

Abstract
Hereditary natural anticoagulant deficiencies are the major cause of genetic thrombophilia in Asia. Given the growing acknowledgment of the risk of venous thromboembolism in Asian populations, we investigated the frequency and mutation spectrums of natural anticoagulant deficiency in Korea. The group of patients consisted of consecutive patients with venous thromboembolism screened for thrombophilia. Genetic tests were performed on suspicion of natural anticoagulant deficiency. For the population group, >3,000 individuals were screened from routine check-ups, and those with a low level (<1st percentile) of natural anticoagulant underwent genetic tests. Mutations were detected by direct sequencing of PROC, PROS1, and SERPINC1, followed by additional multiplex ligation-dependent probe amplification for PROS1 and SERPINC1 for dosage mutations. Among 500 patients screened, 127 were suspected of having a natural anticoagulant deficiency, and this was genetically confirmed in 71: protein C deficiency in 36 (50.7%), antithrombin deficiency in 21 (29.6%), and protein S deficiency in 14 (19.7%). Among 3,129 individuals from the population who were screened, the frequency of natural anticoagulant deficiency was ~1.0%: antithrombin deficiency 0.49%, protein C deficiency 0.35%, and protein S deficiency 0.16%. Two PROC mutations causing type I protein C deficiency were prevalent (Arg211Trp and Met406Ile in patients and Arg211Trp in the population). Two SERPINC1 mutations causing type II antithrombin deficiency, Arg79Cys and Ser158Pro, were prevalent in the population group. This is the first study on the genetic epidemiology of natural anticoagulant deficiencies in Korea. The results demonstrated that the frequencies and spectrum of mutations underlying genetic thrombophilia in Korea are different not only from those in Caucasians but also those in other Asian populations.
Introduction
Thrombophilia is a condition that makes an individual more susceptible to develop thromboembolism. The well-established backgrounds of thrombophilia of genetic origin (genetic or hereditary thrombophilia) are either increased procoagulant activity or hereditary deficiency of natural anticoagulants.1,2 The former includes activated protein C resistance (factor V Leiden mutation) and prothrombin G20210A mutation, both of which are restricted to Western populations. The latter condition includes deficiencies of protein C (PC), protein S (PS) and antithrombin (AT), and is the major culprit of thrombophilia in Asian populations. Of note, thrombophilia from natural anticoagulant deficiency confers a higher risk of venous thromboembolism (VTE) than that from increased procoagulant activity.3,4 In particular, AT deficiency is the first genetic risk factor for VTE identified in humans and confers the highest risk.2,4,5 Thus, diversity in genetic backgrounds is believed to be the major factor accounting for the different risks of VTE in different ethnic groups. Understanding genotype-phenotype correlations and structure-function relationships by identifying causative genetic defects is important for risk assessment in VTE patients and also in the general population.6 Recently, there has been growing acknowledgment of the risk of VTE and the associated socioeconomic burden in Asian countries, spurring research on the genetic risk factors.7,8 There have been several studies on the prevalence of genetic thrombophilia in Asia; however, the laboratory strategy to detect hereditary thrombophilia has varied across studies. Before the wide-spread availability of molecular genetic tests, the strategy was typically dependent on coagulation test results. Recent studies are trying to incorporate molecular genetic tests to confirm the coagulation defect by direct identification of causative mutations with or without screening by coagulation tests. This is particularly because the coagulation test results can be affected by a variety of factors such as underlying disease and medication, leading to false diagnoses of hereditary deficiency.9 However, there are still limited studies showing the prevalence and mutation spectrum of all three types of natural anticoagulants, particularly simultaneously in both patients with VTE and in the general population of a given ethnicity.
In this regard, we investigated the molecular genetic defects of natural anticoagulant deficiencies (PC, PS, and AT) in a large number of consecutive VTE patients and in the general population in Korea screened by coagulation tests. The results revealed unique frequencies and mutation spectrums of natural anticoagulant deficiencies in the group of VTE patients and in the general population. These frequencies and the mutation spectrums in Korea were not only distinct from those in Caucasian populations but also from those in other Asian countries.
Methods
Patients and population
The group of patients consisted of consecutive VTE patients screened for thrombophilia including PC, PS, and AT deficiencies at Samsung Medical Center, Seoul, Korea, from January 2005 to December 2012. For the population group, at least 3,000 individuals visiting the institution for routine check-ups were screened from September 2005 to January 2006 using the same coagulation tests for natural anticoagulant deficiency as those used in the patients. In each group, those suspected of having a natural anticoagulant deficiency underwent molecular genetic tests to confirm the deficiency. In both groups, we excluded those with low levels of multiple (2 or more) natural anticoagulants, especially in association with underlying liver disease or other extrinsic factors. Written informed consent was obtained from the patients in the study, which was approved by the Institutional Review Board of the Samsung Medical Center, Seoul, Korea.
Coagulation tests
The thrombophilia profile tests for VTE patients included screening for genetic thrombophilia: PC activity (Stachrom® Protein C, Diagnostica Stago, Asnieres-Sur-Seine, France), PS free antigen (Liatest® Free Protein S, Diagnostica Stago), and AT activity (Stachrom® ATIII, Diagnostica Stago). All coagulation tests were performed on the STA® coagulation analyzer (Diagnostica Stago). The local reference intervals were determined according to the guidelines from the Clinical and Laboratory Standards Institute (http://www.clsi.org/) as the 2.5–97.5 percentiles (PC activity, 85.6–167.2%; PS free antigen, 69.3–148.2% for males and 56.0–132.6% for females; and AT activity, 81.5–119.3%).10 Tests for PC antigen, PS total antigen and activity, and AT antigen were additionally performed when indicated. Natural anticoagulant deficiency was suspected in VTE patients when the result was below the lower limit of the reference interval (2.5 percentile). Screening for natural anticoagulant deficiency in the population group was performed using the same coagulation tests as those used in the VTE patients. Individuals with levels of PC, PS, or AT below the 1st percentile were selected for molecular genetic tests.
