

Abstract
Protein arginylation by arginyl–transfer RNA protein transferase (ATE1) is emerging as a regulator protein function that is reminiscent of phosphorylation. For example, arginylation of β-actin has been found to regulate lamellipodial formation at the leading edge in fibroblasts. This finding suggests that similar functions of β-actin in other cell types may also require arginylation. Here, we have tested the hypothesis that ATE1 regulates the cytoskeletal dynamics essential for in vivo platelet adhesion and thrombus formation. To test this hypothesis, we generated conditional knockout mice specifically lacking ATE1 in their platelets and in their megakaryocytes and analyzed the role of arginylation during platelet activation. Surprisingly, rather than finding an impairment of the actin cytoskeleton structure and its rearrangement during platelet activation, we observed that the platelet-specific ATE1 knockout led to enhanced clot retraction and in vivo thrombus formation. This effect might be regulated by myosin II contractility since it was accompanied by enhanced phosphorylation of the myosin regulatory light chain on Ser19, which is an event that activates myosin in vivo. Furthermore, ATE1 and myosin co-immunoprecipitate from platelet lysates. This finding suggests that these proteins directly interact within platelets. These results provide the first evidence that arginylation is involved in phosphorylation-dependent protein regulation, and that arginylation affects myosin function in platelets during clot retraction.
Introduction
Arginyl-transfer RNA protein transferase (ATE) is an evolutionarily conserved enzyme. It is represented by four splice variants in mammals (ATE1-1, ATE1-2, ATE1-3 and ATE1-4), which are required for viability and participate in stress responses.1–3 ATE1 specifically catalyzes the transfer of arginine from tRNA to proteins.4 Post-translational arginylation was once thought to play a singular role in the N-end rule pathway of protein degradation.5–7 However, recent evidence shows that protein arginylation also regulates a wide variety of crucial biological processes, which include embryogenesis, cardiovascular development, angiogenesis, and neural crest cell migration.1,8–12 Yet, despite the contribution of ATE1 to these diverse cellular and developmental events, the molecular mechanism by which arginylation alters the function of proteins remains enigmatic.
Previous studies have shown that a large number of proteins that undergo arginylation in vivo are major structural constituents or regulators of the cytoskeleton.13,14 Furthermore, in fibroblasts, arginylation of β-actin regulates lamella formation at the leading edge during cell locomotion.12,15 In platelets, in which β-actin is the predominant isoform, rapid reorganization of the actin cytoskeleton is the earliest event in platelet activation, a process that is essential for the morphological changes that lead to shape change, granule release, and aggregation. We speculated that arginylation within platelets would regulate their cytoskeleton in a way similar to that seen in fibroblasts, and that such regulation would consequently contribute to the biomechanics of platelets in vivo.16,17
To test this hypothesis, we generated mice that contained a platelet- and megakaryocyte-specific deletion of the ATE1 gene and were, therefore, unable to arginylate proteins within their platelets. Contrary to our predictions, we did not observe any obvious defects in the overall platelet morphology or in the amount or distribution of actin filaments. Instead, we observed a significant increase in clot retraction in the ATE1 knockout platelets. Remarkably, this effect correlated with elevated phosphorylation of the myosin regulatory light chain (RLC). This suggests that ATE1-mediated arginylation directly regulates myosin phosphorylation and controls myosin contractility. This is the first demonstration of an effect of arginylation on myosin activity, and the first indication that regulation via arginylation and phosphorylation may be inter-connected in the regulation of essential in vivo functions.
Methods
All of the mice used for this study were maintained in the animal facility at the University of Pennsylvania in accordance with the National Institute of Health guidelines, and under the approved animal protocols of the Institutional Animal Care and Use Committee Review Board of the University of Pennsylvania. Reagents and chemicals are described in the Online Supplementary Methods section.
Animal model
Platelet-specific ATE1 knockout mice were generated by crossing ATE1-floxed mice (ATE1fl/fl) with mice transgenic for the expression of Cre recombinase under the control of the mouse platelet factor-4 promoter (PF4 Cre+).10,18 The platelet-specific targeting was further confirmed by immunoblotting platelet lysates for ATE1. For all experiments shown, ATE1fl/fl PF4 Cre− littermates were used as control mice.
