

Abstract
Neuroligins are postsynaptic cell adhesion molecules that are important for synaptic function through their trans-synaptic interaction with neurexins (NRXNs). The localization and synaptic effects of neuroligin-1 (NL-1, also called NLGN1) are specific to excitatory synapses with the capacity to enhance excitatory synapses dependent on synaptic activity or Ca2+/calmodulin kinase II (CaMKII). Here we report that CaMKII robustly phosphorylates the intracellular domain of NL-1. We show that T739 is the dominant CaMKII site on NL-1 and is phosphorylated in response to synaptic activity in cultured rodent neurons and sensory experience in vivo. Furthermore, a phosphodeficient mutant (NL-1 T739A) reduces the basal and activity-driven surface expression of NL-1, leading to a reduction in neuroligin-mediated excitatory synaptic potentiation. To the best of our knowledge, our results are the first to demonstrate a direct functional interaction between CaMKII and NL-1, two primary components of excitatory synapses.
The processing and integration of information in the brain depends on cell-to-cell communication. Synapses, the sites of communication, are specialized asymmetrical connections between neurons that allow for the efficient transfer of information. Synaptic formation, structure, maturation and maintenance are sustained by a multifarious network of bidirectional cellular adhesion molecules that span the synaptic cleft, aligning the presynaptic active zone and postsynaptic density. In humans, alterations that perturb these cellular organizers are implicated in cognitive disorders, highlighting their critical roles at the synapse. Arguably, the best-characterized synaptic cell adhesion molecules are the presynaptic NRXNs and postsynaptic neuroligins1-5. The trans-synaptic heterophilic interaction between the extracellular domains of neuroligins and NRXNs can induce synapse formation and maturation6-11.
Neuroligins are postsynaptic proteins that contain a single transmembrane domain, a large extracellular acetylcholinesterase-like domain and a short cytoplasmic tail (c-tail) that includes many protein-protein interaction domains5,12-15. Rodents express four neuroligins (NL-1 to NL-4). Despite high sequence conservation among isoforms, subcellular localization and expression patterns vary among neuroligins16. NL-1 is localized predominantly to excitatory synapses, whereas NL-2 is confined primarily to inhibitory synapses6,8,9,17,18. In contrast, NL-3 is expressed at both types of synapses. NL-4 is enriched at glycinergic synapses, with initial studies having focused on the retina19,20.
Consistent with the localization of NL-1 at excitatory synapses, overexpression of NL-1 increases evoked excitatory postsynaptic currents (EPSCs) and synaptogenesis6,21-24. Moreover, NL-1 has been shown to be involved in diverse forms of synaptic plasticity across species25-30. NL-1–mediated synaptic potentiation is diminished after chronic blockade of NMDA receptors (NMDARs) or CaMKII, suggesting that the function of NL-1 is influenced by NMDAR signaling21. Although the regulatory mechanisms of NL-1–mediated, activity-dependent synaptic enhancement are poorly understood, previous reports have implicated the c-tail in the proper trafficking of NL-1 to the synapse, although its terminal PDZ ligand is not essential7,31. Moreover, induction of synaptic plasticity upregulates the surface expression of NL-1 (ref. 32). In addition, high-frequency stimulation increases the mobilization and number of NL-1 clusters in cultured hippocampal neurons33. Taken together, these results strongly suggest that NL-1 trafficking and subcellular localization are governed by synaptic activity, but the underlying mechanisms have remained elusive.
Protein phosphorylation is a principal and swift regulatory mechanism in the organization and functional modulation of receptors at the synapse34. CaMKII is a major component of the excitatory postsynaptic density and a critical mediator of synaptic plasticity. CaMKII-mediated phosphorylation acutely regulates the function and trafficking of postsynaptic substrates in response to synaptic activity35. Although NL-1–mediated synaptic potentiation depends on CaMKII activity, the direct phosphorylation of NL-1 by CaMKII has not been reported.
Here we show that NL-1 is a direct substrate of CaMKII and identify a phosphorylation site, T739, in the intracellular c-tail of NL-1 that is not conserved in other rodent neuroligin isoforms. We demonstrate that NL-1 T739 is specifically phosphorylated by CaMKII but not by other known activity-dependent kinases in the postsynaptic density. Additionally, T739 phosphorylation is upregulated by synaptic activity. A nonphosphorylatable mutant, T739A, markedly reduces the basal and activity-induced surface expression of NL-1 and, consequently, the postsynaptic actions of NL-1. Notably, this study establishes a direct functional and isoform-specific relationship between CaMKII and NL-1, two critical constituents of the excitatory synapse.
RESULTS
NL-1 is phosphorylated at T739 by CaMKII in vitro
NL-1–mediated potentiation of synaptic transmission depends on CaMKII21. Might NL-1 be a substrate for CaMKII? The intracellular domain of NL-1 contains several serine and threonine residues that could potentially be phosphorylated by CaMKII (Fig. 1a). An in vitro kinase assay with CaMKII, [γ-32P]ATP and engineered glutathione S-transferase (GST) fusion proteins containing the c-tail of NL-1 (amino acids 718–843) or GluA1 (positive control) revealed that NL-1 was robustly phosphorylated by CaMKII as assessed by radiography (Fig. 1a,b). Phosphorylation of NL-1 and GluA1 by CaMKII displayed similar reaction kinetics and were run to saturation (Supplementary Fig. 1a,b). We also evaluated phosphorylation by CaMKII on the c-tails of NL-2, NL-3 and NL-4 and found that NL-2 and NL-3 were not phosphorylated, whereas CaMKII phosphorylated human NL-4, albeit at a much lower level than it phosphorylated NL-1 (Fig. 1c), thus indicating that NL-1 is the best neuroligin substrate for CaMKII.
Figure 1.

NL-1 T739 is phosphorylated by CaMKII in vitro. (a) Alignment of the transmembrane domains and c-tails of NL-1 (mouse), NL-2 (rat), NL-3 (human) and NL-4 (human). The CaMKII phosphorylation site boxed in red, and the pT739-Ab epitope is boxed in gray. (b–d) Audioradiography analysis of GST fusion proteins that were incubated with purified CaMKII, PKA or PKC and [γ-32P]ATP (P-32). The arrow denotes autophosphorylated PKA. Total protein was visualized by Coomassie Brilliant Blue (CBB) protein staining, and GST and the GST-GluA1 c-tail served as the negative and positive phosphorylation controls, respectively, in b–d. (e) Electron transfer dissociation MS/MS spectrum of the phosphorylated NL-1 peptide 730RRCSPQRTTpTNDLTHAPEEEIM751 (where p indicates phosphorylation) found only in GST–NL-1 fusion proteins incubated with ATP and purified CaMKII and not those incubated with PKA or PKC. Samples were digested with chymotrypsin and then analyzed using LC/MS/MS method. m/z, mass-to-charge ratio. (f) Extracted ion chromatogram of a quadruply charged ion at m/z 681.30, which corresponds to the phosphorylated NL-1730–751 peptide, as shown in e, for GST–NL-1 without enzyme (red), with PKA (gray), with PKC (green) or with CaMKII (blue). (g) Extracted ion chromatogram of a quadruply charged ion at m/z 661.31, which corresponds to the nonphosphorylated NL-1730–751 peptide in GST–NL-1 without enzyme (red), with PKA (gray), with PKC (green) or with CaMKII (blue). Full-length blots are presented in Supplementary Figure 4 when applicable.
