

Abstract
Alkaline phosphatase (AP) isozymes are present in a wide range of species from bacteria to man and are capable of dephosphorylation and transphosphorylation of a wide spectrum of substrates in vitro. In humans, four AP isozymes have been identified—one tissue-nonspecific (TNAP) and three tissue-specific—named according to the tissue of their predominant expression: intestinal (IAP), placental (PLAP) and germ cell (GCAP) APs. Modulation of activity of the different AP isozymes may have therapeutic implications in distinct diseases and cellular processes. For instance, changes in the level of IAP activity can affect gut mucosa tolerance to microbial invasion due to the ability of IAP to detoxify bacterial endotoxins, alter the absorption of fatty acids and affect ectopurinergic regulation of duodenal bicarbonate secretion. To identify isozyme selective modulators of the human and mouse IAPs, we developed a series of murine duodenal IAP (Akp3-encoded dIAP isozyme), human IAP (hIAP), PLAP, and TNAP assays. High throughput screening and subsequent SAR efforts generated a potent inhibitor of dIAP, ML260, with specificity for the Akp3-, compared to the Akp5- and Akp6-encoded mouse isozymes.
Alkaline phosphatases (APs) are well-studied enzymes known for their ability to dephosphorylate a wide spectrum of substrates.1 In humans, four AP isozymes have been identified, classified on their predominant tissue localization: intestinal (IAP), placental (PLAP) and germ cell (GCAP) APs. The expression of the fourth isozyme, designated as tissue-nonspecific alkaline phosphatase (TNAP), is ubiquitous, at high levels in the developing neural tube, kidney, liver and mineralizing tissues such as cartilage and bone. Its function is intimately associated with mineralization of skeletal and dental tissues, as deficiency in TNAP function in humans and mice leads to a heritable form of rickets/osteomalacia known as hypophosphatasia1. Mice also have four active AP genes: Alpl (encoding TNAP), the Akp5 gene encoding embryonic AP (EAP) and two genes expressed in the gut, Akp3 and Akp6, encoding a duodenal specific IAP (dIAP) and a globally expressed IAP (gIAP), respectively.1
Recent work using Akp3 knockout mice indicates that dIAP facilitates fat absorption2, 3, maintains gut barrier function4-6 and affects the composition of the gut microbiota.7 Many studies in the literature also relate human IAP with diarrhea-predominant diseases, such as inflammatory bowel disease (IBD) or pathogenic infections. Wada et al. reported that infection with Aeromonas sobria hemolysin causes diarrhea; IAP, by binding hemolysin appears to be involved with its pathogenesis.8 In IBD, genetic and environmental factors along with chronic deregulation of the host immune system response to gut flora appear to play key roles in its pathogenesis.9-11
Exogenous purified IAP may be useful therapeutically for these conditions. IAP may detoxify bacterial products such as lipopolysaccharide (LPS), reducing excessive intestinal inflammation12. For example, the naso-duodenal delivery of calf IAP to ulcerative colitis (UC) patients improved clinical and serological measures.13 More recently, we showed that endogenous IAP likely protects the host from IBD, since oral supplementation of IAP ameliorates clinical signs and symptoms of IBD in two mouse models of chronic colitis6 and prevents metabolic syndrome in Akp3−/− mice.14 Despite the ability of IAP enzyme to detoxify LPS, how IAP affects intestinal inflammation has not been fully elucidated. Knowledge of this mechanism would thus be a key factor for the development of a successful therapy for the treatment of IBD patients. More importantly, immunomodulatory therapy of IBD patients is associated with severe side effects.15
In the present study, we describe a multi-pronged screening approach that enabled the identification of dIAP inhibitors. SAR efforts based on parallel testing of analogs against different AP isozymes generated a potent inhibitor of the murine dIAP with IC50 = 540 nM, at least 65-fold more selective against human IAP than TNAP, and >185-fold more selective than PLAP. Furthermore, the inhibitor proved to be selective against the Akp3 encoded dIAP but not the Akp5- or Akp6-encoded EAP and gIAP isozymes. These compounds are likely to be useful tools in probing the functional roles of human and mouse IAPs during the bacterial endotoxins detoxifying process, absorption of fatty acids and bicarbonate secretion.
Identification of isozyme-specific inhibitors was part of a platform-based approach where the entire NIH's small molecule collection (MLSMR) was interrogated against dIAP and hIAP isozymes in parallel, while assessment of selectivity against TNAP and PLAP isozymes was based on the results of prior screening campaigns.17 This parallel screening strategy, using the same CDP Star® luminescent assay format, not only afforded a direct comparison between several high-throughput screens, but also allowed an efficient elimination of the artifacts.
