

Abstract
Background
We describe a novel application of microarray technology for comparative genomics of bacteria in which libraries of entire genomes rather than the sequence of a single genome or sets of genes are arrayed on the slide and then probed for the presence or absence of specific genes and/or gene alleles.
Results
We first adopted a 96-well high throughput working protocol to efficiently isolate high quality genomic DNA. We then optimized conditions to print genomic DNA onto a glass slide with high density (up to 15000 spots) and to sensitively detect gene targets in each genome spot using fluorescently labeled DNA probe. Finally, we created an E. coli reference collection array and probed it for the presence or absence of the hemolysin (hly) gene using a dual channel non-competing hybridization strategy. Results from the array hybridization matched perfectly with previous tests.
Conclusions
This new form of microarray technology, Library on a Slide, is an efficient way for sharing and utilizing large strain collections in comparative genomic analyses.
Background
Bacteria produce a spectrum of genetic variants that contribute to diverse host specificity and pathogenicity. The genetic variants are not only marked by within-species variation in gene sequences, but most importantly, by their specific gene content. Even strains of the same species may differ by as much as 25% in genetic material [1,2]. Horizontal transferred genes from same or related species, different gene alleles, transposon or phage-related sequences, and extrachromosomal elements contribute to these differences. Each difference may be important for an organism's specific life style and virulence potential. The presence or absence of pathogenicity islands [3,4] on the genomes of pathogenic strains is one example of gene content defining biological properties. Comparing gene frequencies among bacterial isolates collected from different sources, e.g., disease causing and commensal isolates, serves as a valuable strategy to gain insight into the relative importance of a gene sequence in pathogenesis, transmission and other biologically significant properties [5]. The populations studied, and the number of isolates are important in determining the significance of observations made and the power to detect associations. These comparisons are currently accomplished by membrane-based dot blot screening a relatively low throughput process.
Identifying the function and biological significance of bacterial genes and their alleles is fundamental to interpreting data derived from genomic studies. Microarray technology has proven to be a powerful tool in this regard. Current DNA microarray platforms are used to gain insights into gene function and gene interactions using two experimental paradigms: 1) mRNA profiling to provide a global survey of gene activity; and 2) comparative genome scans for global surveys of genetic variants [6-8]. Since current arrays contain probe sequences representing all or most genes of a single annotated genome, genome scans are limited to the genetic features present in the arrayed reference strain. Given the substantial differences among the sequence repertoires of various strains of a single species [9], we have yet to reach a truly comprehensive genome scan for any given bacterial species. In addition, the associated cost and complexity of data acquisition of current microarray platform limits these studies to a small number of samples.
While comparative genome scanning has given us numerous insights into both the evolution of pathogens and overall differences between pathogenic and commensal organisms of the same species [8,10,11], the study of larger numbers of strains is required to determine the relative frequency of various genes within a species and to gain insight into their association with pathogenesis or transmission. Large population-based samples are required to minimize the identification of spurious associations that often arise with small and convenient sample comparisons. Bringing the dot blot hybridization technique onto the glass microarray platform will not only increase the capacity of this valuable traditional method but also add an experimental paradigm to current microarray based comparative genomics of bacteria.
For this new experimental paradigm, Library on a Slide, libraries of entire bacterial genomes rather than sequence of a single genome or sets of genes are arrayed on the slide and then probed for presence or absence of specific genes and or gene alleles. In practice, up to 30,000 isolates might be arrayed on a single slide, facilitating global surveys of bacterial populations. In this study, we tested feasibility and robustness of this platform.
Results and discussion
DNA preparation and array printing
Given the already heterogeneous nature of DNA fragments within a total bacterial genomic preparation, for successful printing in the Library on a Slide technique we used highly purified DNA. We tested various DNA purification methods including both organic extraction and non-organic extraction based on membrane or resin. High quality DNA were obtained from all these methods that were suitable for array printing, but bead beating based lysing followed by a commercial DNA purification column worked most consistently well for both Gram negative and Gram positive bacteria (data not shown).