To identify the individual phosphorylated site(s), we generated point mutations of serine/threonine residues on NL-1 that are not conserved in NL-2 and NL-3 and discovered that mutating T739 to alanine (T739A) markedly reduced phosphorylation by CaMKII (Fig. 1d), whereas similar mutations of neighboring threonine residues had little or no effect.
Neither cyclic AMP (cAMP)-dependent protein kinase A (PKA) nor cAMP-dependent protein kinase C (PKC) phosphorylated NL-1 in vitro as robustly as did CaMKII (Fig. 1b). Furthermore, to detect whether PKA or PKC are able to phosphorylate NL-1 T739, we analyzed GST–NL-1 after in vitro kinase reactions using liquid chromatography coupled to tandem mass spectrometry (LC/MS/MS) and found that only CaMKII phosphorylates T739 (Fig. 1e–g). Additionally, using the LC/MS/MS method, we found that CaMKII phosphorylates the threonine in human NL-4 (T718) that is analogous to rodent NL-1 T739 (data not shown), which is not surprising considering the conservation of the CaMKII consensus sequence in human NL-4 and mouse NL-1 (Fig. 1a). Taken together, these results indicate that NL-1 T739 is the dominant and CaMKII-specific phosphorylation site in the intracellular tail of NL-1 and is not conserved in other excitatory synapse-specific neuroligins.
T739 phosphorylation is regulated by CaMKII in situ
To monitor the phosphorylation of NL-1 T739 in vitro and potentially in vivo, we generated a phosphorylation state–specific antibody, pT739-Ab, against residues 735–744 (Fig. 1a). We tested the specificity of pT739-Ab with an in vitro kinase assay in which we incubated GST–NL-1 (wild type or T739A), GST–NL-2, GST–NL-3 and GST–NL-4 c-tail fusion proteins with ATP and CaMKII. We resolved the proteins by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotting revealed that the phosphorylation state–specific antibody specifically recognized only the NL-1 c-tail that is phosphorylated at T739 (Fig. 2a). Notably, the nonphosphorylatable mutant (T739A), as well as the other neuroligin isoforms that we subjected to the same in vitro kinase assay, showed no immunoreactivity with pT739-Ab, highlighting the specificity of pT739-Ab for NL-1 phosphorylated at T739. It is noteworthy that phosphorylated human NL-4 was not efficiently detected by pT739-Ab, which reveals that either NL-4 T718 is not robustly phosphorylated by CaMKII or pT739-Ab is indeed specific for NL-1 phosphorylated at T739. Regardless, the CaMKII consensus sequence (RXXT) in NL-1 and human NL-4 is completely divergent in rodent NL-4 and therefore would not be detected in rodent lysate preparations and is not a concern in this study36. We chose human NL-4 for analysis, as it is used exclusively in the literature because of its implications in cognitive disorders5.
Figure 2.

NL-1 T739 is phosphorylated by CaMKII in vitro and in hererologous cells as detected by a phosphorylation state–specific antibody. (a) Immunoblot analysis with pT739-Ab of GST, GST–NL-1 (wild type or T739A), GST–NL-2, GST–NL-3 and GST–NL-4 that were phosphorylated in vitro with purified catalytic subunits of CaMKII. Immunoblotting with GST-Ab confirmed equal loading of the protein. WB, western blot. (b) Immunoblot analysis of NL-1 (wild type or T739A) transfected in COS cells and treated with a CaMKII inhibitor, KN93, or cotransfected with constitutively active CaMKII (T286D). (c) Cotransfection of NL-1 (wild type or T739A) with CaMKII (T286D) in HEK293T cells. Immunoblots were probed with the antibodies indicated in b and c. Full-length blots are presented in Supplementary Figure 4 when applicable.
To test whether the full-length NL-1 protein is phosphorylated in intact cells and modulated by CaMKII activity, we transfected wild-type NL-1 or NL-1 T739A in COS cells. Immunoblots of cell lysates probed with pT739-Ab indicated that NL-1 was phosphorylated at T739 under basal conditions and that no signal was observable with the phosphodeficient mutant (Fig. 2b). However, after cotransfection with a constitutively active form of CaMKII (T286D) or after incubation with KN93 (a CaMKII inhibitor), the basal-level phosphorylation of NL-1 T739 robustly increased and decreased, respectively (Fig. 2b). We observed analogous regulation in HEK293T cells, another non-neuronal mammalian cell line (Fig. 2c).
T739 phosphorylation is regulated by synaptic activity
The in vitro and in situ results described thus far demonstrate that CaMKII can phosphorylate NL-1 at T739. To test whether this phosphorylation occurs in neurons, we measured NL-1 T739 phosphorylation in cultured cortical neurons after 21 days in vitro (DIV) and found a specific band at approximately 120 kDa, which is the estimated molecular weight of full-length mature NL-1 (Fig. 3a), indicating basal phosphorylation of this site in neurons. The band was substantially reduced when we pre-infected cultures with a lentiviral short hairpin RNA (shRNA) against NL-1, demonstrating the specificity of the antibody in neurons. Notably, total NL-3 protein levels were not reduced, underscoring the specificity of the NL-1 knockdown.
Figure 3.

