR4 or TLR9.
Figure 5. IFNAR signaling is required for TLR7-mediated neuroprotection.
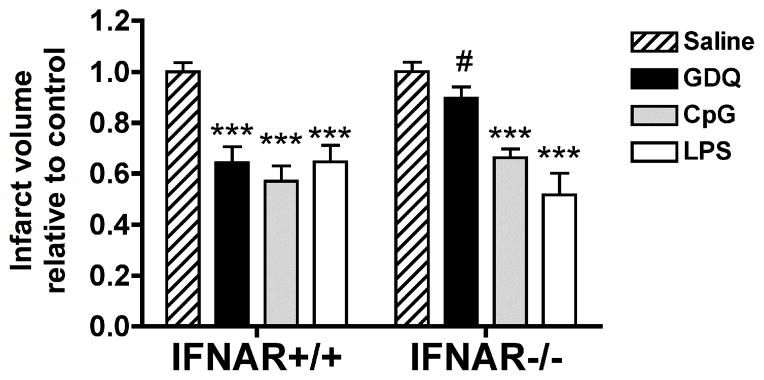
IFNAR+/+ and IFNAR−/− mice were injected with GDQ (N = 6 – 8), CpG (N = 8 – 11), LPS (N = 8 – 11) or saline (N = 6 – 9) 72hr prior to MCAO (45 min). Infarct size was determined 24hr following MCAO. The data are presented as percent damage normalized to saline + MCAO. Two-way ANOVA, Bonferroni post hoc, ***p<0.001 versus saline control for respective genotype; #p<0.05 versus IFNAR+/+ for respective treatment.
Discussion
Mice deficient in either TLR4 or TLR2 exhibit smaller infarcts when subjected to focal cerebral ischemia than wildtype mice, implicating a damaging role for TLR activation in stroke1–5. Inhibiting or altering this TLR damaging effect would provide a potential means of reducing stroke injury. In macrophages, pretreatment with a TLR ligand, including TLR4, 7 and 9 ligands, prior to stimulation with a TLR4 ligand reprograms TLR4 signaling to suppress the NFκB response and to enhance IFNβ7. We have reported evidence of a similar reprogrammed response in LPS and CpG preconditioning induced neuroprotection. In particular, we have shown that LPS preconditioning suppressed NFκB and enhanced IRF3 activation following stroke 26, and preconditioning with either LPS or CpG enhanced the type I IFN genomic response to stroke injury11, 19. This suggests that TLR4 and TLR9 preconditioning-induced neuroprotection reprograms the brain’s damaging TLR4 response to stroke leading to a protective effect. We postulated that a TLR7 ligand would also provide protection from brain ischemia because it induces similar reprogramming of TLR4 signaling in macrophages.
Here we show that systemic administration of the TLR7 ligand, GDQ, prior to stroke reduced ischemic injury and induced the IFN-associate genes (Usp18, Oasl2, Isg15, Ifit1) previously identified following stroke in LPS- and CpG-preconditioned mice11, 19. Thus, as with LPS and CpG preconditioning, GDQ appears to reprogram the TLR response to stroke resulting in enhanced induction of type I IFN gene regulation. The presence of this IFN-dominated response to stroke in the context of GDQ preconditioning is evidence of a potential neuroprotective state that is similar to that induced via TLR4- and TLR9-mediated preconditioning.
The precise molecular mechanism initiated by preconditioning that enables the reprogramming of the TLR response is not clear. While LPS and CpG preconditioning depend on the induction of TNF10, 18, we show a preconditioning dose of GDQ failed to induce TNF, and more importantly, TNF-deficient mice preconditioned with GDQ displayed a similar reduction in infarct size as wildtype mice. Thus, although TLR7 signaling induces reprogramming and provides neuroprotection against brain ischemia, TNF is not required. This suggests that although multiple TLR ligands can induce neuroprotection through genomic reprogramming and induction of type I IFN genes, the molecular pathways leading to the protective phenotype are not identical.