To facilitate high throughput, we adapted a 96 well format DNA isolation kit from MO BIO laboratories to accommodate a large number of strains. This system combines bead beating lysis with a vacuum based membrane column. Its column, however, can be easily clogged by precipitated debris and proteins, which are difficult to avoid during multichannel pipetting. We added an additional step to remove these particles using a 96 well MultiScreen lysate clearing plate before loading the column. To concentrate eluted DNA, we used a MultiScreen PCR plate in a 96 well format. When purified DNA was directly used for printing the array, we observed very weak hybridization signals due to inefficient binding of long DNA molecules. To decrease the viscosity of the DNA solution and to improve the spread and binding of genomic DNA to the glass slides, the bacterial genomic DNA was fragmented. For high throughput operation, DNAs were fragmented to about 2 kb on average by sonication within wells of a 96 well microplate on a microplate horn. Since no probe was inserted into the DNA sample, this fragmentation process did not lead to sample loss or contamination. For convenience, we mixed DNA samples with 2 × commercial printing buffer and printed them onto ammine modified slides. We used solid and stealth pins to produce low and high density microarray, respectively. We found that ~30,000 spots on a 20 × 60 mm glass surface was the maximum density we could reach for a library array for signal detection (data not shown).
Array hybridization and detection
Library on a Slide is based on established cDNA glass microarray fabrication and hybridization techniques, with the novel adaptation that complex total bacterial genomic DNA is printed on the slide instead of homogenous DNA of single genes, and the target fragment (sequence being interrogated by the probe) represent a tiny fraction of the total genome fragments in each spot. Thus, detection sensitivity is a major concern. The hybridization signal is determined by both the target concentration in the spot and the quantity of the fluorescent tag carried by the probe. In standard microarray assays, fluorescent dye is incorporated into the DNA probe by an enzymatic reaction. The longer the probe, the more dye molecules it will eventually carry. We printed a test array with a two fold dilution series of a genomic DNA sample and hybridized it with either 1 kb or 7 kb Cy5 directly-labeled DNA probe. We found no hybridization signal gain beyond 1 ug/ul to 2 ug/ul of spotting concentration, indicating the binding capacity of the glass slide. The use of high DNA concentrations above that limit sometimes resulted in decreased signals due to washing off of DNA that was not directly bound during the hybridization process. Given the limited capacity of the glass surface for immobilizing DNA, 1 kb Cy5 labeled probe generated very weak signals under standard instrument settings. By increasing the laser power and detector sensitivity, measurable signals were obtained but with no valid dynamic range (Figure 1a). When the same array was hybridized with a 7 kb Cy5 labeled DNA probe, the hybridization signal was significantly increased due to a higher number of dye molecules incorporated into the hybridizing probe. A valid dynamic range, i.e. linear response of the signal intensity along the concentration gradient, was shown in the low concentration range (Figure 1b).
Figure 1.
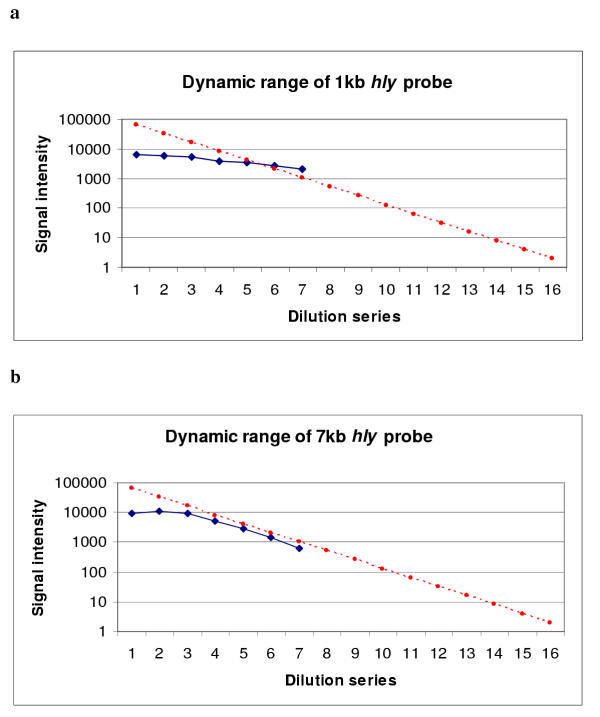
Signal intensities of a two fold genomic DNA dilution series probed with 1 kb (a) or 7 kb (b) direct labeled hly Cy5 probe. The blue dots represent spotting concentrations from 4 μg/ul to 0.125 μg/ul plus a negative control (the last spot in the series). The red line represents the simulated ideal signal responding line for a 2 fold dilution series that covers the whole signal spectrum of the scanner (16 bit image). This ideal responding slope is dictated by the logarithm of the dilution factor [log (0.5) = -0.3]. The background signal (represented by the last blue spot in the series) is much higher in 1 kb hybridization than that of 7 kb hybridization because the laser power and detector sensitivity setting of the scanner was increased in order to obtain analyzable signals.