1536-well high throughput screens of MLMSR library comprising 330,480 compounds against dIAP and hIAP isozymes were conducted at 10 μM compound concentration, as described in PubChem (AID 2544). Ultimately only one compound, hit CID24790981 (Figure 1), was selective against TNAP and PLAP. CID24790981 has an IC50 = 1.82 μM in the dIAP assay and displays excellent selectivity against TNAP and PLAP.
Figure 1.
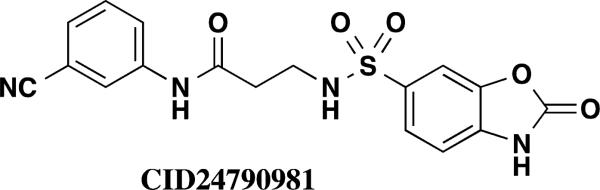
Screening hit
The general SAR strategy we pursued around this scaffold from the screening hit is depicted in Figure 2. We focused on changing the nature and number of the R1 substituents attached to the phenyl ring highlighted in yellow and we investigated changes in the chain length, increasing and decreasing the carbon chain length (n = 0, 1, 2, or 3) highlighted in red. Finally, we investigated if it is possible to replace the hydrogen atom at R2 by alkyl groups highlighted in green.
Figure 2.
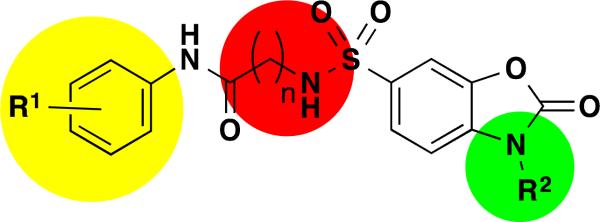
Overall SAR strategy
We developed an efficient synthesis for our lead series of molecules that was straightforward and followed the general methods outlined in Scheme 1. Treatment of the commercially available sulfonyl chloride 1 with the tert-butyl 2-aminoacetate afforded the (sulfonamido)acetic acid 2. Removal of the boc-protecting group of compound 2 with trifluoracetic acid afforded the free acid 3 in excellent yields. Coupling of acid 3 with various amines 4 produced the desired dihydrobenzo[d]oxazole compounds 5 directly.
Scheme 1.

Synthesis of 5, conditions: a. dichloromethane, triethylamine, (70 - 88% yield); b. trifluoroacetic acid, dichloromethane, 0°C warm to RT (100% yield); c. EDC, HOBT, NMM, DMF, (40-55%)
The results of our efforts are summarized in Table 2 below. Initially we focused our SAR on the R1 group in Figure 3, where n = 2. Generally, mono substituents, either electron donating or electron withdrawing, are inactive (Entries 27 to 42). Interestingly, we found the position of substituents on the phenyl ring was critical for activity. For example entry 17 and entry 34 both contain a di-methyl substituent on the phenyl ring, and this scaffold greatly prefers the 2,5 substitution pattern of entry 17 over the 3,4 substitution pattern of entry 34. We found the most active compounds contain either a 2,5-di-methylphenyl, 3,4-di-methoxyphenyl, 4-methoxybenzyl, 3-cyanobenzyl or methylene-di-oxybenzyl substituents (Entries 17 – 21). Most of these compounds display little activity against TNAP or PLAP with IC50 values >100 μM. Methylation of the nitrogen atom of the dihydrobenzo[d]oxazole heterocycle completely eliminates the activity (Entry 34). We next focused our attention on increasing the chain length by one carbon atom, where n = 3. Generally these compounds are less active than comparable compounds in the n = 2 series of molecules. (Entries 50 – 61). The series of compounds where n = 0 produced very disappointing results as all compounds are inactive with IC50 values ranging from 40 to >100 μM. (Entries 1-8 and 16).
Table 2.