We have recently published that LPS and CpG preconditioning depend on the transcription factors IRF3 and IRF711, 19, which are key modulators of the type I IFN response27, 28. Thus, we postulated that since TNF was not required for GDQ-preconditioning and TLR7 stimulation leads to robust production of IFNα, the mechanism underlying TLR7 preconditioning may be based on interferon regulation. We found that our preconditioning dose of GDQ induced a significant increase in serum IFNα, and that the increase in IFNα was functionally relevant since IFNAR−/− mice were not protected by GDQ preconditioning. Importantly, the IFNAR−/− mice could be protected by preconditioning with either LPS or CpG, implying that the mechanism of protection involving IFNAR is unique to TLR7. Further, we report that IRF7 is required for GDQ-induced neuroprotection. We suggest this occurs through TLR7-driven activation of IRF7 and subsequent induction of IFNα because IRF7−/− mice failed to induce IFNα and were unable to be protected against ischemia in response to GDQ. These results implicate a new mechanism of TLR-induced preconditioning in which TLR7 initiates a pathway of protection driven by IRF7 induction of IFNα and activation of the type I IFN receptor culminating in a reprogrammed TLR response to injury.
The mechanism by which IFNβ is involved in the TLR7-mediated reprogramming of the response to ischemic injury is unclear. However, work in macrophages may provide some insight. Similar to our current results, it has previously been shown that TLR4 signaling in response to LPS was altered following IFNα treatment, wherein type I IFN and IRF gene regulation were enhanced29. The alteration of TLR4 signaling was induced by pretreatment of macrophages with IFNα, which resulted in increased TRIF as well as downstream molecules IKKβ and IRF7. Such regulation is similar to our findings showing the effect of GDQ preconditioning on the genomic response to stroke injury. In addition, systemic IFNα can induce central nervous system upregulation of IRF genes30, suggesting that IFNα may be able to cross the blood-brain-barrier (BBB) to elicit these responses. Thus, in our model, GDQ preconditioning-induced neuroprotection may occur through the induction of systemic IFNα that in turn crosses the BBB to affect the brain’s endogenous TLR4 response to ischemia.
In conclusion, we describe the novel finding that tolerance to ischemic brain injury can be induced by prior systemic administration of the TLR7 ligand, GDQ. TLR7-mediated preconditioning results in new IFN-associated gene regulation in response to ischemic injury, which mirrors the TLR-reprogrammed response to stroke that we have previously reported for TLR4 and TLR9 preconditioning11, 19. These findings support the postulate that TLR reprogramming is an endogenous process capable of providing protection against subsequent TLR-mediated stroke injury. However, in contrast to TLR4 and TLR9 preconditioning that depend on the pro-inflammatory cytokine TNF10, 18, TLR7-induced neuroprotection is independent of TNF. Instead, TLR7-induced neuroprotection relies on a novel mechanism of IRF7-mediated induction of IFNα and signaling through the type I IFN receptor. These findings demonstrate that at least two different pathways participate in TLR-induced protection against ischemic injury, providing two distinct targets for the development of therapeutic interventions against stroke injury.
Acknowledgments
We would like to thank Dr. Keri Vartanian for her thoughtful comments on the manuscript.
Source of Funding: NIH R01 NS062381-01 and MRF Tartar Trust Fellowship (OHSU).
Footnotes
Disclosures: None.