In using probes ranging in size from a few hundred base pairs to 2 kb, signal amplification is necessary for detecting the target on the Library on a Slide. We tested the ability of both DNA dendrimer (3DNA reagent) and Tyramine Signal Amplification System (TSA) to increase detection sensitivity. A 3DNA dendrimer is a signal amplification molecule made from DNA. Each 3DNA molecule contains an average of 375 fluorescent dye molecules and can bind to any sized DNA probe with a capture sequence at its end. Theoretically, a dendrimer probe can generate signal intensity equivalent to that produced by a 10 kb directly-labeled DNA probe – assuming a dye incorporation rate of one per 25 bp during enzymatic reaction. However, we were not able to reach that signal level, although a 1 kb dendrimer probe generated a much higher signal than a 1 kb directly-labeled probe (Figure 2). Initially, the dendrimer probe was prepared using dsDNA fragment. However we were not able to obtain consistent strong signals with the dendrimer probe. We therefore prepared ssDNA dendrimer probe using a ssDNA fragment generated by λ exonulcease treatment. The single stranded dendrimer probe eliminated probe self hybridization, enhancing probe and target hybridization kinetics, and thus generated better and more consistent hybridization results on the Library on a Slide.
Figure 2.

A test array of the E. coli J96 genomic DNA (five different spotting concentrations with five replicates each) was hybridized with a Cy3 direct labeled 1 kb hly gene probe prepared with random priming (a), with a single stranded 1 kb hly gene fragment with a 5' capture sequence and detected by Cy3 DNA Dendrimer (b), and with fluorescein labeled 1 kb hly probe and detected with Tyramide Signal Amplification (TSA) system (c).
TSA is an enzyme-based secondary signal amplification system. The probe was first labeled with either fluorescein or biotin and the hybridized probe was then recognized by antibody-horseradish peroxidase conjugate which catalyzes the deposition of Cy3 or Cy5 labeled tyramide reagent. The TSA system produced much stronger signals than the dendrimer probe (Figure 2). Despite an elevated background compared to the dendrimer probe, as well as the need for extra incubation and washing steps, the TSA system was the best choice for our Library on a Slide hybridization.
E. coli test library array
As a proof of principle, we created a test Library on a Slide using the E. coli ECOR collection [12]. We created low density and high density version arrays, with ~2,000 and ~15,000 spots respectively, on a 22 mm × 60 mm surface by replicate spotting of these strains. Our goal was to screen these isolates for the presence or absence of E. coli virulence genes and compare them to previous results obtained by other methods. We present the results of hemolysin gene (hly) hybridizations as an example.
To detect the presence or absence of a gene sequence in each genome spot on the array, we compared signals of immobilized sample genomes to a positive control. It was therefore critical that the same number of copies of each genome be compared. Although all genomic DNA samples were suspended in the spotting buffer at the same concentration before arraying, they still could differ in genome copy number per spot due to genome size and plasmid content variations. In addition, exact amounts of DNA fixed in each spot could vary due to technical limitations during the printing and post-print processes. To insure equality, we took advantage of the multiplex labeling and detection features of the microarry platform and mutichannel laser scanner for measuring DNA quantity on the printed spots, employing a dual channel non-competing hybridization strategy. One channel detected signal from a quantification probe and the other for the probe of interest. We used the 16s ribosomal RNA gene, present in all strains of the E. coli species in the same copy number, as our quantification probe and was labeled with Cy5 dye. The other probe contained the DNA sequence of interest, hly, and was labeled with Cy3 dye. Since the genome quantification probe and the gene probe of interest recognize different target sequences, they can be used in the same hybridization process.
The hybridization result of each probe was obtained by scanning the slide at a different wavelength, since they were labeled with non interfering dyes that excite at different wave lengths (Figure 3). The 16s rRNA gene probe recognizes the same number of target sequences per genome of every sample. Therefore, its hybridization signal intensity was considered an indicator of genome quantity and used for hly hybridization signal adjustment using the Cy3/Cy5 signal ratio. The adjusted signal to the positive control ratio was determined and used to determine the presence or absence of the probe of interest, defined on the basis of a cutoff point established in our previously study [13]. Using a 50% cutoff point, twelve strains were identified as hly gene positive; this was 100% congruent with previous established known results based on our previous dot blot and Southern hybridization experiments (unpublished results).
Figure 3.