Phosphorylation of NL-1 T739 in neurons. (a) Detection of NL-1 T739 phosphorylation in DIV 21 cortical neurons with or without expression of a lentiviral shRNA, either scrambled (scrambled knockdown (KD)) or targeting NL-1 (NL-1 KD). (b) Regulation of NL-1 T739 phosphorylation under control conditions (DMSO) or in the presence of BCC with or without pretreatment of AP5 and NBQX (AP5/NBQX) in DIV 21 cortical neurons. Immunoblots were probed with the antibodies indicated. (c) Phosphorylated NL-1 (pNL-1) to total NL-1 (means ± s.e.m.) normalized to DMSO control (n = 7) with AP-5/NBQX pretreatment plus BCC (P > 0.05, n = 4) or with BCC treatment alone (P < 0.0006, n = 7). (d) Total NL-1 (means ± s.e.m.) normalized to actin control (n = 7) with AP5/NBQX pretreatment plus BCC (P > 0.05, n = 4) or with BCC treatment alone (P = 0.0156, n = 7). (e) Detection of NL-1 T739 phosphorylation in DIV 21 cortical neurons with or without expression of a lentiviral shRNA, either scrambled or targeting CaMKII. (f) Phosphorylated to total NL-1 (means ± s.e.m.) normalized to no virus treatment (n = 4) with scrambled knockdown (P > 0.05, n = 4) or CaMKII knockdown (P = 0.0286, n = 4). (g) Total CaMKII (means ± s.e.m.) normalized to actin control (n = 4) with scrambled knockdown (P > 0.05, n = 4) or CaMKII knockdown (P = 0.029, n = 4). (h) Detection of NL-1 T739 phosphorylation in adult wild-type or NL-1 knockout (KO) brains. Arrows in a and b denote the NL-1–specific band. IP, immunoprecipitation. *P < 0.05, ***P < 0.001. Full-length blots are presented in Supplementary Figure 4 when applicable.
To examine whether T739 phosphorylation is modulated by synaptic activity, we enhanced synaptic activity by treating cortical cultures with bicuculline (BCC), a GABAA receptor (GABAAR) antagonist, for 2 h, which induced a sevenfold increase in NL-1 T739 phosphorylation relative to treatment with a dimethylsulphoxide (DMSO) control (Fig. 3b,c). The induction of phosphorylation with BCC was efficiently blocked with a 1-h pretreatment of AP5 (D(–)-2-amino-5-phosphonovaleric acid) and NBQX (6-nitro-2,3-dioxo-1,4-dihydrobenzo[f]quinoxaline-7-sulfonamide), which are NMDAR and AMPAR antagonists, respectively (Fig. 3b,c), thus indicating that NL-1 T739 phosphorylation is dynamically regulated by changes in neuronal activity. Notably, BCC treatment also caused a reduction in total NL-1 protein that was prevented with AP5 and NBQX pretreatment (Fig. 3d), a likely result of NL-1 N-terminal shedding, as has been observed previously37,38.
Next we explored whether CaMKII phosphorylates NL-1 T739 in neurons. To examine this possibility, we used a knockdown approach in cultured cortical neurons and found that a 75% reduction in CaMKII levels resulted in a 60% reduction in T739 phosphorylation levels without effecting total NL-1 levels (Fig. 3e–g). Taken together with our in vitro and in situ results, this finding indicates that CaMKII is the dominant kinase that phosphorylates NL-1 T739 in neurons.
To investigate whether T739 phosphorylation occurs in vivo, we immunoprecipitated phosphorylated NL-1 from wild-type and NL-1 knockout brains and detected a 120-kDa band in the wild-type brain only (Fig. 3h). This band indicates that a subset of endogenous NL-1 is phosphorylated at T739 in an intact mouse brain. Collectively these data highlight that NL-1 T739 is phosphorylated in live animals and dynamically regulated by synaptic activity in neurons.
NL-1 T739A reduces functional synapses
Does T739 function in the trafficking of NL-1 to the cell surface? We examined the surface expression of NL-1 (wild type or T739A) expressed in cultured hippocampal neurons with immunofluorescence confocal microscopy. As previously described, we performed these experiments on a triple knockdown background to reduce the levels of endogenous neuroligins using exogenous chained microRNAs against NL-1, NL-2 and NL-3 (NLmiRs) to prevent the potential masking of neuroligin mutation phenotypes by dimerization with wild-type protein23. We labeled surface and intracellular receptors with an anti-hemagglutinin (anti-HA) tag that was inserted downstream of the signal peptide on NL-1. Notably, T739A greatly reduced the surface expression of NL-1 (Fig. 4a,b), suggesting that phosphorylation regulates the forward trafficking or stabilization at the plasma membrane of NL-1.
Figure 4.

T739A reduces the surface expression and synaptic enhancement of NL-1. (a) Hippocampal neurons coexpressing NLmiRs and NL-1 (wild type or T739A). Surface receptors were labeled with anti-HA and Alexa 555–conjugated secondary antibody (red). After fixation and permeabilization, internal receptors were visualized with anti-HA and Alexa 647–conjugated secondary antibody (pseudocolored white) (scale bar, 50 μm). The images below each row are enlargements of the boxes areas. (b) NL-1 T739A levels (P = 0.0001, n = 36; means ± s.e.m.) normalized to NL-1 (n = 34). (c) Neurons transfected as in a. VGLUT1 was labeled with anti-VGLUT1 and Alexa 647–conjugated secondary antibody (pseudocolored white), and PSD-95 was visualized with anti–PSD-95 and Alexa 555–conjugated secondary antibody (red). The images in c are shown at the same magnification as the bottom rows in a. (d) VGLUT1 levels (means ± s.e.m.) normalized to NL-1 (n = 24) for NLmiRs (P = 0.0001, n = 23) and NL-1 T739A (P = 0.0001, n = 24). (e) PSD-95 levels (means ± s.e.m.) normalized to NL-1 (n = 24) for NLmiRs (P = 0.0001, n = 23) and NL-1 T739A (P = 0.0001, n = 24). (f) Colocalized VGLUT1 and PSD-95 puncta (means ± s.e.m.) normalized to NL-1 (n = 24) for NLmiRs (P = 0.0001, n = 23) and NL-1 T739A (P = 0.0182, n = 24). (g) Representative mEPSC traces of hippocampal neurons expressing GFP, NLmiRs or NLmiRs and NL-1 (wild type or T739A) (vertical scale bar, 15 pA; horizontal scale bar, 500 ms). (h) Spontaneous mEPSC mean amplitudes (error bars, s.e.m.) of cells transfected with GFP (n = 9), NLmiRs (P > 0.05, n = 9), NLmiRs plus NL-1 (P = 0.0028, n = 9) or NLmiRs plus NL-1 T739A (P = 0.0003, n = 9) as compared to GFP-transfected cells. NL-1 T739A had no change in mEPSC amplitude when compared to NL-1 (not significant (NS, P > 0.05), n = 9). (i) Spontaneous mEPSCs (mean frequencies ± s.e.m.) of cells transfected with GFP (n = 9), NLmiRs (P = 0.0031, n = 9), NLmiRs plus NL-1 (P = 0.0012, n = 9) or NLmiRs plus NL-1 T739A (P > 0.05, n = 9) as compared to GFP-transfected cells. NL-1 T739A reduces mEPSC frequency when compared to NL-1 (P = 0.0001, n = 9). *P < 0.05 **P < 0.01, ***P < 0.001.