SAR elucidation of dihydrobenzo[d]oxazole series (n = chain length)
| Entry | R1 | R2 | n | IC50 (μM) | ||
|---|---|---|---|---|---|---|
| muIAP | TNAP | PLAP | ||||
| 1 | 3-OMe-benzyl | H | 0 | 40 | >100 | >100 |
| 2 | 2,4-di-OMe-benzyl | H | 0 | 89 | >100 | >100 |
| 3 | 4-OMe-benzyl | H | 0 | 98 | >100 | >100 |
| 4 | 3-CN-phenyl | H | 0 | >100 | >100 | >100 |
| 5 | 4-CN-phenyl | H | 0 | >100 | >100 | >100 |
| 6 | 2,5-di-Me-phenyl | 0 | >100 | >100 | >100 | |
| 7 | 3,4-di-OMe-phenyl | H | 0 | >100 | >100 | >100 |
| 8 |
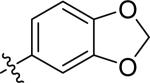
|
H | 0 | >100 | >100 | >100 |
| 9 ML260 |
2,5-di-Me-phenyl | H | 1 | 0.54 | 35 | >100 |
| 10 | 3-OMe-benzyl | H | 1 | 0.73 | 74 | >100 |
| 11 | 4-OMe-benzyl | H | 1 | 2.1 | >100 | >100 |
| 12 | 4-CN-phenyl | H | 1 | 4.7 | 81 | >100 |
| 13 | 3-CN-phenyl | H | 1 | >100 | >100 | >100 |
| 14 | 2,4-di-OMe-benzyl | H | 1 | >100 | >100 | >100 |
| 15 |
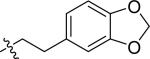
|
H | 1 | >100 | >100 | >100 |
| 16 |
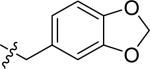
|
H | 1 | >100 | >100 | >100 |
| 17 | 2,5-di-Me-phenyl | H | 2 | 0.4 | >100 | >100 |
| 18 | 3,4-di-OMe-phenyl | H | 2 | 0.53 | 37 | >100 |
| 19 | 3-CN-benzyl | H | 2 | 2 | >100 | >100 |
| 20 | 4-OMe-benzyl | H | 2 | 2.3 | >100 | >100 |
| 21 |
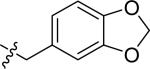
|
H | 2 | 2.6 | >100 | >100 |
| 22 | 3,4-di-hydroxybenzyl | H | 2 | 7.7 | >100 | >100 |
| 23 |
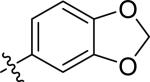
|
H | 2 | 12 | >100 | >100 |
| 24 | 3-F-benzyl | H | 2 | 66 | >100 | >100 |
| 25 |
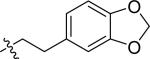
|
H | 2 | 19 | >100 | >100 |
| 26 |
|
H | 2 | 44 | >100 | >100 |
| 27 | 3-trifluoromethylbenzyl | H | 2 | 70 | >100 | >100 |
| 28 | benzyl | H | 2 | 81 | >100 | >100 |
| 29 | 3,4-dimethylphenyl | H | 2 | 84 | >100 | >100 |
| 30 | 4-chlorobenzyl | H | 2 | 88 | >100 | >100 |
| 31 | 4-methylbenzyl | H | 2 | 89 | >100 | >100 |
| 33 | 4-fluorobenzyl | H | 2 | >100 | >100 | >100 |
| 34 | 2,5-di-Me-phenyl | CH3 | 2 | >100 | >100 | >100 |
| 35 | 3-MeO-benzyl | H | 2 | >100 | >100 | >100 |
| 36 | 2-MeO-benzyl | H | 2 | >100 | >100 | >100 |
| 37 | 2-F-phenyl | H | 2 | >100 | >100 | >100 |
| 38 | 4-OH-benzyl | H | 2 | >100 | >100 | >100 |
| 39 | 4-CF3-benzyl | H | 2 | >100 | >100 | >100 |
| 40 | 3-F-benzyl | H | 2 | >100 | >100 | >100 |
| 41 | 2-Cl-benzyl | H | 2 | >100 | >100 | >100 |
| 42 | 3-Cl-benzyl | H | 2 | >100 | >100 | >100 |
| 43 |
|
H | 0 | >100 | >100 | >100 |
| 44 | 3,4-di-OMe-phenylethyl | H | 3 | 1.5 | 96 | >100 |
| 45 | 2-MeO-benzyl | H | 3 | 2 | >100 | >100 |
| 46 | 4-HO-phenethyl | H | 3 | 4.2 | >100 | >100 |
| 47 | 4-MeO-phenethyl | H | 3 | 7.4 | >100 | >100 |
| 48 | 2,4-di-MeO-phenyl | H | 3 | 6.3 | >100 | >100 |
| 49 | 3-F-benzyl | H | 3 | 13 | >100 | >100 |
| 50 | 2,4-di-MeO-benzyl | H | 3 | 37 | >100 | >100 |
| 51 |
|
H | 3 | 45 | >100 | >100 |
| 52 | 3,4-di-MeO-benzyl | H | 3 | 78 | >100 | >100 |
| 53 | 2-Cl-phenyl | H | 3 | >100 | >100 | >100 |
| 54 | 2-Me-benzyl | H | 3 | >100 | >100 | >100 |
| 55 | 3,4-di-OMe-phenyl | H | 3 | 1.5 | >100 | >100 |
Figure 3.