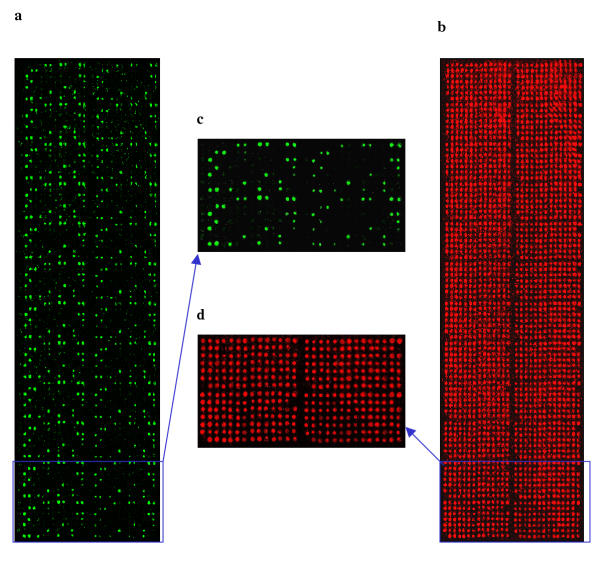
An E. coli reference collection (ECOR) library array was probed for the presence or absence of the hemolysin gene (hly). A green fluorescence labeled hly probe (a) and a red fluorescence labeled quantification probe, the16s rRNA gene, (b) were simultaneously used in hybridization. The array has 2352 spots representing 24 replications of 72 ECOR strains plus controls. (c) and (d) are four sub-grids of (a) and (b), respectively, each with 98 spots.
When the adjusted percentage signal intensity relative to the positive control of these strains was plotted, the two clusters around positive and negative control strains were more narrowly defined (Figure 4) than using the unadjusted intensities. Therefore, the normalization process led to more robust classification since these two clusters were more separated. It was also of interest to note that two strains had a hybridization signal intensity almost twice the positive control, one of which was a control strain known to have two copies of hly gene clusters. The other also appeared to have two copies of hly gene cluster based on previous Southern hybridization (data not shown).
Figure 4.
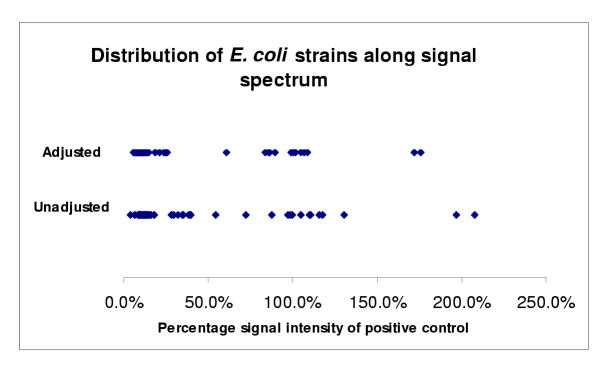
Scatter plots of average percentage signal intensities relative to the positive control of hly probed ECOR strains and controls from Library on a Slide hybridization. The top plot was based on the 16sRNA probe signal adjusted percentage of hly signal compared to positive control and the bottom plot on the unadjusted percentage of hly signal compared to positive control. Strains with >50% signal intensities were proven positive (i.e. contain hly gene) previously by Southern hybridization. Signal variability of hly containing strains may be due to the variability of hybridizations and/or sequence variations among hly genes.
Conclusions
Library on a Slide provides an extremely high throughput and robust platform for comparative genomics studies. Printing and probing the Library on a Slide is similar to current cDNA microarray fabrication and hybridization techniques; the most significant difference is that complex total bacterial genomic DNA is printed on the slide whereas single homogenous DNA molecules are printed on the slide in cDNA array techniques. The technical challenge for Library on a Slide is the sensitivity of target detection in each complex genome spot. Our study demonstrates that Library on a Slide is a viable screening platform with currently available array detection technology. The adaptations of fluorescent probes also provide ways for multiplex probing and in-spot DNA quantification beyond that in traditional dot blot hybridization. With the development of more dyes and more capable scanners, multiple gene screening can be accomplished in a single experiment. We are in the process of adapting Library on a Slide technology in our study of Escherichia coli, Group B Streptococcus, and Haemophilus influenzae. It will enhance our ability to screen large bacterial populations to identify genes associated with virulence and transmission.
While we currently use the Library on a Slide for determining absence or presence of a gene or part of a gene, improvement in hybridization signal and glass surface chemistry will enable us to scan for finer sequence variations. For example, it is possible to print the array on a three-dimensional gel matrix [14] that can be used to perform an array primer extension for detecting a single base mutation.