Does T739 phosphorylation function in NL-1–mediated synaptogenesis by regulating NL-1 surface expression? To examine this possibility, we used immunofluorescence confocal microscopy and coexpressed NL-1 (wild type or T739A) or GFP with NLmiRs in cultured hippocampal neurons and assayed endogenous VGLUT1 and PSD-95, which are presynaptic and postsynaptic markers, respectively. Knockdown of endogenous neuroligins nearly eliminated VGLUT1 and PSD-95 staining, which resulted in a profound decrease in colocalization of these synaptic markers, an anatomical measure of synapse number (Fig. 4c–f). Given the time course of knockdown in a developing neuronal culture, these results are consistent with a role of neuroligins in synapse assembly or maintenance. T739A expression only partially rescued VGLUT1 and PSD-95 recruitment and colocalization but did so to a consistently lesser extent than did expression of NL-1 (Fig. 4c–e).
Moreover, when we compared miniature postsynaptic currents (mEPSCs) in cultured hippocampal neurons transfected with NLmiRs and NL-1 (wild type or T739A), T739A did not enhance the mEPSC frequency, as we saw with expression of NL-1 (Fig. 4g,i). In agreement with previous results, knockdown of endogenous neuroligins (with NLmiRs) resulted in a reduction in mEPSC frequency23 (Fig. 4g,i). Furthermore, we observed no difference in mEPSC amplitudes between NL-1 and NL-1 T739A or between GFP- and NLmiR-transfected cells (Fig. 4g–i). Notably, neuroligin overexpression induced a reduction in mEPSC amplitude (Fig. 4g,h), a likely outcome of exogenous neuroligin expression increasing cell size. Together these results implicate a role for T739 phosphorylation in NL-1–mediated synaptogenesis, a likely consequence of reduced surface expression.
T739A mitigates activity-induced effects of NL-1
NL-1 mobilization and surface expression can be dynamically modulated by high-frequency stimulation and synaptic plasticity32,33. Does T739 phosphorylation function in activity-induced increases in NL-1 surface expression? To investigate this possibility, we used immunofluorescence confocal microscopy to compare the surface expression of NL-1 (wild type and T739A) with or without 2 h of BCC treatment (the same duration used to induce T739 phosphorylation; Fig. 3b,c) when expressed in cultured hippocampal neurons (using a setup analogous to that in Fig. 4). BCC induced a modest increase in NL-1 surface expression (Fig. 5a,b). Notably, the activity-driven increase in NL-1 surface expression was eliminated in the T739A mutant (Fig. 5a,b).
Figure 5.
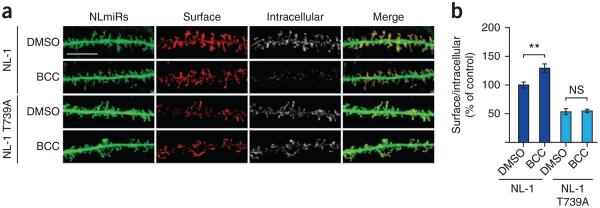
Activity-dependent increase in NL-1 surface expression is diminished in NL-1 T739A. (a) Hippocampal neurons coexpressing NLmiRs and NL-1 (wild type or T739A), as in Figure 4a, that were treated for 2 h with DMSO or BCC. Scale bar, 25 μm. (b) Levels of NL-1 plus BCC (P = 0.0027, n = 34), NL-1 T739A + DMSO (n = 28) and NL-1 T739A + BCC (P > 0.05, n = 28) normalized to NL-1 + DMSO (n = 36) (means ± s.e.m.). **P < 0.01.
NL-1–induced potentiation is diminished by T739A
Postsynaptic expression of NL-1 results in a robust enhancement of excitatory postsynaptic currents21,23,39-41. Might T739 function in NL-1–induced synaptic potentiation? To test this possibility, we expressed NL-1 (wild type or T739A) in organotypic hippocampal slice cultures using sparse biolistic transfection, which allows for the simultaneous recording of evoked excitatory currents in both transfected and neighboring control cells. The magnitude of the evoked current in the transfected cell compared to the untransfected control cell functions as a readout of the experimental manipulation on synaptic strength. We previously saw no significant effect of activity block or the T739A mutant compared to the wild-type protein with high levels of NL-1 expression (data not shown)23. We wondered whether these high expression levels might have obscured any effect of activity and decided to reduce the expression level by placing the NL-1 gene after the internal ribosomal entry site (IRES). Under reduced expression levels, NL-1 was still able to robustly potentiate both AMPAR and NMDAR currents, but at these reduced levels, NL-1 T739A did not potentiate AMPAR (Fig. 6a,b) or NMDAR (Fig. 6c,d) currents beyond those in neighboring control cells. Notably, the differences in synaptic strength between NL-1 and NL-1 T739A are not the result of changes in glutamate release, as paired-pulse ratios of AMPAR currents, a measure of presynaptic release probability, were unchanged in the transfected cells as compared to the untransfected cells (Supplementary Fig. 2a–c).
Figure 6.

Synaptic enhancement by NL-1 is reduced by either glutamate receptor blockade or the T739A mutation. (a) Low-level postsynaptic expression of NL-1 (n = 17) results in an enhancement of AMPAR-mediated currents to a greater extent than does the expression of NL-1 in the presence of AP5 and NBQX (P = 0.0439, n = 15) or the expression of NL-1 T739A (P = 0.0470, n = 12). Expression of NL-1 T739A in the presence of AP5 and NBQX did not further reduce AMPAR currents (P > 0.05, n = 9). The data are plotted as the percentage of control (mean ± s.e.m.) comparing transfected cells to simultaneously recorded, neighboring untransfected cells. All expression is on the background of the NLmiRs. (b) Scatter plots showing the individual conditions summarized in a. Open circles represent individual paired recordings, and filled circles represent the means ± s.e.m. The traces show representative currents for each condition, with the transfected cell in color and the control cell in black (vertical scale bars, 30 pA; horizontal scale bars, 20 ms). (c) A similar result to that in a is shown for NMDAR-mediated currents, with a greater enhancement by low-level postsynaptic expression of NL-1 (n = 17) than by NL-1 expression in the presence of AP5 and NBQX (P = 0.0363, n = 15) or the expression of NL-1 T739A (P = 0.0599, n = 12). Expression of NL-1 T739A in the presence of AP5 and NBQX did not further reduce NMDAR currents (P > 0.05, n = 7). The bar graph is as described in a. (d) Scatter plots showing the individual conditions summarized in c. Open circles represent individual paired recordings, and filled circles represent the means ± s.e.m. Traces show representative currents for each condition, with the transfected cell in color and the control cell in black (vertical scale bars, 60 pA; horizontal scale bars, 100 ms). *P < 0.05.