Preparation of platelets for functional analysis, immunoblotting, and immunoprecipitation
Mouse blood was collected as previously described.19 For immunoblotting, 1×108 murine, washed platelets were lysed in RIPA buffer with protease inhibitor cocktail (Sigma). For lysates prepared for probing pRLC, we used the method described by Getz et al., which is to terminate the platelet activation by adding ice-cold 0.6 N HCIO4, followed by centrifugation at 13,000 g for 10 min at 4°C.20 The pellet was solubilized in RIPA buffer, and the lysate was fractionated on a 4–12% bis-tris gel prior to immunoblotting. Immunoprecipitation was done by lysing 1.5×108 murine, washed platelets in HEPES-Tyrode buffer. After centrifugation at 14,000 g at 4°C for 10 min, the supernatant was incubated with anti-non-muscle myosin IIA antibody, under gentle rotation at 4°C overnight. The immunocomplex was captured by incubating with 50 μL rProtein G agarose slurry, and centrifugation at 4°C for 30 min. This immunoprecipitate was then fractionated by sodium dodecylsulfate polyacrylamide gel electrophoresis and immunoblotted with either anti-myosin or anti-ATE1 antibodies. Methods for platelet aggregometry, clot retraction, platelet spreading, platelet adhesion, coagulation assays, and in vivo thrombosis studies are described in the Online Supplementary Methods.
Biochemical fractionation of actin and the distribution of G-actin and F-actin
Murine, washed platelets (1.5×108) in HEPES-Tyrode buffer were stimulated with 1 U/mL thrombin at room temperature. The reactions were stopped by adding 1/5 volume 5X PHEM buffer at a final concentration of 1% Triton X-100, 2 μM phalloidin, 10 nM hirudin, and protease inhibitors at pH 7.2. The G-actin in the soluble fraction was separated from the F-actin in the insoluble fraction by ultracentrifugation at 100,000 g for 1 h at 4°C. The insoluble fraction was lysed in RIPA buffer. The two fractions were run on a 4–12% bis-tris gel prior to immunoblotting for actin. The relative G-actin and F-actin levels were quantified based on the intensity of their blots using Image J Software.
Mass spectrometry analysis of arginylated proteins
Whole platelet pellets were subjected to trypsin digestion, liquid chromatography-mass spectrometry/mass spectrometry analysis, and the data were processed to identify arginylated sites as described by Wong et al.13 The full list of peptides arginylated in platelets is presented in Online Supplementary Table S1. Raw data files and data analysis parameter files can be found at http://fields.scripps.edu/published/arginylation_platelets/.
Statistical methods
The Student t-test was used to determine whether the two sets of data generated from ATE1 knockout mice and their littermate control mice were significantly different. P values <0.05 were considered statistically significant. The statistical analyses were performed using Excel (Microsoft Corp.).
Results
Generation of mice lacking ATE1 in platelets
To test whether ATE1 is expressed in platelets, we analyzed the presence of individual ATE1 isoforms in wild-type mouse platelets by reverse transcriptase polymerase chain reaction (Figure 1A).2 We found that ATE1-2 was the predominant platelet isoform, that small amounts of ATE1-1 were detectable, and that there were no detectable mRNA transcripts of either ATE1-3 or ATE1-4. Immunoblotting with an antibody that recognizes all four isoforms of ATE1 indicated that the overall amount of ATE1 in platelets is similar to that found in mouse fibroblasts when normalized to total protein levels (Figure 1B).21
Figure 1.
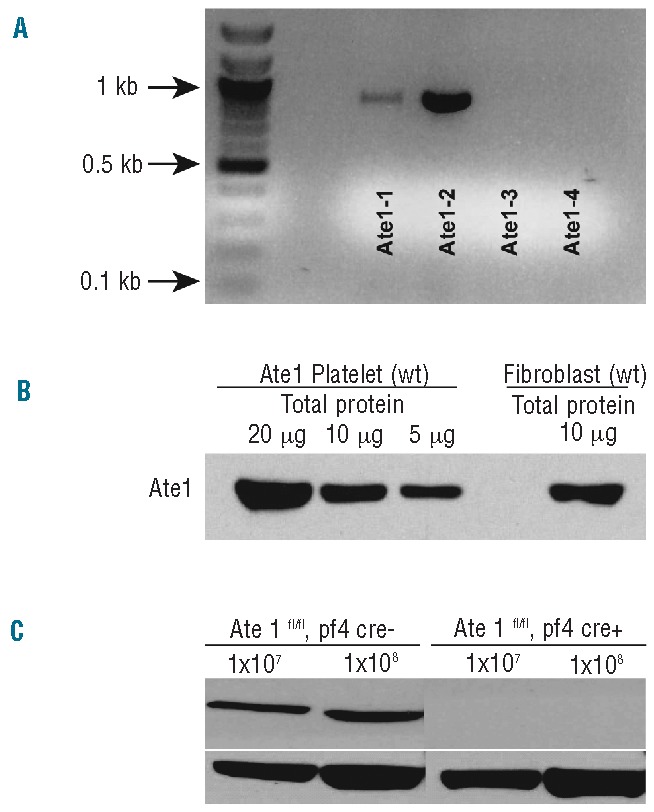
ATE1 expression in mouse platelets and its abolishment in ATE1 knockout animals. (A) Reverse transcriptase polymerase chain reaction of RNA extracted from wild-type (WT) mouse platelets with ATE1 isoform-specific primer sets.2 ATE1-2 was the dominant isoform, while ATE1-3 and ATE1-4 were not detectable in platelets. (B) Comparisons of ATE1 levels in platelets and fibroblasts. (C) Immunoblot of the lysates from ATE1-deleted (Knockout) platelets or ATE1-floxed PF4 CRE− (control) platelets verified the absence of ATE1 protein in the platelets of the mouse knockout model.