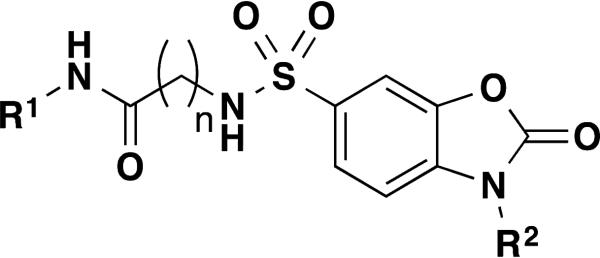
General structure for dihydrobenzo[d]oxazoles
Finally, we investigated a series of compounds where n = 1. We synthesized 8 analogs with the most potent analog containing the 2,5-di-methylphenyl group, entry 9. Consideration of the potency and in vitro selectivity data presented herein we nominated compound 9 as our probe molecule ML260 as it was the most potent dIAP inhibitor in this series while having excellent selectivity against both PLAP and TNAP. The multiple dose response titrations of ML260 against dIAP, TNAP and PLAP are shown in Figure 4.
Figure 4.

Potency and selectivity of ML260. The potency of the inhibitor against dIAP (o), PLAP (+) and TNAP (x) was assessed by 16pt dose-response testing in 1536-well format in duplicate in 1xAssay Buffer, containing 100 mM DEA, pH 9.8, 0.02 mM ZnCl2, 1 mM MgCl2, and 1:250, 1:1000 and 1:2000 diluted enzymes, respectively, in a total volume of 4 μL/well. The reactions were started by addition of the CDP-star substrate to the final concentrations of 200 μM for the dIAP reaction, and 250 μM for the PLAP and TNAP reactions, with the luminescence intensity measured after 30 min incubation at room temperature.
When the relative IAP selectivity was further tested using p-nitrophenyl phosphate as a substrate the Akp3-encoded dIAP was inhibited selectively over murine EAP and gIAP isozymes as well as the hIAP isozyme with an IC50 equivalent to 3.8 μM (Figure 5). Further analysis, using pNPP as a substrate revealed that ML260 inhibits dIAP competitively with an apparent Ki = 3.2 μM (Figure 6). For this reason, a concentration of 10 μM was selected to inhibit dIAP activity expressed by transfected Cos-1 cells. Figure 7 shows that the dIAP activity expressed in these cells could be fully inhibited with ML260 at 10 μM.
Figure 5.

Selectivity of intestinal isozyme inhibition by ML260. Dose-dependency of inhibition of dIAP (IC50 = 3.8 μM), versus inhibition of EAP (IC50 = 21.1 μM), gIAP (IC50 = 21 μM) and hIAP (IC50 = 27.6 μM), as indicated.
Figure 6.

Mechanism of dIAP inhibition by ML260. Double reciprocal plots of residual dIAP activity versus the concentration of pNPP, constructed for each of the indicated ML260 concentrations.
Figure 7.

CHO cells expressing dIAP protein, stained with ELF97 in the absence (A) or presence of 10 μM ML260 (C). (B) and (D) are phase contrast views of (A) and (C) respectively. Bar = 50 μm.
In conclusion, we have discovered the first selective murine IAP inhibitor, ML260, which represents a tool compound to further explore the functional role of dIAP in a variety of disease states, in particular as a gut mucosal defense factor18 and as a mediator in fatty acid absorption. Current efforts to examine the effects of IAP inhibition in these models are underway. 19
Supplementary Material
Table 1.