Molecular genetic analyses
Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit following the standard protocols (Promega, Madison, WI, USA). Molecular genetic diagnosis of natural anticoagulant deficiency was performed by detecting causative mutations of the PROC gene (PC deficiency), PROS1 (PS deficiency), and SERPINC1 (AT deficiency). Point mutations were detected by direct sequencing of all coding exons and flanking intronic sequences of each gene on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using a BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems). Multiplex ligation-dependent probe amplification (MLPA) experiments were additionally performed to detect large dosage mutations when no point mutations had been detected (SALSA MLPA kit P112 PROS1 and P227 SERPINC1; MRC Holland, Amsterdam, the Netherlands), as previously described.11 Mutations identified are described following the recommendations from the Human Genome Variation Society (http://www.hgvs.org/mutnomen/), along with the conventional numbering. For validation of novel mutations, population frequencies were obtained by targeted sequencing involving 100 control chromosomes of Korean descent. For novel missense mutations, functional predictions were also performed using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT algorithms (http://sift.bii.a-star.edu.sg/), and the degree of conservation was determined by aligning amino acid sequences across different species using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Results
Natural anticoagulant deficiency and causative mutations among the patients with venous thromboembolism
A total of 500 patients were diagnosed with VTE and screened for thrombophilia during the study period. Among them, 127 patients (25.4%) were suspected of having a specific natural anticoagulant deficiency (PC deficiency in 62 patients, PS deficiency in 37, and AT deficiency in 28 patients) and underwent molecular genetic tests to confirm the deficiency. As a result, molecular genetic tests identified causative mutations in 71 patients of a total of 500 (14.2%) with VTE: PROC mutations (PC deficiency) in 36 patients, PROS1 mutations (PS deficiency) in 14 patients, and SERPINC1 mutations (AT deficiency) in 21 patients. The mutation detection rate was 58.1% in PC deficiency (36/62), 37.8% in PS deficiency (14/37), and 75% in AT deficiency (21/28), with an overall mutation detection rate of 55.9%. Overall 43 mutations (18 unique mutations) were detected in PROC in 36 patients, all in a heterozygous state (Figure 1A). Seven patients (19.4%) had two (double) mutations (Table 2). Among the PROC mutations, two were novel (Arg264Gly and Gly266Arg) (Table 1 and Figure 1A). Two missense mutations, Arg211Trp and Met406Ile, accounted for 53.5% of all mutations (32.6% and 20.9%, respectively). A total of 14 mutations (12 unique mutations) were detected in PROS1 in 14 patients, all in a heterozygous state (Figure 1B). No patient had two mutations. Among the PROS1 mutations, six were novel (Ser194Glnfs*14, Arg316His, Ala325Glu, Gln548His, Gln572*, and Lys591*) (Table 1 and Figure 1B). Overall, 22 mutations (20 different mutations) were detected in SERPINC1 in 21 patients, all in a heterozygous state (Figure 1C). One patient (4.8%) had two mutations (Table 2). Among the SERPINC1 mutations, seven were novel [Ala126Asp, Met284Arg, Leu331Profs*16, Pro353Ala, c.1154-2A>G, c.1153+5G>T, and c.(?_1)_(1395+_?)dup (duplication of all 1–7 exons)] (Table 1 and Figure 1C). No predominant mutations were observed in PROS1 or in SERPINC1. The distribution of mutations by type in the patient group demonstrated a predominance of missense mutations in PROC (81.4%) (Figure 2). Large deletions/duplications were not observed in PROC, while they accounted for 14.3% and 18.2% of mutations in PROS1 and SERPINC1, respectively. The frequency of all the novel mutations was 0% in the general population. Among the eight novel missense mutations, all mutated residues were highly conserved across species, and all mutations were predicted to be damaging except for Arg316His (Table 1 and Online Supplementary Figures). Among the eight patients with double mutations, four were considered to have severe PC deficiency from compound heterozygous PROC mutations based on coagulation test results with or without family studies (Table 2).
Figure 1.
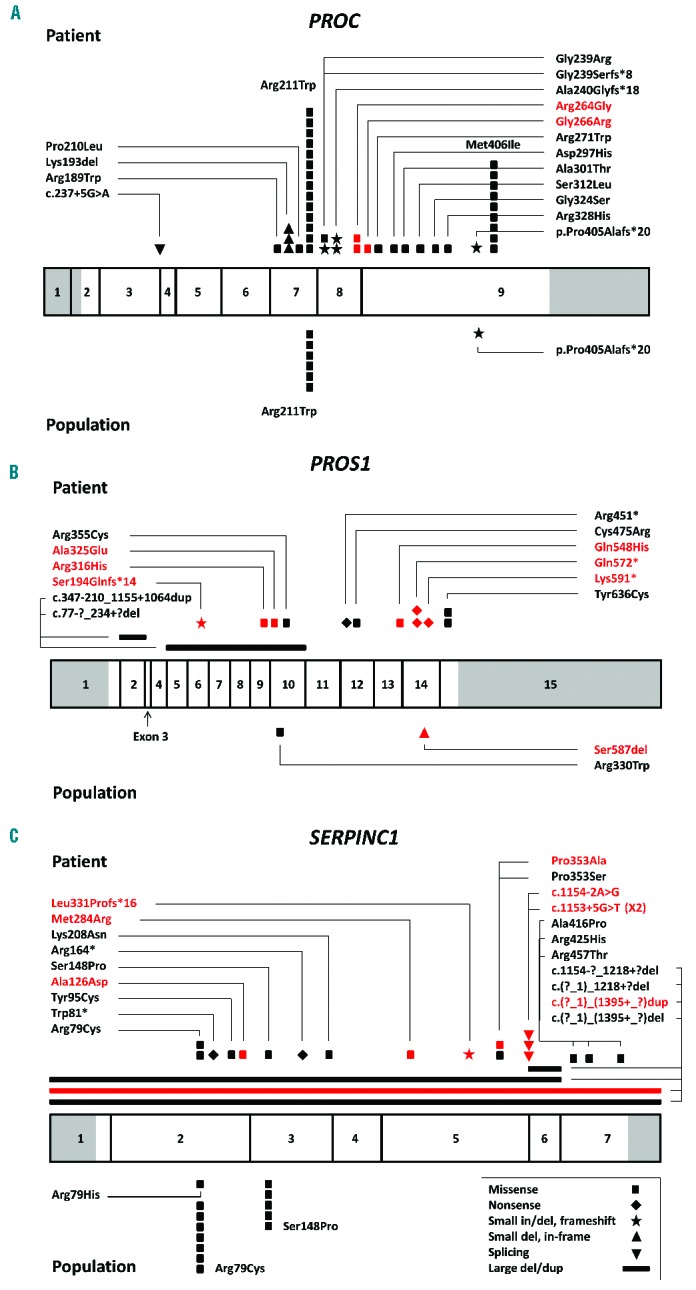
(A) PROC mutations, (B) PROS1 mutations, and (C) SERPINC1 mutations in the group of patients and in the population group (above and below the bars, respectively). The bars represent the genes, and the numbers indicate the exons (sized to the scale) with non-coding sequences shaded. Novel mutations are marked in red (both description and symbol).