Because global ATE1 deletion leads to embryonic lethality in mice, to analyze ATE1 function in platelets, we engineered mice lacking ATE1 specifically in their megakaryocytes and in their platelets.1 This was achieved by pairing ATE1fl/fl mice with an established mouse line that expresses Cre recombinase under the megakaryocyte-specific PF4 promoter.18 The resulting mice, ATE1fl/fl PF4 Cre+ mice had normal complete blood counts, despite completely lacking ATE1 protein in their platelets. This was confirmed by western blots (Figure 1C). These mice and their littermate control siblings, ATE1fl/fl PF4 CRE− mice, were indistinguishable and exhibited no spontaneous hemorrhages or thromboses.
Loss of platelet ATE1 has no effect on the actin cytoskeleton
Reorganization of the platelet cytoskeleton has been shown to contribute to the regulation of platelet adhesion, secretion, and aggregation. Given that the deletion of ATE1 causes defects in spreading and lamella formation in fibroblasts, we anticipated that platelets lacking ATE1 would have similar defects.15 However, we found that thrombin-stimulated platelets that lack ATE1 spread normally upon fibrinogen, and extended normal amounts of lamellipodia and filopodia (Figure 2A and 2B.) Immunostaining of fully spread control and ATE1-null platelets showed no differences in the distribution of β-actin or in the overall quantity of phalloidin-stained actin stress fibers (Figure 2C). In addition, the footprint area of fully spread platelets was unaffected by the loss of ATE1 (Figure 2D). These data demonstrate that, in contrast to the situation in fibroblasts, ATE1 is not essential for actin assembly in platelets. Accordingly, we were also unable to detect any contribution of ATE1 in platelet aggregation, secretion, or adhesion (Figure 3).
Figure 2.
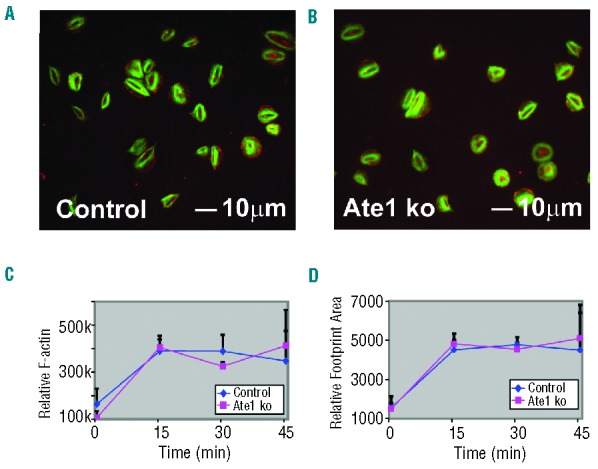
Platelet spreading on fibrinogen in response to thrombin. (A and B) Control and ATE1 knockout (ko) platelets were allowed to spread on fibrinogen-coated coverslips in the presence of thrombin. The cells were fixed and stained with phalloidin to detect F-actin (green) and antibodies were directed to detect β-actin (red). F-actin was concentrated centrally in the cortex of the spread platelets while β-actin was detected mostly at the edges. (C) Relative F-actin levels of platelets were assessed by flow cytometry staining with Alexa 488-Phalloidin. (D) The relative area (footprint size) of spread platelets is shown. The mean ± standard deviation are shown.
Figure 3.