Summary of characteristic of initial hit CID24790981
| Entry | IC50 (μM) | |||
|---|---|---|---|---|
| dIAP | huIAP | TNAP | PLAP | |
| 1A | 1.82 | >100 | >100 | >100 |
Acknowledgements
This work was supported by NIH grants X01-MH077602-01 and R01 DE12889 to JLM and an NIH Molecular Libraries grant (U54HG005033-03) to the Conrad Prebys Center for Chemical Genomics at the Sanford Burnham Medical Research Institute, one of the comprehensive centers of the NIH Molecular Libraries Probe Production Centers Network (MLPCN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Millán JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Wiley-VCH Verlag GmbH & Co.; Weinheim, Germany: 2006. [Google Scholar]
- 2.Narisawa S, Hoylaerts MF, Doctor KS, Fukuda MN, Alpers DH, Millán JL. A novel phosphatase upregulated in Akp3 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293:G1068–1077. doi: 10.1152/ajpgi.00073.2007. [DOI] [PubMed] [Google Scholar]
- 3.Nakano T, Inoue I, Koyama I, Kanazawa K, Nakamura K, Narisawa S, Tanaka K, Akita M, Masuyama T, Seo M, Hokari S, Katayama S, Alpers DH, Millán JL, Komoda T. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G1439–1449. doi: 10.1152/ajpgi.00331.2006. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg RF, Austen WG, Jr., Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millán JL, Hodin RA. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3551–3556. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen KT, Malo MS, Beasley-Topliffe LK, Poelstra K, Millán JL, Mostafa G, Alam SN, Ramasamy S, Warren HS, Hohmann EL, Hodin RA. A role for intestinal alkaline phosphatase in the maintenance of local gut immunity. Dig. Dis. Sci. 2011;56:1020–1027. doi: 10.1007/s10620-010-1396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramasamy S, Nguyen DD, Eston MA, Alam SN, Moss AK, Ebrahimi F, Biswas B, Mostafa G, Chen KT, Kaliannan K, Yammine H, Narisawa S, Millán JL, Warren HS, Hohmann EL, Mizoguchi E, Reinecker HC, Bhan AK, Snapper SB, Malo MS, Hodin RA. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm. Bowel Dis. 2011;17:532–542. doi: 10.1002/ibd.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, Chen KT, Moss AK, Ramasamy S, Faruqui A, Hodin S, Malo PS, Ebrahimi F, Biswas B, Narisawa S, Millán JL, Warren HS, Kaplan JB, Kitts CL, Hohmann EL, Hodin RA. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut. 2010;59:1476–1484. doi: 10.1136/gut.2010.211706. [DOI] [PubMed] [Google Scholar]
- 8.Wada A, Wang AP, Isomoto H, Satomi Y, Takao T, Takahashi A, Awata S, Nomura T, Fujii Y, Kohno S, Okamoto K, Moss J, Millán JL, Hirayama T. Placental and intestinal alkaline phosphatases are receptors for Aeromonas sobria hemolysin. Int. J. Med. Microbiol. 2005;294:427–435. doi: 10.1016/j.ijmm.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 11.Mizoguchi A, Mizoguchi E, Bhan AK. Immune networks in animal models of inflammatory bowel disease. Inflamm Bowel Dis. 2003;9:246–259. doi: 10.1097/00054725-200307000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Poelstra K, Bakker WW, Klok PA, Hardonk MJ, Meijer DK. A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab Invest. 1997;76:319–327. [PubMed] [Google Scholar]
- 13.Lukas M, Drastich P, Konecny M, Gionchetti P, Urban O, Cantoni F, Bortlik M, Duricova D, Bulitta M. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm Bowel Dis. 2010;16:1180–1186. doi: 10.1002/ibd.21161. [DOI] [PubMed] [Google Scholar]
- 14.Kaliannan K, Hamarneh SR, Economopoulos KP, Nasrin Alam S, Moaven O, Patel P, Malo NS, Ray M, Abtahi SM, Muhammad N, Raychowdhury A, Teshager A, Mohamed MM, Moss AK, Ahmed R, Hakimian S, Narisawa S, Millan JL, Hohmann E, Warren HS, Bhan AK, Malo MS, Hodin RA. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2013;110:7003–7008. doi: 10.1073/pnas.1220180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozuch PL, Hanauer SB. Treatment of inflammatory bowel disease: a review of medical therapy. World J Gastroenterol. 2008;14:354–377. doi: 10.3748/wjg.14.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozlenkov A, Le Du M-H, Cuniasse P, Ny T, Hoylaerts MF, Millán JL. Residues determining the binding specificity of uncompetitive inhibitors to tissue-nonspecific alkaline phosphatase. J. Bone Miner. Res. 2004;19:1862–1872. doi: 10.1359/JBMR.040608. [DOI] [PubMed] [Google Scholar]
- 17.Chung TD, Sergienko E, Millán JL. Assay format as a critical success factor for identification of novel inhibitor chemotypes of tissue-nonspecific alkaline phosphatase from high-throughput screening. Molecules. 2010;15:3010–3037. doi: 10.3390/molecules15053010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moss AK, Hamarneh SR, Mohamed MM, Ramasamy S, Yammine H, Patel P, Kaliannan K, Alam SN, Muhammad N, Moaven O, Teshager A, Malo NS, Narisawa S, Millan JL, Warren HS, Hohmann E, Malo MS, Hodin RA. Intestinal alkaline phosphatase inhibits the proinflammatory nucleotide uridine diphosphate. Am J Physiol Gastrointest Liver Physiol. 2013;304(6):G597–604. doi: 10.1152/ajpgi.00455.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynes M, Narisawa S, Millán JL, Widmaier EP. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301(6):R1738–1747. doi: 10.1152/ajpregu.00235.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.