A previous report highlighted that NL-1–mediated postsynaptic enhancement is dependent on synaptic activity and, specifically, CaMKII activation21. Notably, with the reduced expression level, we found that activity blockade with AP5 and NBQX mirrored the diminished postsynaptic enhancement seen with expression of NL-1 T739A (Fig. 6a–d). Cells expressing NL-1 T739A with the same drug treatment did not display a further decrease in synaptic strength (Fig. 6a–d).
Sensory experience induces NL-1 T739 phosphorylation
We then examined whether NL-1 T739 phosphorylation is modulated by synaptic activity in vivo. We initially discovered that T739 is robustly phosphorylated in the visual cortex, which is an area of sensory-evoked synaptic refinement42 (Fig. 7a). To accurately identify changes in phosphorylation levels between varying conditions and brain samples, we used protein lysate concentrations that did not saturate the amount of antibody we used for immunoprecipitation (Supplementary Fig. 3a,b). To test whether T739 phosphorylation is regulated by synaptic activity in vivo, we used a well-established dark-rearing paradigm that involves depriving mice of light for 5 d (postnatal day (P) 21–P26) and re-exposing them to light for 2 h (dark rearing plus light rearing). This procedure induces anatomical, functional and synaptic reassembly and refinement of cortical circuits in the developing visual cortex42,43. Dark-reared mice had a reduction in T739 phosphorylation (Fig. 7b,c) compared to their light-reared littermates. Furthermore, dark-reared mice that were subjected to 2 h of light rearing had an increase in NL-1 T739 phosphorylation when compared to either light-reared or dark-reared littermates (Fig. 7b,c). These results show that increases or decreases in neuronal activity can modulate NL-1 T739 phosphorylation in vivo.
Figure 7.
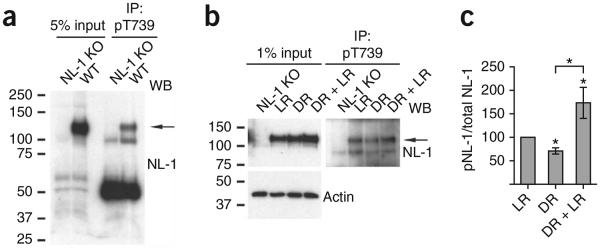
Synaptic activity dynamically regulates T739 phosphorylation in vivo. (a) Detection of NL-1 T739 phosphorylation in adult wild-type or NL-1 knockout visual cortex. (b) Detection of NL-1 T739 phosphorylation in the visual cortex of wild-type mice that were either light reared for 26 d (LR), light reared to P21 and then subjected to 5 d of dark rearing from P21 to P26 (DR) or light reared and then subjected to 5 d of dark rearing (P21–P26) and finally exposed to 2 h of light stimulus at P26 (DR + LR). When animals were light reared, they were maintained on a normal day and night cycle, whereas dark-reared animals were in complete darkness for 5 d. Also shown is the regulation of NL-1 T739 phosphorylation in light-reared, dark-reared or dark-reared plus light-reared P26 visual cortices. Adult NL-1 knockout visual cortices served as negative controls, and actin served as a total protein control. Nonsaturating protein concentrations (0.25 mg) were used, as described in Supplementary Figure 3a,b. (c) pNL-1 (immunoprecipitated) to total NL-1 (input) (means ± s.e.m.) normalized to light-reared control (n = 7) for the dark-reared (P = 0.0156, n = 7) and dark-reared plus light-reared (P = 0.0469, n = 7) conditions. All comparisons between conditions were using littermates. *P < 0.05. Full-length blots are presented in Supplementary Figure 4 when applicable.
DISCUSSION
Our findings indicate a direct and previously unidentified functional interplay between NL-1 and CaMKII, two hallmarks of excitatory synapses, which results in the activity-dependent phosphorylation of NL-1 at residue T739. Furthermore, using a combination of techniques, we found that a nonphosphorylatable mutant, T739A, displayed diminished basal and activity-induced NL-1–mediated synaptic potentiation.
Notably, NL-3, the other neuroligin isoform that is known to localize to excitatory synapses and that can also dimerize with NL-1, does not undergo the same protein modification. This is also the case for NL-2, although its endogenous localization is restricted to inhibitory synapses (Fig. 1c). When aligning the c-tail sequences of NL-1, NL-2 and NL-3, the analogous residue in NL-3 is an alanine and not a threonine and contains a glycine insertion within the RXXT CaMKII consensus motif of NL-1. Comparably, NL-2 contains a 12-amino-acid insertion in the corresponding region that is not contained in NL-1, NL-3 or NL-4. Therefore, the sequences of NL-2 and NL-3 are sufficiently divergent from that of NL-1 such that the consensus sequence is not conserved and is unsuitable for CaMKII phosphorylation. To the best of our knowledge, this is the first reported biochemical distinction between the c-tails of NL-1 and NL-3. Conventional domains on the c-tail such as the PDZ ligand and the gephyrin binding domain are conserved among all neuroligin isoforms (Fig. 1a)15. A newly discovered critical residue, which is important in regulating neuroligin-mediated postsynaptic effects at excitatory synapses, is evolutionarily preserved between NL-1 and NL-3 (NL-1 E747 and NL-3 E740; Fig. 1a)23. Additionally, a recent study found that Y782 is a phosphorylation site on the c-tail of NL-1 (ref. 44). This residue is conserved among all neuroligins (NL-2 Y770, NL-3 Y772 and NL-4 Y760; Fig. 1a) and may be an evolutionarily conserved phosphorylation site. The importance of the phosphorylation of NL-1, but not NL-3, by CaMKII in this region may underscore differences in synaptogenic strength, mobility or subcellular localization between NL-1 and NL-3. Alternatively, it is possible that through dimerization with NL-1, T739 phosphorylation could regulate the cellular or surface localization of NL-3. All of these possibilities certainly deserve attention in future studies.
Our findings show that CaMKII phosphorylates NL-1 T739 in vitro, in heterologous cells and in neurons. PKA and PKC, two activity-dependent kinases with similar consensus sequences and localizations to CaMKII, are unable to phosphorylate NL-1 T739 in vitro (Fig. 1e–g). Moreover, knockdown of CaMKII in heterologous cells (Fig. 2b) and neurons (Fig. 3e–g) results in a marked decrease in T739 phosphorylation. Likewise, overexpression of constitutively active CaMKII in heterologous cells results in a substantial increase in T739 phosphorylation (Fig. 2b,c). Furthermore, the previous finding that NL-1 synaptic potentiation is diminished with chronic blockade of CaMKII again supports CaMKII-specific regulation of NL-1 (ref. 21). However, we cannot definitively rule out the possibility that other kinases phosphorylate T739.