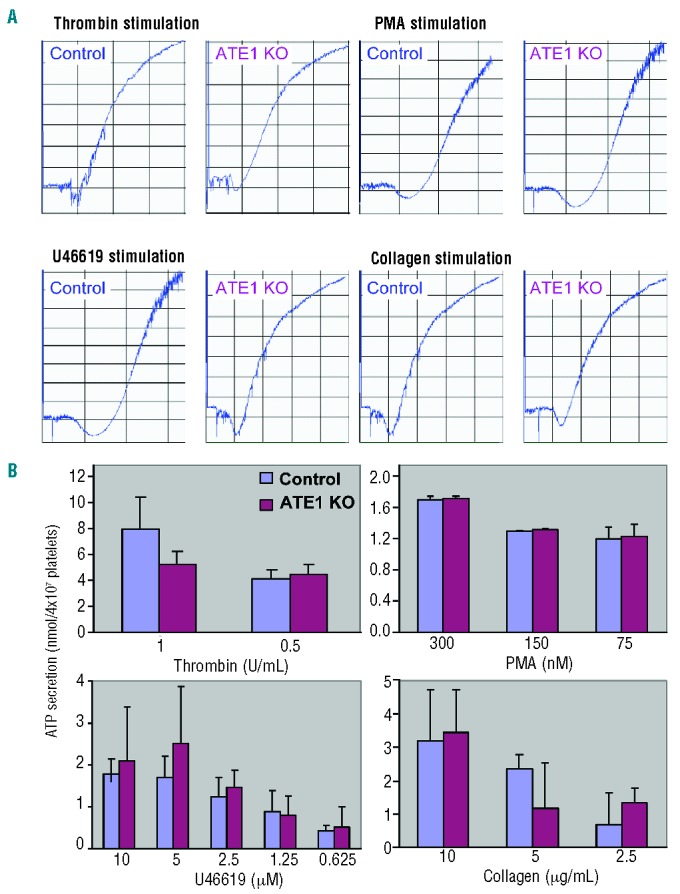
Platelet aggregation and secretion. (A) Aggregation tracings of platelets derived from control and ATE1 knockout (ko) mice after stimulation with various agonists. (B) Platelets derived from control or ATE1 KO platelets were analyzed for secretion of ATP using a luceriferase assay.
Platelets lacking ATE1 undergo accelerated clot retraction
It is notable that many cytoskeletal proteins other than β-actin are also prominent targets of ATE1, and collectively, these cytoskeletal proteins participate in generating contractile forces in different cell types.22–24 To test the possibility that ATE1 affects the ability of platelets to generate contractile forces, we analyzed the effect of ATE1 knockout on platelet clot retraction. Remarkably, platelet-rich plasma derived from ATE1fl/fl PF4 Cre+ mice had significantly enhanced clot retraction when compared with platelet-rich plasma derived from ATE1fl/fl PF4 Cre− littermate control mice (P<0.03) (Figure 4A and 4B). Given that clot retraction is driven by myosin, which is a motor protein that interacts with actin and plays an essential role in the contractile behavior of cells, we hypothesized that the observed accelerated clot retraction in ATE1-null platelets is due to enhanced myosin-mediated force applied to the existing actin cables.18
Figure 4.
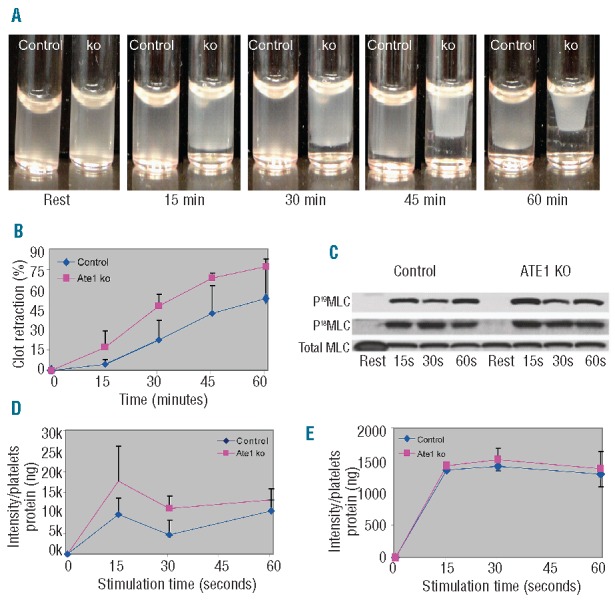
Increased clot retraction and phosphorylation of myosin in platelets lacking ATE1. (A) Platelet-rich plasma from ATE1 knockout (ko) and control mice was stimulated with thrombin at 37°C to induce clotting. Clot formation and retraction were continuously photographed before and after thrombin stimulation at 15 min intervals. Representative images at each time point show increased clot retraction in ATE1 ko platelets as compared to control platelets. (B) Quantification of clot retraction at each time point was measured as the percentage of the retracted area as compared to the initial total area of the clot (n=5.) Deletion of ATE1 significantly increased clot retraction (P<0.05, except at 60 min). The mean ± standard deviation are shown. (C) Phosphorylation of RLC at residues Ser19 and Thr18 was undetectable in resting platelets, but increased following stimulation with thrombin. Phosphorylation of RLC at either Ser19 (D) or Thr18 (E) was quantified based on the immunoblots of platelets stimulated with thrombin. The level of Ser19-RLC phosphorylation was significantly increased in platelets lacking ATE1 as compared to that in the controls (P<0.05) at 15 seconds (P<0.03) and at 30 seconds (P<0.05) after agonist stimulation. In contrast, Thr18-RLC phosphorylation remained the same in both groups. The mean ± standard deviation are shown.