A single Library on a Slide can also be constructed with isolates from several related species, or species that are part of a microbial ecosystem such as rumen. Such platform will enable us to examine the extent of shared genetic elements across species – especially horizontally transferred virulence factors and antibiotic resistance genes. More importantly, comprehensive Library on a Slide can be produced in large quantities and made available to other investigators. Library on a Slide will be an efficient and cost effective way for sharing and utilizing large strain collections in various comparative genomics studies.
Determining the gene content of isolates and correlating these with epidemiological and clinical information is of considerable interest to epidemiologists and microbial population geneticists. Library on a Slide provides an additional experimental paradigm complementary to current array-based comparative genomics of bacteria. Current gene microarray-based applications aim at parallel examination of a maximum number of DNA or RNA features of a single (or few) organism samples. Library on a Slide aims at applications that screen a maximum number of samples for the presence (or variation) of specific genetic elements of interest. Both serve the same purpose, that is, to understand the organism's phenotype in relation to its underling genotype.
Methods
DNA isolation and arraying
QIAGEN Genomic-tip 20/G (Qiagen, CA), UltraClean microbial DNA kit (MoBio, CA), and Wizard Genomic DNA purification kit (Promega, WI) with an additional phenol extraction step were used to isolate DNA if limited number of strains were involved. For DNA isolation from a large number of strains, the UltraClean-htp 96 well microbial DNA kit (Mobio) combined with MultiScreen Plate (Millipore, MA) was used. The DNA concentration was determined by UV absorbance (260 nm) reading. DNA was fragmented using Sonicator 3000 with a plate horn (Misonix, NY) at amplitude setting of 10 for 8 min (rest 1 min for every 1 min on). A VersArray ChipWriter Compact system (Bio-Rad, CA) was used to spot DNA onto SuperAmine glass slides (TeleChem, CA) using either solid for low density printing and stealth pin for high density printing.
Probe labeling and array hybridization
Random priming was used to incorporate Cy3, Cy5, fluorescein, or biotin into dsDNA probes using the BioPrime DNA labeling system (Invitrogen, CA) with appropriate dNTP mixtures. To prepare ssDNA used as dendrimer probe, DNA was first amplified by a pair of gene-specific primers. One primer had a manufacture specified capture sequence at 5' end and the other had a phosphorylated 5' end. The dsDNA PCR product was then treated with λ exonuclease (Strandase Kit from Novagen, WI) to digest one strand of duplex DNA from the 5' phosphorylated end to generate ssDNA probe. All labeled probes were cleaned with a Qia-quick PCR purification kit (Qiagen). To prepare the hybridization mixture, 500 ng probes and 2 ug denatured salmon sperm DNA were mixed with 1.25 × HybIt buffer (Telechem) to a final volume of 50 ul for each slide. The probes were denatured at 95°C for 3 min and pipetted onto arrays, cover slips were applied, and the slides were placed in a hybridization chamber (Corning, NY). Arrays were incubated at 63°C water bath for 18–24 hr, and subsequently washed according to the manufacture's suggestion. A 3DNA Submicro Expression Array Detection Kit (Genisphere, PA) was used for subsequent dendrimer hybridization and a MICROMAX TSA labeling and detection kit (PerkinElmer, MA) was used for TSA signal amplification. In both cases, manufacture's protocols were followed. Detailed information of these two labeling and detection systems can be found at http://www.genisphere.com/array_detection_faqs.html and http://las.perkinelmer.com/catalog/Category.aspx?CategoryName=MICROMAX, respectively.
Array scanning and data acquisition
Arrays were scanned with a VersArray ChipReader (Bio-Rad, CA) at 10 μm resolution and variable photomulipier tube (PMT) voltage settings to obtain the maximal signal intensities with no saturation. In case of comparing signals of different hybridization conditions, the PMT and sensitivity setting of the scanner were kept at the same level. The resulting images were analyzed using either accompanied VersArray Analyzer software or ImageQuant Version 5.2 (Molecular Dynamics). To determine the presence or absence of hly gene (Cy3 signal) on the ECOR array, the percentage signal intensity relative to the positive control of each strain was calculated both with and without DNA concentration adjustment based on 16s rRNA gene hybridization signal (Cy5 signal). The unadjusted percentage was calculated as Cy3 signal of the sample dividing by the average Cy3 signal of the positive controls. The adjusted percentage was calculated as Cy3/Cy5 signal ratio of the sample timing the average Cy5 signal of the positive control then dividing by average Cy3 signal of the positive control. Based on an early study [13], 50% was used as cutoff point for differentiating hly positive and negative strain. The 50% threshold was the optimal breakpoint for classifying for the presence or absence of hly gene. It was established by examining the sensitivity and specificity of different classification criteria [13].