In addition to the proper trafficking of NL-1 under basal conditions, there is literature suggesting that NL-1 mobility is upregulated after increases in activity, showing an increase in NL-1 surface expression specifically32,33. Indeed, we were able to reproduce these findings, as we detected a significant rise in NL-1 surface expression after elevated network activity. Notably this activity-triggered increase was absent for NL-1 T739A, which is suggestive of a model in which activity promotes the phosphorylation of NL-1 T739 by CaMKII, further stabilizing or inducing the forward trafficking of NL-1 to the plasma membrane. It is tempting to speculate that this increase in surface expression might lead to activity-dependent synaptogenesis or spine growth. Notably, previous methods to assess activity-driven NL-1 mobility have varied in the stimuli or drug treatment protocol used, whether plasticity was induced and the techniques used to gauge surface expression32. It is possible there are different mechanisms of increasing NL-1 surface expression, which are examined under the varying protocols. Because of these caveats, we will refrain from concluding that T739 is the master switch in promoting activity-driven NL-1 surface expression. Additionally, there may be activity-dependent mechanisms that are independent of increasing NL-1 surface expression that contribute to the activity-dependent potentiation of NL-1. These possibilities, taken together with our results, still lead us to conclude that T739 is a prominent modulator of activity-induced NL-1 surface expression.
The molecular mechanism that regulates NL-1 surface expression through the phosphorylation of T739 by CaMKII remains to be elucidated. The substitution of T739 to an alanine (a phosphodeficient mutation) decreased the forward trafficking or stabilization of NL-1 at the cell surface. This result suggests that phosphorylation could promote a protein-protein interaction or break a retention interaction in the secretory pathway, thereby promoting or stabilizing NL-1 at the plasma membrane. The substitution of T739 to an aspartate also reduced surface expression of NL-1 (data not shown). This finding supports a dynamic role for T739 phosphorylation and is consistent with the nonphosphorylated threonine being critical at some stage of the trafficking process. Alternatively, the phosphomimetic may be unable to sterically mimic the phospho group, as is often seen in the literature45.
A previous report by our group failed to see an activity-dependent effect on NL-1–induced potentiation in organotypic slice cultures23. A key distinction between the previous and current studies is that we previously used high expression levels of NL-1, whereas in the current study we reduced total NL-1 expression levels and observed a significant activity-dependent effect of NL-1, as well as a decrease in postsynaptic currents by T739A, neither of which we observed with high expression levels. These results suggest that relative neuroligin levels may be critical when assessing the trafficking or phenotypes of other neuroligin mutations. Furthermore, this finding may underlie the apparent inconsistencies in the literature that varied in promoter strength, method and efficiency of plasmid entry.
Recent studies have identified an activity-dependent proteolytic cleavage of the extracellular domain of NL-1 that is regulated by the activation of NMDARs and, subsequently, CaMKII37,38. How might CaMKII, or synaptic activity more generally, induce increases in NL-1 surface expression and cleavage? We did not directly assess NL-1 cleavage in the present study, but we did observe a reduction in endogenous NL-1 protein levels after 2 h of BCC treatment that was blocked in the presence of AP5 and NBQX (Fig. 3b,d), with the most conceivable explanation being activity-induced NL-1 cleavage. As previously discussed, if network activity (a mechanism to activate CaMKII) promotes the surface expression of NL-1, it would present matrix metalloprotease 9 (MMP9) with more NL-1 substrate on the plasma membrane to cleave. Although increasingly complex hypotheses of how activity can regulate both NL-1 surface expression and cleavage are plausible, we deem this explanation to be the most parsimonious on the basis of the literature and our results.
The relationship between neuroligins and the etiology of autism remains unclear. Mutations in the extracellular domains of NL-3 and NL-4 have been associated with the disease, with a sole mutation having been found in the cytoplasmic tail of NL-4 (refs. 46-48). Although we cannot relate NL-1 T739 phosphorylation directly to the pathogenesis of autism, the principle of post-translational modifications modulating neuroligin function and subcellular localization may contribute indirectly to the pathology. Notably, NL-3 R451C, the most intensely studied neuroligin mutation in autism, displays secretory trafficking deficits and is retained in the endoplasmic reticulum, thus underlining the importance of the proper targeting of neuroligins and the drastic phenotypes seen by a single point mutant on localization49. We believe our findings elucidate a new avenue of future research, which may focus on how point mutations affect post-translational modifications that perturb neuroligin function.
Our present results further advance the neuroligin field and introduce a compelling new acute and local mechanism of neuroligin regulation: protein phosphorylation. Although protein phosphorylation has been shown to positively or negatively regulate numerous neuronal proteins, to our knowledge, this is the first isoform-specific phosphorylation site described on any neuroligin protein. Such a mechanism of regulation may also be observed in other neuroligin isoforms or in other recently discovered postsynaptic organizer molecules such as leucine-rich repeat transmembrane neuronal proteins (LRRTMs) and may contribute to isoform specificities seen in imaging and electrophysiological experiments50. Furthermore, the expanding association of neuroligins with cognitive disorders, particularly the implications of improper neuroligin trafficking, leads to speculation that better understanding of neuroligin function and regulation may provide therapeutic strategies for addressing synaptic dysfunction in clinical applications.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Plasmids, antibodies and neuronal cultures
Mouse pCAG-HA–NL-1 (or T739A)-IRES-mCherry, pCAG-eGFP, pCAG-NLmiRs-GFP and pCMV-CaMKII T286D plasmids were used for biochemical, electrophysiological (dispersed hippocampal cultures) and imaging experiments23. pCAG-IRES–NL-1 (or T739A), in which the protein was subcloned under the IRES to reduce expression, was used for organotypic slice culture experiments. All NL-1 constructs were RNA interference proof, as described previously23. Point mutations were made using PCR-based mutagenesis (QuikChange Site-Directed Instruction Manual). The primers used to construct NL-1 T737A were forward (FOR) 5′-GTGCAGCCCTCAGCGCGCGACCACCAACGACCTAAC-3′ and reverse (REV) 5′-GTTAGGTCGTTGGTGGTCGCGCGCTGAGGGCTGCAC-3′, those used for NL-1 T738A were FOR 5′-GCAGCCCTCAGCGCACGGCCACCAACGACCTAACCC-3′ and REV 5′-GGGTTAGGTCGTTGTGG CCGTGCGCTGAGGGCTGC-3′, and those used for NL-1 T739A were FOR 5′-CTCAGCGCACGACCGCCAACGACCTAACC-3′ and REV 5′-GGTTAG GTCGTTGGCGGTCGTGCGCTGAG-3′. GST c-tail constructs were amplified with synthetic primers containing EcoR1 and XhoI flanking regions from pCAG–NL-1 (mouse), pCAG–NL-2 (rat), pCAG–NL-3 (human) and pCAG–NL-4 (human) plasmids and subcloned into the GST fusion vector pGEX-6P-1 (GE Healthcare). To generate the phosphorylation state–specific antibody (pT739-Ab), rabbits were immunized with the synthetic phosphopeptide Ac-CQRTT(pT)NDLTH-amide corresponding to amino acids 735–744 of NL-1 generated by New England Peptide. Sera were collected and affinity purified with an antigen phosphopeptide. All immunoblotting using NL-1 T739-Ab began with 1 h of blocking in 5% PhosphoBLOCKER (CELL BIOLABS, INC) at room temperature, followed by blocking in 1% PhosphoBLOCKER in the primary and secondary antibody incubations. The antibodies used in the study were anti–NL-1 4C12 (Synaptic Systems, 1:1,000), anti-GST (Bethyl Laboratories, 1:50,000), anti-HA rat (Roche, 1:1,000), anti-HA rabbit (Abcam, 1:1,000), anti–NL-3 (Neuromab, 1:1,000), anti-CaMKII (Thermo Scientific, 1:1,000), anti–PSD-95 (Neuromab, 1:1,000), anti-VGLUT1 (Millipore, 1:5,000) and anti-actin (ABM, 1:5,000). Primary cultured neurons were isolated from male and female embryonic day 18 Sprague-Dawley rats. Cortical neurons were used for biochemical experiments, as the cortex provides sufficient material for protein analysis. Hippocampal cultures were used for immunocytochemistry and electrophysiology experiments. The use and care of animals used in this study followed the guidelines of the US National Institutes of Health Animal Research Advisory Committee.