Myosin arginylation in platelets correlates with changes in myosin regulatory light chain phosphorylation
To test the hypothesis that myosin is a target of ATE1, we first analyzed whether myosin was normally arginylated within activated platelets. Analysis of thrombin-stimulated platelets by mass spectrometry demonstrated that both the heavy and the essential light chain subunits of myosin were arginylated (Online Supplementary Table S1).
Myosin activity is controlled by phosphorylation of its RLC. In platelets, as in many other non-muscle cells, myosin RLC can be phosphorylated at two distinct amino acid residues in response to thrombin stimulation: Ser19 and Thr18.20,25,26 To test whether enhanced clot retraction in ATE1-null platelets correlates with changes in myosin RLC phosphorylation, we probed thrombin-stimulated platelets with phospho-Ser19 and phospho-Thr18 antibodies. Remarkably, Ser19 phosphorylation was enhanced in ATE1-null platelets, while Thr18 phosphorylation was normal (Figure 4C–E, P<0.05). We further analyzed whether there was a direct interaction between ATE1 and platelet myosin. In addition, we observed that ATE1 co-immunoprecipitates with myosin in platelet lysates (Figure 5). These findings show that loss of ATE1 in the platelets of ATE1fl/fl PF4 Cre+ mice causes increased phosphorylation of myosin RLC. Collectively, our evidence suggest that ATE1 regulates myosin activation and clot retraction.
Figure 5.
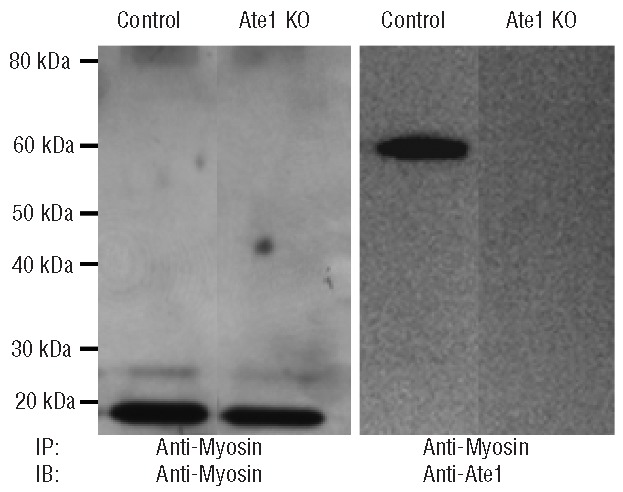
ATE1 and myosin co-immunoprecipitate in murine platelets. Myosin was immunoprecipitated from lysates of ATE1 knockout (KO) and control platelets. The immunoprecipitates were fractionated by polyacrylamide gel electrophoresis and immunoblotted with myosin or ATE1 antibodies. On the left, an anti-myosin immunoblot demonstrates that myosin was equally immunoprecipitated from control and ATE1 knockout platelets. The immunoblot on the right demonstrates that the anti-myosin immunoprecipitates also contain ATE1.
Loss of platelet ATE1 enhances stable thrombus formation in vivo
Since actin-dependent myosin contractility is thought to contribute to thrombus stability in the arterial system,37 we analyzed the role of platelet ATE1 in supporting thrombus formation and thrombus stability in vivo. This was initially analyzed by comparing the tail bleeding times of ATE1fl/fl PF4 Cre+ and ATE1fl/fl PF4 Cre− mice. We observed that mice lacking ATE1 in their platelets stopped bleeding more rapidly than control mice in this assay (Figure 6A.) This difference was not due to an alteration in the function of coagulation factors in the plasma, since the prothrombin times and activated partial thromboplastin times were identical in the ATE1fl/fl PF4 Cre+ mice and the ATE1fl/fl PF4 Cre− mice (Figure 6B). We further analyzed the ability of platelets lacking ATE1 to form thrombi in vivo by inducing a chemical injury to the carotid artery of ATE1fl/fl PF4 Cre+ and ATE1fl/fl PF4 Cre− mice. Stimulation of thrombus formation by FeCl3-induced injury of the carotid artery showed that that the Ate1fl/fl PF4 Cre+ mice formed thrombi faster than their littermate controls (Figure 6C and 6D.) Thus, ATE1 deletion in platelets causes enhanced clotting in vivo. This accelerated thrombus formation is likely due to increased myosin light chain phosphorylation and thereby enhanced myosin activation facilitating clot retraction and clot stability.