Table 2.
Genetic and coagulation test results in eight patients with double mutations.
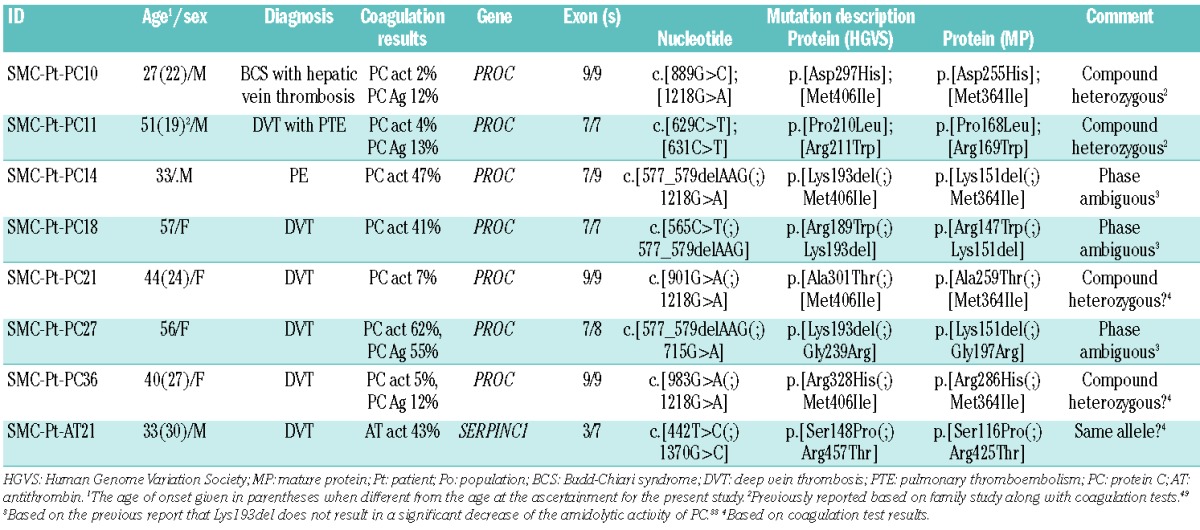
Table 1.
Novel mutations causing hereditary thrombophilia from natural anticoagulant deficiency identified in 19 study subjects.
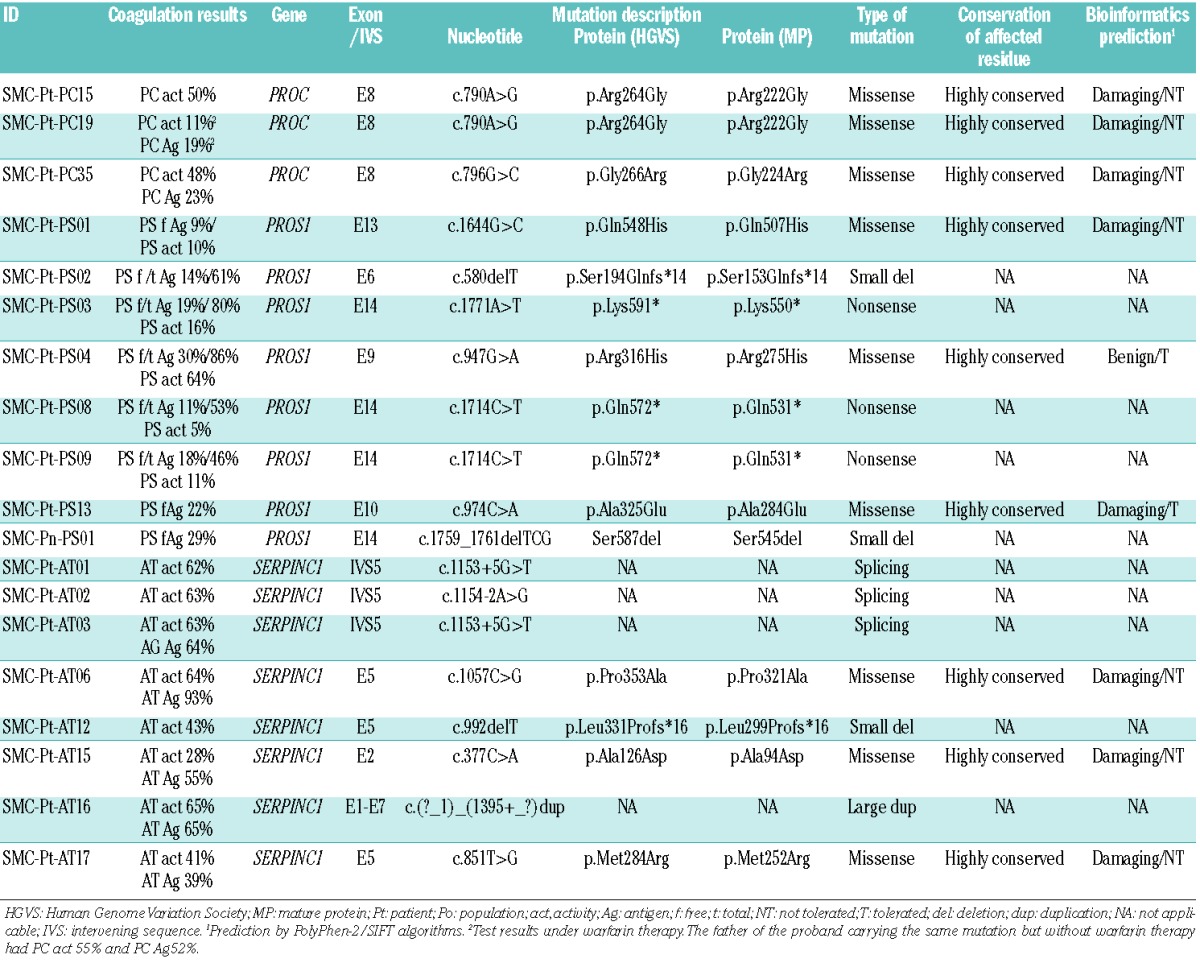
Figure 2.