GSt fusion protein production and in vitro phosphorylation
Using the protocol provided by GE Healthcare, GST fusion proteins were purified from BL21 bacterial cells transformed with pGEX–NL-1, pGEX–NL-2, pGEX–NL-3, pGEX–NL-4 or pGEX-GluA1. 50-ml cultures were grown at 37 °C to an absorbance at 600 nm (A600) of 1.1–1.2 after adding 1 ml of overnight cultures. To induce protein expression, 50 μM isopropyl β-D-1-thiogalactopyranoside was added to the cultures, and they were grown at 16 °C for 10–12 h. The cells were then lysed with a sonicator in a Tris-buffered saline (TBS) buffer containing protease inhibitors (Roche), 100 μg ml−1 lysozyme, 1 mM dithiothreitol (DTT) and 0.2 mM ethylenediaminetetraacetic acid (EDTA). The sonicated lysate was incubated with a 10:1 ratio of glutathione–Sepharose 4B (GE Healthcare) for 1 h at 4 °C and subsequently washed extensively with TBS buffer. For PKA in vitro phosphorylation, GST fusion proteins were phosphorylated in 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.0), 20 mM MgCl2, 50 μM ATP and 1 pmol of [γ-32P]ATP (3,000 Ci mmol−1) with 50 ng of purified PKA catalytic subunit (Promega). For PKC phosphorylation, reactions were performed in 20 mM HEPES, pH 7.4, 1.67 mM CaCl2, 1 mM DTT, 10 mM MgCl2, 50 μM ATP and 1 pmol of [γ-32P]ATP (3,000 Ci mmol−1) with 10 ng of purified PKC (Promega). For CaMKII phosphorylation, reactions were executed in 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, 2.4 μM calmodulin, 2 mM CaCl2, 100 μM ATP and 1 pmol of [γ-32P]ATP (3,000 Ci mmol−1) with 25 ng of recombinant CaMKII-α (Calbiochem). All in vitro kinase assays were performed at 30 °C for 30 min, except when stated otherwise. The reactions were halted with addition of SDS-PAGE sample buffer and incubation at 65 °C for 5 min. The proteins were resolved by SDS-PAGE and visualized by autoradiography. For in vitro kinase assays analyzed by immunoreactivity, SDS-PAGE–separated proteins were transferred to polyvinylidene difluoride membranes and assayed for phosphorylation (or total protein) by immunoblotting with their respective antibodies.
Mass spectrometry
Samples were alkylated with iodoacetamide and digested with chymotrypsin overnight at 25 °C. Peptides were analyzed by a nano-LC/MS/MS system with an Ultimate 3000 high-performance LC (HPLC) instrument (Thermo-Dionex) connected to an Orbitrap Elite mass spectrometer (Thermo Scientific) through an Easy-Spray ion source (Thermo Scientific). Peptides were separated on an ES800 Easy-Spray column (75-mm inner diameter, 15-cm length, 3-μm C18 beads; Thermo Scientific) at a flow rate of 300 nl min−1 with a 25-min linear gradient of 2–27% mobile phase B (mobile phase A: 2% acetonitrile, 0.1% formic acid; mobile phase B: 98% acetonitrile, 0.1% formic acid). The Thermo Scientific Orbitrap Elite mass spectrometer was operated in positive nano-electrospray mode. Mass spectrometry data were acquired in both profile and data-dependent modes. The resolution of the survey scan was set at 60,000 at m/z 400 with a target value of 1 × 106 ions. The m/z range for survey scans was 300–2,000. The isolation window for MS/MS fragmentation was set to 1.9, and the top two most abundant ions were selected for product ion analysis. The ion trap–enhanced scan rate was used for MS/MS data acquisition with the decision tree procedure activated. The dynamic exclusion was 9 s, and early expiration was abled. Xcalibur RAW files were converted to peak list files in mgf format using Mascot Distiller (version 2.4.3.3). The database search was performed using Mascot Daemon (2.4.0) against a house-built database containing NCBI human, GST NL-1 and NL-1 T739A sequences.