Figure 6.
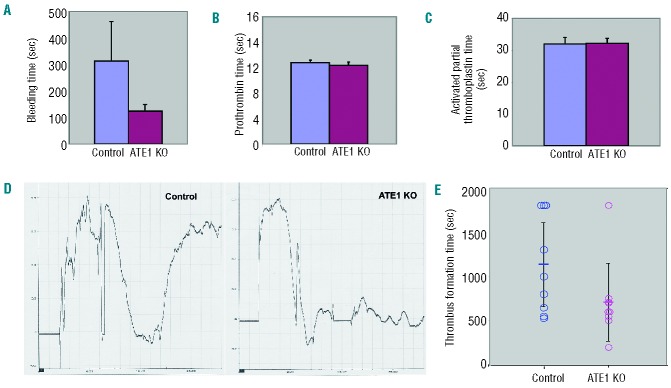
Accelerated in vivo thrombus formation in mice with platelets that lack ATE1. (A, B, and C) Mice lacking ATE1 in their platelets had shorter tail bleeding times than their littermate controls (P<0.05), but the prothrombin time (PT) and activated partial thromboplastin time (aPTT) were identical in these two genotypes (P>0.05 for both PT and aPTT). (D) Carotid injury was induced by the application of FeCl3-soaked filter paper for 2 min, and arterial blood flow was monitored by a Doppler ultrasound probe. As shown by the representative tracings, ATE1 KO platelets were more likely to occlude faster and form stable thrombi. (E) The mean time of the thrombus formation is indicated by the short horizontal bar in the graph. Thrombus formation was quicker in the mice lacking ATE1 in their platelets (P=0.024).
Discussion
Our results demonstrate that ATE1 deletion in platelets causes a significant effect on clot retraction, potentially because of an increase in the phosphorylation of the myosin RLC on Ser19, which in turn stimulates actomyosin contractility. This finding provides the first evidence that arginylation of a platelet protein has a cellular and organism effect. It is also the first demonstration of a likely functional connection between arginylation and phosphorylation, which are two global biological regulators.
It was previously shown that arginylation of β-actin, but not γ-actin, regulates lamella formation at the leading edge of fibroblasts during cell locomotion.12,15 Deletion of ATE1 caused a 2.5-fold reduction in polymeric actin and a dramatic collapse of the leading edge. As in all eukaryotic cells, actin is highly abundant in platelets. However, what is unique to platelets is that β-actin is five times more abundant than γ-actin.27,28 Surprisingly, unlike in fibroblasts, there were no obvious effects of the ATE1 knockout on the actin cytoskeleton structure in platelets.12,15 This difference can be explained by the fact that in platelets, unlike in fibroblasts, β-actin does not appear to be a prominent arginylation target, as we determined by mass spectrometry. Moreover, since our genetic approach only impaired the arginylation in mature megakaryocytes, proteins with long-half lives that were synthesized in immature megakaryocytes could still exist in their arginylated state in circulating platelets. Consistent with this, we found that some arginylated proteins were still present in the platelets of ATE1fl/fl PF4 Cre+ mice. This was evidenced by mass spectrometry analysis of platelet preparations from these mice. Since arginylation-dependent signaling is expected to be tightly and temporally regulated, we predict that such long-lived arginylated proteins likely play a structural rather than regulatory role, and contribute to the overall platelet architecture rather than to platelet-dependent signaling events.
It is notable that we have observed arginylation of myosin’s essential light chain, but not arginylation of myosin’s RLC, which is regulated by phosphorylation. The effect of the arginylation of other polypeptides in the myosin complex on the phosphorylation of the RLC is likely achieved via an arginylation-dependent conformational change in the myosin neck region that indirectly affects the RLC phosphorylation state. For example, arginylation of this region within myosin may inhibit the binding of the myosin light chain kinase, thereby impairing its ability to phosphorylate the myosin residue Ser19. This arginylation-induced conformational effect on myosin is likely very specific, since it does not affect Thr18 phosphorylation. Mapping of the arginylation sites on the three-dimensional structure of the myosin motor complex (Figure 7) suggests that the arginylated sites in the essential light chain are exposed on the protein’s surface. We hypothesize that the addition of a bulky Arg residue to these sites significantly affects the conformation and protein-protein binding in this region.