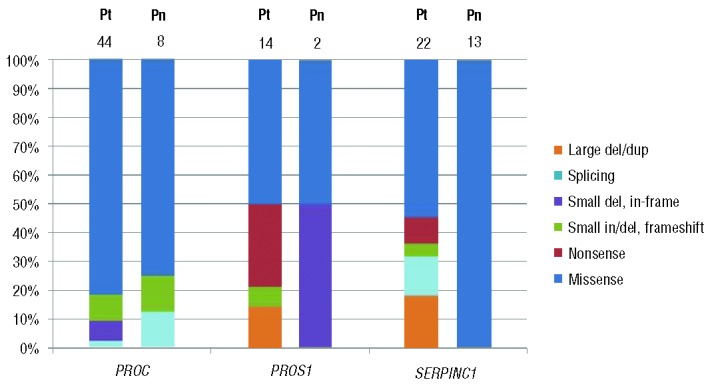
The distribution of mutation types in each gene in the patient (Pt) and population (Pn) groups. The numbers at the top of each bar represent the number of mutations observed.
Natural anticoagulant deficiency and causative mutations in the population group
A total of 3,129 individuals from the general population were screened for PC, PS, or AT deficiency by coagulation tests. This population group consisted of 1,873 males and 1,256 females (male-to-female ratio, 1.49:1) with a mean age [± standard deviation (SD)] of 52.5 years (±9.5). The values of PC activity, PS free antigen, and AT activity were successfully obtained in 2,953, 3,033, and 3,046 individuals with the mean ± SD being 120.7±20.3, 101.0±22.2, and 102.8±10.2, respectively. We selected individuals with a level of any one of the three measurements below the 1st percentile of the normal population. For PC deficiency, 31 individuals had PC activity below the 1st percentile (<75%). Molecular genetic tests were performed in 21 individuals and seven had a single heterozygous mutation of PROC: Arg211Trp in six out the seven cases (85.7%) and a frameshift mutation in the other case (Figure 1A). For PS deficiency, 32 individuals had a level of free PS antigen below the 1st percentile (<60% in males and <51% in females). Molecular genetic tests were performed in 13 individuals and two each had a single mutation in PROS1 (Figure 1B). For AT deficiency, 30 individuals had AT activities below the 1st percentile (<74%). Molecular genetic tests were performed in 26 individuals and 13 had a single heterozygous mutation of SERPINC1 (50%, 13/26). All SERPINC1 mutations in the population group were missense mutations, with two predominant mutations (Arg79Cys in 7 individuals and Ser148Pro in 5) (Figure 1C and Figure 2). None had double mutations or large dosage mutations on MLPA in the population group. In the population group, one novel mutation was detected in PROS1 (Ser587del), with a population frequency of 0% (Table 1). The mutation detection rates were 33.3% in PC deficiency (7/21), 15.4% in PS deficiency (2/13), and 50% in AT deficiency (13/26). Collectively, 22 individuals were found to have genetically proven natural anticoagulant deficiencies in our sample of the Korean population. The population frequencies of PC, PS, and AT deficiencies were thought to be at least 0.24% (7/2,953), 0.066% (2/3,033), and 0.43% (13/3,046), respectively. Taking into consideration the individuals without samples available for genetic analyses, the extrapolated frequencies were approximately 0.35%, 0.16%, and 0.49% for PC, PS, and AT deficiencies, respectively (1.0% when all 3 natural anticoagulants were combined). According to a review of the medical records of those 22 individuals with genetically proven hereditary natural anticoagulant deficiency, none had a relevant clinical history of thromboembolism.
Discussion
In this study, we determined the frequencies and mutation spectrums of genetically confirmed natural anticoagulant deficiencies underlying genetic thrombophilia in Korea. The results demonstrated that AT deficiency is frequent both in VTE patients and in the general population in Korea. In particular, the frequency of AT deficiency in the population group was higher (0.49%) than that in previous population studies.9,12–16 In the group of patients with VTE, the frequency of genetically confirmed natural anticoagulant deficiency was 14.2% (71/500). PC deficiency was most frequent (50.7%), followed by AT deficiency (29.6%) and PS deficiency (19.7%). Among the 43 PROC mutations in the group of patients, two missense mutations, Arg211Trp and Met406Ile, accounted for more than half (53.5%) of all mutations (Figure 1A). In the corresponding population group, Arg211Trp was more predominant (accounting for 85.7%), while Met406Ile was not observed. Both Arg211Trp and Met406Ile (or Arg169Trp and Met364Ile) were first reported in Japanese patients with VTE and cause type I PC deficiency.17,18 Arg211Trp is a recurrent mutation occurring at a CpG hotspot at the α thrombin cleavage site of the heavy chain. It was reported to account for ~10% of PROC mutations in Japan and has also been described in Caucasian patients with VTE.19–21 Met406IIle occurs at a non-CpG site of the trypsin-like serine protease domain and has been described exclusively in Japan, accounting for ~8% of PROC mutations in Japanese VTE patients.21 Thus, both Arg211Trp and Met406Ile of PROC are recurrent in Korea and Japan, but much enriched in Korea (32% and 21% in Korea versus 10% and 8% in Japan, respectively). The unusually high frequency of AT deficiency in our population group was due to two prevalent missense mutations, Arg79Cys and Ser148Pro of SER-PINC1 (Figure 1C). By contrast, these mutations were observed in only three of the group of