Authors' contributions
All authors participated in study design and interpretation of the results. LZ and US carried out the experiments. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
This research was support in part by a Bioinformatics Program University of Michigan Medical School Pilot Grant awarded by Howard Hughes Medical Institute (LZ), and NIH grants AI054406 (LZ), DK 055496 (CFM), AI51675 (BF) and DC005840 (JRG). We thank the University of Michigan Office of the Vice President for Research and School of Public Health for their assistance in establishing a microarray facility.
Contributor Information
Lixin Zhang, Email: lxzhang@umich.edu.
Usha Srinivasan, Email: usha@umich.edu.
Carl F Marrs, Email: cfmarrs@umich.edu.
Debashis Ghosh, Email: ghoshd@umich.edu.
Janet R Gilsdorf, Email: gilsdorf@med.umich.edu.
Betsy Foxman, Email: bfoxman@umich.edu.
References
- Bergthorsson U, Ochman H. Heterogeneity of genome sizes among natural isolates of Escherichia coli. J Bacteriol. 1995;177:5784–5789. doi: 10.1128/jb.177.20.5784-5789.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthorsson U, Ochman H. Distribution of chromosome length variation in natural isolates of Escherichia coli. Mol Biol Evol. 1998;15:6–16. doi: 10.1093/oxfordjournals.molbev.a025847. [DOI] [PubMed] [Google Scholar]
- Lee CA. Pathogenicity islands and the evolution of bacterial pathogens. Infect Agents Dis. 1996;5:1–7. [PubMed] [Google Scholar]
- Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Foxman B, Manning SD, Tallman P, Marrs CF. Molecular epidemiologic approaches to urinary tract infection gene discovery in uropathogenic Escherichia coli. Infect Immun. 2000;68:2009–15. doi: 10.1128/IAI.68.4.2009-2015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington CA, Rosenow C, Retief J. Monitoring gene expression using DNA microarray. Curr Opin Microbiol. 2000;3:285–291. doi: 10.1016/S1369-5274(00)00091-6. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JR, Musser JM. Evolutionary genomics of pathogenic bacteria. Trends Microbiol. 2001;9:547–553. doi: 10.1016/S0966-842X(01)02228-4. [DOI] [PubMed] [Google Scholar]
- Schoolnik GK. Functional and comparative genomics of pathogenic bacteria. Curr Opin Microbiol. 2002;5:20–26. doi: 10.1016/S1369-5274(02)00280-1. [DOI] [PubMed] [Google Scholar]
- Dougan G, Haque A, Pickard D, Frankel G, O'Goara P, Wain J. The Escherichia coli gene pool. Curr Opin Microbiol. 2001;4:90–94. doi: 10.1016/S1369-5274(00)00170-3. [DOI] [PubMed] [Google Scholar]
- Welch RA, Burland V, Plunkett G, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HL, Donnenberg MS, Blattner FR. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci USA. 2002;99:17020–4. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittam TS, Bumbaugh AC. Inferences from whole-genome sequences of bacterial pathogens. Curr Opin Genet Dev. 2002;12:719–25. doi: 10.1016/S0959-437X(02)00361-1. [DOI] [PubMed] [Google Scholar]
- Ochman H, Selander RK. Standard reference strains of Escherichia coli from nature populations. J Bacteriol. 1984;157:690–693. doi: 10.1128/jb.157.2.690-693.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Gillespie BW, Marrs CF, Foxman B. Optimization of a Fluorescent-based phosphor imaging dot blot DNA hybridization assay to assess E. coli virulence gene profiles. J Microbiol Method. 2001;44:225–233. doi: 10.1016/S0167-7012(01)00222-6. [DOI] [PubMed] [Google Scholar]
- Barsky VE, Kolchinsky AM, Lysov YuP, Mirzabekov AD. Biological microchips with hydrogel-immobilized nucleic acids, proteins, and other compounds: properties and applications in genomics. Mol Biol. 2002;36:437–455. doi: 10.1023/A:1019804005711. [DOI] [PubMed] [Google Scholar]