Figure 7.
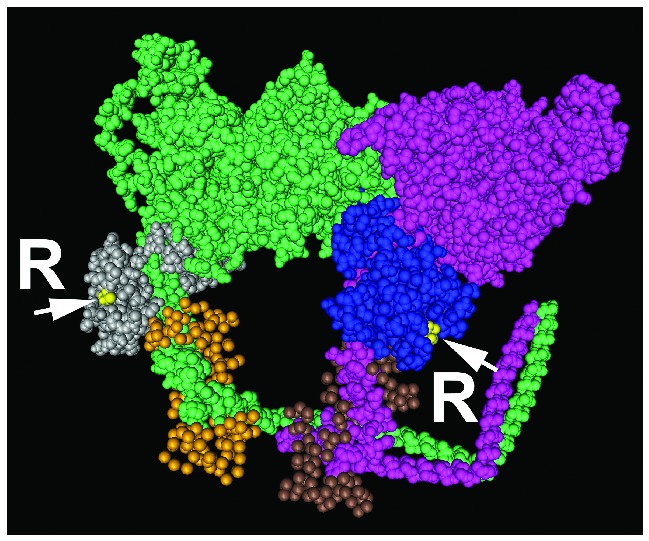
RLC regulation of the 19th amino acid phosphorylation site by ATE1 arginylation of the essential light chain (ELC). Structural model of the myosin head and neck region (pink and green for two myosin heavy chain fragments) with the bound light chains (PDB identifier 1I84). The arginylated sites (denoted by yellow dots, arrows, and letters R) are found in the myosin ELC (blue and gray), adjacent to the RLC (brown and gold). It is likely that arginylation in these regions affects myosin conformation and/or binding of the enzymatic machinery that regulates myosin motor activity.
Since both phosphorylation and arginylation have emerged in recent studies as essential modifications and global regulators of protein activity, our finding that these two post-translational modifications directly affect each other in vivo constitutes a major and important discovery. It has been previously found that arginylation facilitates Arg-methylation.8 Our finding that it also regulates phosphorylation suggests that arginylation plays an important role in the hierarchical network of post-translational modifications and likely underlies major regulatory pathways. It is also notable that arginylation and phosphorylation add oppositely charged chemical groups to the protein surface. This suggests that they confer different chemical properties that may neutralize each other. We hypothesize that this phenomenon is the first of many examples in which arginylation and phosphorylation will be found to interact and contribute to major regulatory events in cell signaling.
In summary, we have found that the loss of ATE1 in platelets, in contrast to its effect in fibroblasts, leads to post-translational defects in myosin arginylation, phosphorylation, and function. Loss of platelet ATE1 also results in accelerated clot retraction ex vivo and thrombus formation in vivo. Our findings provide the first evidence that myosin may be regulated by arginylation in platelets. How arginylation of myosin affects the cytoskeleton in non-hematopoietic cells is currently an open question. These findings should lead to a better understanding of the vital role of arginylation-mediated regulation of signaling in general.
Supplementary Material
Acknowledgments
This work was supported in part by funds from the NIH PO1 HL40387 (to CSA), R01 HL083392 (to CSA), R01 HL110110 (to CSA), P41 RR011823 (to JRY) and GM103533 (to JRY).