patients. Both Arg79Cys and Ser148Pro (or Arg47Cys and Ser116Pro), which were first described in Japan, result in loss of the heparin binding ability of the AT molecule and cause type IIb deficiency (a qualitative defect).22,23 Arg79Cys occurs at a CpG hotspot and has been reported to be recurrent, while Ser148Pro does not involve a CpG hotspot, and no other patient has been described to carry this mutation in the literature since the first report.24 Type II AT deficiency is further divided into three types based on the location of the molecular defect: IIa (thrombin-binding site), IIb (heparin-binding site), and IIc (pleiotropic). Among these, type IIb AT deficiency is known to be less thrombogenic and is less common in patients with VTE than in the general population.25 Our results, revealing a striking difference in the proportion of type IIb mutations between the patients and the population group [13.6% (3/22) versus 100% (13/13), respectively], are in line with the previous observations. In our study, PS deficiency was the least frequent natural anticoagulant deficiency both in the patients and in the population group and there were no predominant mutations (Figure 1B). As for the population-specific prevalent mutations, frequent Lys196Glu (K196E) of PROS1 in Japan results in PS deficiency being most frequent in both VTE patients and the general population (Table 3).26–28 Although the sensitivity of coagulation tests for detecting heterozygous Lys196Glu mutation is limited,27,29,30 earlier studies involving only coagulation tests also demonstrated a high frequency of PS deficiency in the Japan population.14,15,31 Lys196Glu was not observed in our study or in other previous studies, indicating that it is a unique mutation in Japan. In China, two recent studies found two predominant mutations of PROC, Lys173del and Arg189Trp, both in VTE patients and in the general population (Table 3).32,33 In our study, Lys173del and Arg189Trp were observed in three and one patients, respectively, but not in the population group (Figure 1A). Of note, all three patients with heterozygous Lys173del also had another PROC mutation (Table 2). Since Lys173del does not result in a significant decrease of the amidolytic activity of PC, it is plausible that any heterozygous carriers of Lys173del in the patient or population group would have been missed in this study.33 Studies from Western countries have shown that PC deficiency is the most frequent natural anticoagulant deficiencies in VTE patients, followed by PS and AT deficiency, with limited reports on predominant mutations.2,9 Ala416Ser (or Ala384Ser) of SERPINC1 (antithrombin Cambridge II) was reported to be frequent both in VTE patients and in the general population in Britain, while it was not detected in China.34,35 Collectively, the frequency and mutation spectrum underlying genetic thrombophilia were different among Asian countries due to the presence of population-specific prevalent mutations possibly from founder effects.
Table 3.
Population frequencies of natural anticoagulant deficiencies in Asian countries.
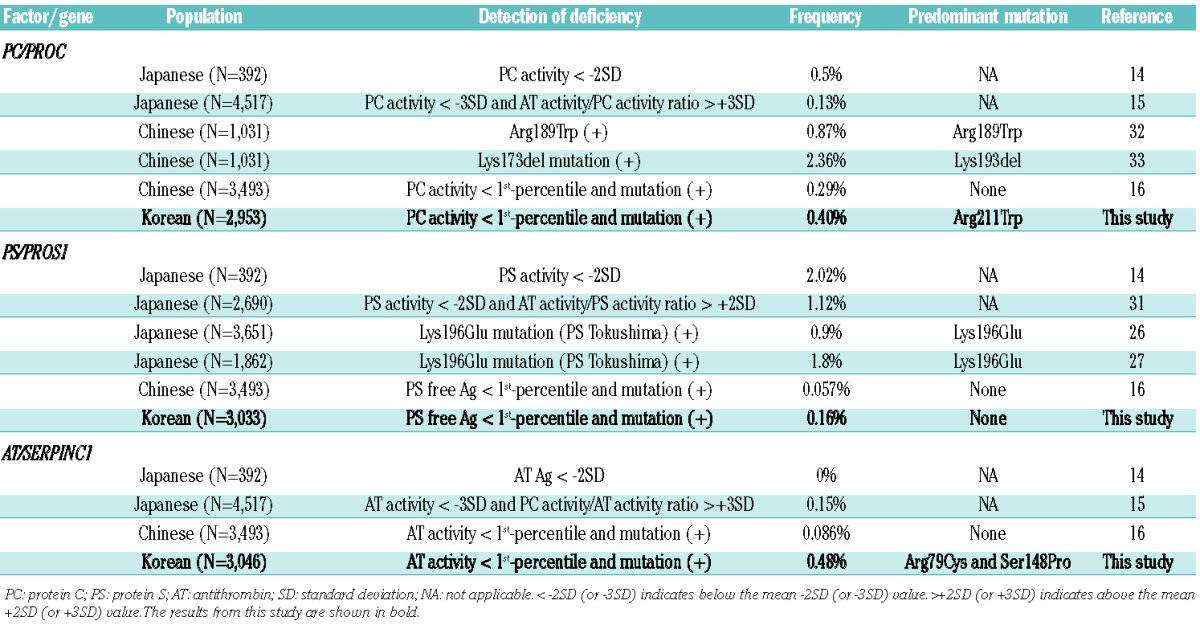
The frequency of genetically confirmed natural anticoagulant deficiency in consecutive (unselected) VTE patients in our series was 14.2%. The frequency of natural anticoagulant deficiency in VTE patients of Western ethnicity was reported to be ~8%.2,9 In Asian countries, the frequency had been reported to be 28.3% in Japan, based on coagulation tests, which was similar to the 25.4% with suspected natural anticoagulant deficiency in our study.14 In a recent study by Miyata et al., in which natural anticoagulant deficiency was detected by molecular genetic analyses without screening by coagulation tests, 32% of VTE patients had genetically confirmed natural anticoagulant deficiency.28 In addition to the presence of prevalent Lys196Glu of PROS1 in Japan, it was suggested that this high frequency could have been due to a selection bias in patient recruitment which had possibly enriched the study population with patients with natural anticoagulant deficiency. Likewise, the 14.4% frequency in the present study was lower than the frequency of 24.1% in our previous study on selected patients with (idiopathic) pulmonary embolism.36 The population frequencies of natural anticoagulant deficiency in Western ethnic populations have been described as 0.2–0.45% for PC deficiency, 0.03–1.3% for PS deficiency, and 0.02–0.25% for AT deficiency.9,37–42 The differences in the methodological strategy used to screen for and confirm the deficiency (e.g., type/frequency of tests, family study, and cutoffs) could also have affected the figures. In particular, earlier studies were based only on coagulation tests, while recent studies tend to involve molecular genetic tests to confirm the hereditary nature of the deficiency.