References
- 1.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 2.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, et al. Toll-like receptor 2 mediates cns injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, et al. Pivotal role for neuronal toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke. 2008;39:1314–1320. doi: 10.1161/STROKEAHA.107.498212. [DOI] [PubMed] [Google Scholar]
- 5.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 6.Brea D, Blanco M, Ramos-Cabrer P, Moldes O, Arias S, Perez-Mato M, et al. Toll-like receptors 2 and 4 in ischemic stroke: Outcome and therapeutic values. J Cereb Blood Flow Metab. 2011 doi: 10.1038/jcbfm.2010.231. advance on-line publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broad A, Kirby JA, Jones DE. Toll-like receptor interactions: Tolerance of myd88-dependent cytokines but enhancement of myd88-independent interferon-beta production. Immunology. 2007;120:103–111. doi: 10.1111/j.1365-2567.2006.02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, et al. Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:959–967. doi: 10.1038/sj.jcbfm.9600086. [DOI] [PubMed] [Google Scholar]
- 9.Ridder DA, Schwaninger M. Nf-kappab signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, et al. Endotoxin preconditioning protects against the cytotoxic effects of tnfalpha after stroke: A novel role for tnfalpha in lps-ischemic tolerance. J Cereb Blood Flow & Metab. 2007;27:1663–1674. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- 11.Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: A critical role for irf3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veldhuis WB, Derksen JW, Floris S, van der Meide PH, de Vries HE, Schepers J, et al. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow & Metab. 2003;23:1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6. [DOI] [PubMed] [Google Scholar]
- 13.Liu H, Xin L, Chan BPL, Teoh R, Tang BL, Tan YH. Interferon beta administration confers a beneficial outcome in a rabbit model of thromboembolic cerebral ischemia. Neurosci Lett. 2002;327:146–148. doi: 10.1016/s0304-3940(02)00371-3. [DOI] [PubMed] [Google Scholar]
- 14.Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, et al. Preconditioning with a tlr2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 16.Hickey EJ, You X, Kaimaktchiev V, Stenzel-Poore M, Ungerleider RM. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents the cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 18.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: A new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. Journal of Neuroscience. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type i interferon response to mature and induce cd4+ th1 immunity with poly ic as adjuvant. J Exp Med. 2009;206:1589–1602. doi: 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurological Research. 1997;19:641–648. doi: 10.1080/01616412.1997.11740874. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, et al. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods. 2002;117:207–214. doi: 10.1016/s0165-0270(02)00114-0. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD. Chronic behavioral testing after focal ischemia in the mouse: Functional recovery and the effects of gender. Exp Neurol. 2004;187:94–104. doi: 10.1016/j.expneurol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Schon MP, Schon M, Klotz KN. The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a tlr7- and tlr8-independent fashion. J Invest Dermatol. 2006;126:1338–1347. doi: 10.1038/sj.jid.5700286. [DOI] [PubMed] [Google Scholar]
- 25.Reiter MJ, Testerman TL, Miller RL, Weeks CE, Tomai MA. Cytokine induction in mice by the immunomodulator imiquimod. J Leukoc Biol. 1994;55:234–240. doi: 10.1002/jlb.55.2.234. [DOI] [PubMed] [Google Scholar]
- 26.Vartanian KB, Stevens SL, Marsh B, Williams-Karnesky RL, Lessov NS, Stenzel-Poore MP. Lps preconditioning redirects tlr signaling following stroke: Trif-irf3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation. 2011 doi: 10.1186/1742-2094-8-140. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, et al. Myd88-dependent and myd88-independent pathways in synergy, priming, and tolerance between tlr agonists. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (irf)-3 and irf-7 in activation of the interferon-beta gene promoter. Eur J Biochem. 2004;271:3693–3703. doi: 10.1111/j.1432-1033.2004.04310.x. [DOI] [PubMed] [Google Scholar]
- 29.Siren J, Pirhonen J, Julkunen I, Matikainen S. Ifn-alpha regulates tlr-dependent gene expression of ifn-alpha, ifn-beta, il-28, and il-29. J Immunol. 2005;174:1932–1937. doi: 10.4049/jimmunol.174.4.1932. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Campbell IL, Zhang H. Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry. 2008;13:293–301. doi: 10.1038/sj.mp.4002013. [DOI] [PubMed] [Google Scholar]