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, et al. An essential role of N-terminal arginylation in cardiovascular development. Science. 2002;297(5578):96–9 [DOI] [PubMed] [Google Scholar]
- 2.Rai R, Kashina A. Identification of mammalian arginyltransferases that modify a specific subset of protein substrates. Proc Natl Acad Sci USA. 2005;102(29):10123–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpio MA, Lopez Sambrooks C, Durand ES, Hallak ME. The arginylation-dependent association of calreticulin with stress granules is regulated by calcium. Biochem J. 2010;429(1):63. [DOI] [PubMed] [Google Scholar]
- 4.Soffer RL. Enzymatic modification of proteins. V. Protein acceptor specificity in the arginine-transfer reaction. J Biol Chem. 1971;246(6):1602–6 [PubMed] [Google Scholar]
- 5.Ciechanover A, Ferber S, Ganoth D, Elias S, Hershko A, Arfin S. Purification and characterization of arginyl-tRNA-protein transferase from rabbit reticulocytes. Its involvement in post-translational modification and degradation of acidic NH2 termini substrates of the ubiquitin pathway. J Biol Chem. 1988;263(23):11155–67 [PubMed] [Google Scholar]
- 6.Ferber S, Ciechanover A. Role of arginine-tRNA in protein degradation by the ubiquitin pathway. Nature. 1987;326(6115):808–11 [DOI] [PubMed] [Google Scholar]
- 7.Eriksson AC, Whiss PA. Measurement of adhesion of human platelets in plasma to protein surfaces in microplates. J Pharmacol Toxicol Methods. 2005;52(3):356–65 [DOI] [PubMed] [Google Scholar]
- 8.Saha S, Kashina A. Posttranslational arginylation as a global biological regulator. Dev Biol. 2011;358(1):1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaji H, Kaji A. Protein modification by arginylation. Chem Biol. 2011;18(1):6–7 [DOI] [PubMed] [Google Scholar]
- 10.Rai R, Wong CC, Xu T, Leu NA, Dong DW, Guo C, et al. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development. 2008;135(23):3881–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saha S, Wang J, Buckley B, Wang Q, Lilly B, Chernov M, et al. Small molecule inhibitors of arginyltransferase regulate arginylation-dependent protein degradation, cell motility, and angiogenesis. Biochem Pharmacol. 2012;83(7):866–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurosaka S, Leu NA, Zhang F, Bunte R, Saha S, Wang J, et al. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS Genet. 2010;6(3):e1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong CC, Xu T, Rai R, Bailey AO, Yates JR, 3rd, Wolf YI, et al. Global analysis of posttranslational protein arginylation. Plos Biol. 2007;5(10):e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu T, Wong CC, Kashina A, Yates JR., 3rd Identification of N-terminally arginylated proteins and peptides by mass spectrometry. Nat Protoc. 2009;4(3):325–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR, 3rd, Mogilner A, et al. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 2006;313(5784):192–6 [DOI] [PubMed] [Google Scholar]
- 16.Lam WA, Chaudhuri O, Crow A, Webster KD, Li TD, Kita A, et al. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat Mater. 2011;10(1):61–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrlicher A, Hartwig JH. Cell mechanics: contracting to stiffness. Nat Mater. 2011; 10(1):12–3 [DOI] [PubMed] [Google Scholar]
- 18.Leon C, Eckly A, Hechler B, Aleil B, Freund M, Ravanat C, et al. Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood. 2007;110(9):3183–91 [DOI] [PubMed] [Google Scholar]
- 19.Lian L, Wang Y, Flick M, Choi J, Scott EW, Degen J, et al. Loss of pleckstrin defines a novel pathway for PKC-mediated exocytosis. Blood. 2009;113(15):3577–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Getz TM, Dangelmaier CA, Jin J, Daniel JL, Kunapuli SP. Differential phosphorylation of myosin light chain (Thr)18 and (Ser)19 and functional implications in platelets. J Thromb Haemost. 2010;8(10):2283–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Han X, Saha S, Xu T, Rai R, Zhang F, et al. Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chem Biol. 2011;18(1):121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haling JR, Monkley SJ, Critchley DR, Petrich BG. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood. 2011;117(5):1719–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki-Inoue K, Hughes CE, Inoue O, Kaneko M, Cuyun-Lira O, Takafuta T, et al. Involvement of Src kinases and PLCgamma2 in clot retraction. Thromb Res. 2007;120(2):251–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randriamboavonjy V, Isaak J, Fromel T, Viollet B, Fisslthaler B, Preissner KT, et al. AMPK alpha2 subunit is involved in platelet signaling, clot retraction, and thrombus stability. Blood. 2010;116(12): 2134–40 [DOI] [PubMed] [Google Scholar]
- 25.Stull JT, Tansey MG, Tang DC, Word RA, Kamm KE. Phosphorylation of myosin light chain kinase: a cellular mechanism for Ca2+ desensitization. Mol Cell Biochem. 1993;127–128: 229–37 [DOI] [PubMed] [Google Scholar]
- 26.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273(5272):245–8 [DOI] [PubMed] [Google Scholar]
- 27.Hartwig JH. Mechanisms of actin rearrangements mediating platelet activation. J Cell Biol. 1992;118(6):1421–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landon F, Huc C, Thome F, Oriol C, Olomucki A. Human platelet actin. Evidence of beta and gamma forms and similarity of properties with sarcomeric actin. Eur J Biochem. 1977;81(3):571–7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.