Molecular genetic tests are critical not only to rule out transient or acquired deficiency but also to understand the structure-function relationship and precise molecular pathophysiology (particularly relevant to thrombotic risk assessment in AT deficiency). For example, when a research group assessed the frequency of AT deficiency in the same population, the frequency decreased from 0.25% assessed only by coagulation tests to 0.17% when further molecular genetic confirmation was introduced.12,13 In our study, we first screened for natural anticoagulant deficiency by a panel of PC activity, PS free antigen, and AT activity tests, which are recommended as the first screening assays with high sensitivity and reproducibility, and low interference, despite the limitations regarding certain rare types of mutant molecules.43–45 We then selected individuals with a value <1st percentile for molecular genetic tests. For PS and AT deficiency, we additionally performed MLPA experiments when no point mutations were detected on direct sequencing analyses, considering the significant occurrence of large dosage mutations in PROS1 and SERPINC1.11 The figures we obtained are, therefore, conservative estimations of population frequencies of natural anticoagulant deficiency. A recent study from China adopted the same strategy as in our study (below 1st percentile of coagulation tests followed by molecular confirmation) on 3,493 individuals from the general population, and found that the frequency of natural anticoagulant deficiency was 0.43% (PC deficiency 0.29%, AT deficiency 0.086%, and PS deficiency 0.057%) without any prevalent mutations (Table 3).16 Thus, our findings of a population frequency of natural anticoagulant deficiency of ~0.49% and of AT deficiency with three recurrent missense mutations (Arg211Trp of PROC and Arg79Cys and Ser148Pro of SERPINC1) are distinct from the findings in previous studies in Asian countries as well as in those in Western populations.
The mutation detection rate for confirmation of natural anticoagulant deficiency in our study was 55.9% (71/127) in the group of patients and 36.7% (22/60) in the population group. In both groups, the rate was highest in SER-PINC1 (75% and 50% in patients and in the general population, respectively), followed by PROC (55.9% and 36.7%, respectively) and PROS1 (37.8% and 15.4%, respectively). The overall mutation detection rate (3 genes combined) and the order were similar to those recently reported in a large cohort of patients from Germany (overall 65.8%, with SERPINC1 84%, PROC 69.2%, and PROS1 43.2%).46 The overall mutation detection rate in the population group in our study was lower than that in the group of patients (36.7% versus 56.7%), but higher than the 15.8% in a population study in China recently reported by Zhu et al.16 In addition, the results from the study by Zhu et al. demonstrated the highest mutation detection rate in PROC (31.3%), followed by SERPINC1 (9.7%) and PROS1 (6.3%). This striking difference was in part due to the presence of two prevalent missense mutations (Arg79Cys and Ser148Pro) in the Korean population. Interestingly, however, two of three SERPINC1 mutations in the Chinese population were also Arg79Cys and Ser148Pro. Large dosage mutations were frequent in AT deficiency and PS deficiency in our group of patients (18.2% in SERPINC1 and 14.3% in PROS1) (Figure 2), and they were more frequent than in previous studies (9.1% and 6.4%, respectively).47,48 Although we did not perform MLPA experiments for PROC, the number of cases with large dosage mutations causing PC deficiency would have been quite limited when considering the rarity of these mutations in PROC (0.6%).47,48 None of the population group had large dosage mutations. A total of 16 novel mutations were identified in this study, two in PROC, seven in PROS1, and seven in SERPINC1 (Table 1).48 The large duplication mutation involving all exons (1 through 7) of SERPINC1 was detected in Patient SMC-Pt-AT16 with type I AT deficiency. We could not delineate the extent of the duplicated genomic segment in this case, but it could have been large enough to involve a significant extragenic region.11
Eight patients had double mutations (7 with PC deficiency and 1 with AT deficiency) (Table 2). Among seven patients with two PROC mutations, four patients were considered to have compound heterozygous mutations (severe PC deficiency) based on coagulation test results (PC activity 2–7%) with or without family studies (SMC-Pt-PC10, 11, 21, and 36). Three patients had Lys193del plus a missense mutation (SMC-Pt-PC14, 18, and 27). Although the PC activities of these three patients were 41–62%, the phase of the two mutations could not be inferred without testing their parents because of the minimal decrease of the amidolytic activity of PC in Lys193del carriers.33 The two SERPINC1 mutations in Patient SMC-Pt-AT21 were inferred to be on the same allele (cis) considering the AT activity of 43%. Of note, the mean age of onset of disease in four patients with severe deficiency from compound heterozygous mutations was lower than that in the other four patients (23 versus 39 years). Collectively, the proportion of severe deficiency in our series of patients was estimated to be 11% for PC deficiency and 0% for both PS and AT deficiency. Indeed, while severe PC deficiency with relatively preserved PC activity is not rare and has heterogeneous clinical manifestations, severe or homozygous PS/AT deficiencies are functionally deleterious, typically leading to a lethal phenotype or purpura fulminans.25,49,50 Life-compatible cases of severe AT deficiency were typically carrying two hypothrombogenic type IIb mutations (heparin-binding site), which could be relevant in Korea where these mutations are highly prevalent.25
In conclusion, this is the first large study on the genetic epidemiology of natural anticoagulant deficiencies in Korea. The results demonstrated that the frequencies and mutation spectrums of natural anticoagulant deficiencies underlying genetic thrombophilia in Korea are distinct from those in other Asian populations as well as in Western populations.
Supplementary Material
Acknowledgments
This study was supported by grant #SBRI C-A5-106 from Samsung Biomedical Research Institute, Seoul, Korea and by Diagnostica Stago, Asnieres, France. We would like to thank Dr. Schlegel and Dr. Morboeuf for their support and helpful discussion.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.De Stefano V, Finazzi G, Mannucci PM. Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood. 1996;87(9):3531–44 [PubMed] [Google Scholar]
- 2.Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344(16):1222–31 [DOI] [PubMed] [Google Scholar]
- 3.Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost. 2009;101(1):93–9 [PubMed] [Google Scholar]
- 4.Lijfering WM, Brouwer JL, Veeger NJ, Bank I, Coppens M, Middeldorp S, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood. 2009;113(21):5314–22 [DOI] [PubMed] [Google Scholar]
- 5.Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965;13:516–30 [PubMed] [Google Scholar]
- 6.Dahlback B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112(1):19–27 [DOI] [PubMed] [Google Scholar]
- 7.Angchaisuksiri P. Venous thromboembolism in Asia--an unrecognised and under-treated problem? Thromb Haemost. 2011;106(4): 585–90 [DOI] [PubMed] [Google Scholar]
- 8.White RH, Keenan CR. Effects of race and ethnicity on the incidence of venous thromboembolism. Thromb Res. 2009;123 (Suppl 4):S11–7 [DOI] [PubMed] [Google Scholar]
- 9.De Stefano V, Rossi E, Paciaroni K, Leone G. Screening for inherited thrombophilia: indications and therapeutic implications. Haematologica. 2002;87(10):1095–108 [PubMed] [Google Scholar]
- 10.Jang JH, Seo JY, Bang SH, Park IA, Kim HJ, Kim SH. Establishment of reference intervals for von Willebrand factor antigen and eight coagulation factors in a Korean population following the Clinical and Laboratory Standards Institute guidelines. Blood Coagul Fibrinolysis. 2010;21(3):251–5 [DOI] [PubMed] [Google Scholar]
- 11.Kim HJ, Kim DK, Yoo KY, You CW, Yoo JH, Lee KO, et al. Heterogeneous lengths of copy number mutations in human coagulopathy revealed by genome-wide high-density SNP array. Haematologica. 2012;97(2): 304–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tait RC, Walker ID, Islam SI, McCall F, Conkie JA, Mitchell R, et al. Influence of demographic factors on antithrombin III activity in a healthy population. Br J Haematol. 1993;84(3):476–80 [DOI] [PubMed] [Google Scholar]
- 13.Tait RC, Walker ID, Perry DJ, Islam SI, Daly ME, McCall F, et al. Prevalence of antithrombin deficiency in the healthy population. Br J Haematol. 1994;87(1):106–12 [DOI] [PubMed] [Google Scholar]
- 14.Suehisa E, Nomura T, Kawasaki T, Kanakura Y. Frequency of natural coagulation inhibitor (antithrombin III, protein C and protein S) deficiencies in Japanese patients with spontaneous deep vein thrombosis. Blood Coagul Fibrinolysis. 2001;12(2):95–9 [DOI] [PubMed] [Google Scholar]
- 15.Sakata T, Okamoto A, Mannami T, Matsuo H, Miyata T. Protein C and antithrombin deficiency are important risk factors for deep vein thrombosis in Japanese. J Thromb Haemost. 2004;2(3):528–30 [DOI] [PubMed] [Google Scholar]
- 16.Zhu T, Ding Q, Bai X, Wang X, Kaguelidou F, Alberti C, et al. Normal ranges and genetic variants of antithrombin, protein C and protein S in the general Chinese population. Results of the Chinese Hemostasis Investigation on Natural Anticoagulants Study I Group. Haematologica. 2011;96(7): 1033–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda M, Sugo T, Sakata Y, Murayama H, Mimuro J, Tanabe S, et al. A thrombotic state due to an abnormal protein C. N Engl J Med. 1988;319(19):1265–8 [DOI] [PubMed] [Google Scholar]
- 18.Miyata T, Zheng YZ, Sakata T, Tsushima N, Kato H. Three missense mutations in the protein C heavy chain causing type I and type II protein C deficiency. Thromb Haemost. 1994;71(1):32–7 [PubMed] [Google Scholar]
- 19.Grundy C, Chitolie A, Talbot S, Bevan D, Kakkar V, Cooper DN. Protein C London 1: recurrent mutation at Arg 169 (CGG----TGG) in the protein C gene causing thrombosis. Nucleic Acids Res. 1989;17(24):10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsay W, Greengard JS, Montgomery RR, McPherson RA, Fucci JC, Koerper MA, et al. Genetic mutations in ten unrelated American patients with symptomatic type 1 protein C deficiency. Blood Coagul Fibrinolysis. 1993;4(5):791–6 [PubMed] [Google Scholar]
- 21.Sakata T, Kario K, Katayama Y, Matsuyama T, Kato H, Miyata T. Studies on congenital protein C deficiency in Japanese: prevalence, genetic analysis, and relevance to the onset of arterial occlusive diseases. Semin Thromb Hemost. 2000;26(1):11–6 [DOI] [PubMed] [Google Scholar]
- 22.Koide T, Odani S, Takahashi K, Ono T, Sakuragawa N. Antithrombin III Toyama: replacement of arginine-47 by cysteine in hereditary abnormal antithrombin III that lacks heparin-binding ability. Proc Natl Acad Sci USA. 1984;81(2):289–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okajima K, Abe H, Maeda S, Motomura M, Tsujihata M, Nagataki S, et al. Antithrombin III Nagasaki (Ser116-Pro): a heterozygous variant with defective heparin binding associated with thrombosis. Blood. 1993;81(5): 1300–5 [PubMed] [Google Scholar]
- 24.Antithrombin Mutation Database (http://www1.imperial.ac.uk/departmentofmedicine/divisions/experimentalmedicine/haematology/coag/antithrombin/).
- 25.Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008; 14(6):1229–39 [DOI] [PubMed] [Google Scholar]
- 26.Kimura R, Honda S, Kawasaki T, Tsuji H, Madoiwa S, Sakata Y, et al. Protein S-K196E mutation as a genetic risk factor for deep vein thrombosis in Japanese patients. Blood. 2006;107(4):1737–8 [DOI] [PubMed] [Google Scholar]
- 27.Kimura R, Sakata T, Kokubo Y, Okamoto A, Okayama A, Tomoike H, et al. Plasma protein S activity correlates with protein S genotype but is not sensitive to identify K196E mutant carriers. J Thromb Haemost. 2006; 4(9):2010–3 [DOI] [PubMed] [Google Scholar]
- 28.Miyata T, Sato Y, Ishikawa J, Okada H, Takeshita S, Sakata T, et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res. 2009;124(1):14–8 [DOI] [PubMed] [Google Scholar]
- 29.Yamazaki T, Sugiura I, Matsushita T, Kojima T, Kagami K, Takamatsu J, et al. A phenotypically neutral dimorphism of protein S: the substitution of Lys155 by Glu in the second EGF domain predicted by an A to G base exchange in the gene. Thromb Res. 1993; 70(5):395–403 [DOI] [PubMed] [Google Scholar]
- 30.Hayashi T, Nishioka J, Shigekiyo T, Saito S, Suzuki K. Protein S Tokushima: abnormal molecule with a substitution of Glu for Lys-155 in the second epidermal growth factor-like domain of protein S. Blood. 1994;83 (3):683–90 [PubMed] [Google Scholar]
- 31.Sakata T, Okamoto A, Mannami T, Tomoike H, Miyata T. Prevalence of protein S deficiency in the Japanese general population: the Suita Study. J Thromb Haemost. 2004;2(6):1012–3 [DOI] [PubMed] [Google Scholar]
- 32.Tang L, Guo T, Yang R, Mei H, Wang H, Lu X, et al. Genetic background analysis of protein C deficiency demonstrates a recurrent mutation associated with venous thrombosis in Chinese population. PLoS One. 2012;7(4):e35773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang L, Lu X, Yu JM, Wang QY, Yang R, Guo T, et al. PROC c.574_576del polymorphism: a common genetic risk factor for venous thrombosis in the Chinese population. J Thromb Haemost. 2012;10(10):2019–26 [DOI] [PubMed] [Google Scholar]
- 34.Corral J, Hernandez-Espinosa D, Soria JM, Gonzalez-Conejero R, Ordonez A, Gonzalez-Porras JR, et al. Antithrombin Cambridge II (A384S): an underestimated genetic risk factor for venous thrombosis. Blood. 2007;109(10):4258–63 [DOI] [PubMed] [Google Scholar]
- 35.Zhang GS, Tang YM, Tang MQ, Qing ZJ, Shu C, Tang XQ, et al. Antithrombin Cambridge II(A384S) mutation frequency and antithrombin activity levels in 120 of deep venous thrombosis and 150 of cerebral infarction patients in a single center in Southern China. Blood Coagul Fibrinolysis. 2010;21(6):588–91 [DOI] [PubMed] [Google Scholar]
- 36.Lee M, No HJ, Jang SY, Kim N, Choi SH, Kim H, et al. Hereditary thrombophilia in Korean patients with idiopathic pulmonary embolism. Yonsei Med J. 2012;53(3):571–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miletich J, Sherman L, Broze G., Jr Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med. 1987;317(16):991–6 [DOI] [PubMed] [Google Scholar]
- 38.Tait RC, Walker ID, Reitsma PH, Islam SI, McCall F, Poort SR, et al. Prevalence of protein C deficiency in the healthy population. Thromb Haemost. 1995;73(1):87–93 [PubMed] [Google Scholar]
- 39.Dykes AC, Walker ID, McMahon AD, Islam SI, Tait RC. A study of protein S antigen levels in 3788 healthy volunteers: influence of age, sex and hormone use, and estimate for prevalence of deficiency state. Br J Haematol. 2001;113(3):636–41 [DOI] [PubMed] [Google Scholar]
- 40.Beauchamp NJ, Dykes AC, Parikh N, Campbell Tait R, Daly ME. The prevalence of, and molecular defects underlying, inherited protein S deficiency in the general population. Br J Haematol. 2004;125(5): 647–54 [DOI] [PubMed] [Google Scholar]
- 41.Koster T, Rosendaal FR, Briet E, van der Meer FJ, Colly LP, Trienekens PH, et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study). Blood. 1995;85(10):2756–61 [PubMed] [Google Scholar]
- 42.Faioni EM, Valsecchi C, Palla A, Taioli E, Razzari C, Mannucci PM. Free protein S deficiency is a risk factor for venous thrombosis. Thromb Haemost. 1997;78(5):1343–6 [PubMed] [Google Scholar]
- 43.Kottke-Marchant K, Comp P. Laboratory issues in diagnosing abnormalities of protein C, thrombomodulin, and endothelial cell protein C receptor. Arch Pathol Lab Med. 2002;126(11):1337–48 [DOI] [PubMed] [Google Scholar]
- 44.Marlar RA, Adcock DM, Madden RM. Hereditary dysfunctional protein C molecules (type II): assay characterization and proposed classification. Thromb Haemost. 1990;63(3):375–9 [PubMed] [Google Scholar]
- 45.Goodwin AJ, Rosendaal FR, Kottke-Marchant K, Bovill EG. A review of the technical, diagnostic, and epidemiologic considerations for protein S assays. Arch Pathol Lab Med. 2002;126(11):1349–66 [DOI] [PubMed] [Google Scholar]
- 46.Caspers M, Pavlova A, Driesen J, Harbrecht U, Klamroth R, Kadar J, et al. Deficiencies of antithrombin, protein C and protein S - practical experience in genetic analysis of a large patient cohort. Thromb Haemost. 2012;108 (2):247–57 [DOI] [PubMed] [Google Scholar]
- 47.Cooper PC, Goodeve AC, Beauchamp NJ. Quality in molecular biology testing for inherited thrombophilia disorders. Semin Thromb Hemost. 2012;38(6):600–12 [DOI] [PubMed] [Google Scholar]
- 48.Human Genome Mutation Database (HGMD® Professional) [Internet]. BIOBASE GmbH. Available at: http://www.biobase-international.com/product/hgmd
- 49.Kim HJ, Kim DK, Koh KC, Kim JY, Kim SH. Severe protein C deficiency from compound heterozygous mutations in the PROC gene in two Korean adult patients. Thromb Res. 2008;123(2):412–7 [DOI] [PubMed] [Google Scholar]
- 50.Tripodi A, Franchi F, Krachmalnicoff A, Mannucci PM. Asymptomatic homozygous protein C deficiency. Acta Haematol. 1990;83(3):152–5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.