Immunoblotting
HEK293T or COS cells were transfected using Lipofectamine 2000 reagent (Invitrogen). 1–2 d after transfection, cells were washed in PBS and collected in a TBS buffer containing 150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA and protease (Roche) and phosphatase (Sigma) inhibitors. After centrifugation, pelleted cells were lysed directly with SDS-PAGE sample buffer and subjected to western blotting. KN93 (Tocris) or DMSO (Sigma) treatment occurred 2 h before cell isolation. DIV 21 cortical neurons were isolated and lysed with the same protocol as was used for the HEK293T and COS cells. Treatment with 40 μM bicuculline (Tocris) or DMSO began 2 h before cell isolation or 1 h after treatment with AP5 (Tocris) and NBQX (Tocris). For brain immunoprecipitations, wild-type or NL-1 knockout brains were homogenized in TEVP buffer (20 mM Tris (pH 7.5), 0.32 M sucrose, 5 mM EDTA and protease and phosphatase inhibitors). Homogenates were centrifuged at 800g for 10 min, then the nuclear pellet was discarded and the supernatant was centrifuged at 9,400g for 15 min. The P2 membrane fraction was resuspended in TEVP buffer supplemented with 1% SDS and 150 mM NaCl, gently sonicated and incubated at 37 °C for 15 min. The lysate was then neutralized with tenfold TEVP buffer supplemented with 2% Triton X-100 and 150 mM NaCl for a minimum of 15 min at 4 °C. The neutralized lysate was incubated with pT739-Ab and protein A–Sepharose beads (GE Healthcare) at 4 °C overnight. Samples were immunoblotted with NL-1 4C12 antibody after three washes in TEVP buffer. To knock down NL-1 or CaMKII in neurons, we subcloned a short hairpin targeting the rodent NL-1 sequence GGAAGGTACTGGAAATCTATA (previously characterized)6 or the CaMKII sequence TCCTCTGAGAGCACCAACA under the ubiquitin promoter in a FUGW lentivirus vector. To produce the virus, the hairpin targeting vector, a viral packaging vector and a VSVG envelope vector were cotransfected into HEK-293FT cells using FUGENE (Roche). Supernatants were collected 48 h after transfection, centrifuged at 82,000g to pellet the lentivirus and resuspended in PBS. Cortical neurons (in a six-well dish) were infected with 3–5 μl of concentrated virus for 7–10 d.
Immunocytochemistry
Cultured hippocampal neurons (DIV 10–12) were grown on glass coverslips precoated with poly-D-lysine (Sigma) and cotransfected with NLmiRs and HA–NL-1 (wild type or T739A) with Lipofectamine 2000 and analyzed at DIV 14–16. To label surface protein, transfected live cells were labeled with anti-HA (rat) for 10 min at room temperature. Cells were washed in PBS and then fixed in 4% paraformaldehyde and 4% sucrose in PBS for 8 min. The cells were incubated with Alexa 555–conjugated (red) anti-rat secondary antibody (Molecular Probes). After surface staining, cells were washed extensively and permeabilized in 0.25% Triton X-100, blocked in 10% normal goat serum and labeled with anti-HA (rabbit, 1:200). After being washed with PBS, cells were labeled with Alexa 647–conjugated (blue) anti-rabbit secondary antibody (shown in white; 1:500) and mounted with a ProLong Gold Antifade kit (Molecular Probes). Experiments that involved treating cultures with DMSO or 40 μM BCC had drug application 2 h before surface staining. For endogenous VGLUT1 and PSD-95 staining, cells were fixed, permeabilized and blocked as described above and then labeled with anti-VGLUT1 (guinea pig, 1:5,000) and anti–PSD-95 (mouse, 1:100). After primary antibody incubation, the cells were washed extensively and labeled with Alexa 647–conjugated (blue) anti–guinea pig secondary antibody (shown in white; 1:500) and Alexa 555–conjugated (red; 1:500) anti-mouse secondary antibody and mounted as described above. All primary and secondary antibody incubations were performed in the BioWave Pro system (Pelco). Neurons were imaged with a 63× objective on a Zeiss LSM 510 confocal microscope. For analysis, images from three dendrites per neuron from at least seven neurons per experiment were collected and quantified by normalizing the fluorescence intensity of surface-expressed NL-1 (or T739A) with the fluorescence intensity of total NL-1 (or T739A) in each cell using MetaMorph Version 7 (Universal Imaging). The fluorescence intensities of VGLUT1 and PSD-95 were measured similarly with the number of colocalized puncta obtained from three to four regions of 30 μm per cell. Cell selection was not done in the blind and was based on positive transfection, but all data analyses were done in the blind. Three independent experiments were performed per experiment, and the number of neurons (n) per condition is shown in the corresponding figure legend.
Electrophysiology
Whole-cell voltage clamp recordings of mEPSCs were performed in cultured hippocampal neurons. The time, duration and cotransfection protocol are analogous to the procedures used in the immunocytochemistry experiments. Recordings were made at 20–25 °C using glass patch electrodes filled with an internal solution consisting of 140 mM CsMeSO4, 5 mM NaCl, 10 mM HEPES, 10 mM ethylene glycol tetraacetic acid (EGTA), 4 mM Mg-ATP and 0.3 mM Na-GTP with a pH of 7.4 and Osm of 290 and an external solution containing 140 mM NaCl, 4 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 μM tetrodotoxin and 5 mM glucose. Cells cotransfected with NLmiRs and NL-1 (wild type or T739A) were visualized with fluorescence, mEPSCs were measured at −60 mV, and only cells with capacitances of 60 pF or above were analyzed to reduce variability. mEPSCs were measured offline with customized software (IGOR). Dual whole-cell recordings were made similarly using an internal solution containing 120 mM CsMeSO4, 20 mM CsCl, 4 mM NaCl, 10 mM HEPES, 4 mM NaCl, 0.4 mM EGTA, 0.3 mM CaCl2, 4 mM Mg-ATP, 0.3 mM Na-GTP and 5 mM QX-314 and an external solution containing 125 mM NaCl, 2.5 mM KCl, 4 mM MgCl2, 4 mM CaCl2, 1.25 mM NaH2PO4, 25 mM NaHCO3 and 11 mM glucose bubbled continuously with 95% O2 and 5% CO2. Recordings were made in the presence of picrotoxin (100 μM) to block inhibitory currents at −70 mV (AMPA) and +40 mV (NMDA). AMPAR-mediated currents were measured at the peak of the current, whereas NMDAR-mediated currents were measured 100 ms after the simulation. Slices pretreated with AP5 and NBQX had the drugs washed out before recordings were made. The paired-pulse ratio was determined by providing two pulses separated by 40 ms. The ratio was expressed as the peak current of the second EPSC over the peak current of the first EPSC. Cell selection was not done in the blind and was based on positive transfection, but all data analyses were done in the blind. At least three independent experiments were performed per condition, and the number of neurons per mEPSC or dual whole-cell recording (n) per condition is shown in the corresponding figure legend.
Dark rearing experiments
Male and female C57BL/6J littermates were maintained in a traditional light and dark cycle (12 h light, 12 h dark) from P0 to P26 (light reared), were relocated to complete darkness from P21 to P26 (dark reared) or were transferred to darkness from P21 to P26 and shifted back to light for 2 h before euthanization (dark reared plus light reared). The primary visual cortex was macrodissected in the dark for the dark-reared condition or in the light for the light-reared and dark-reared plus light-reared conditions. Homogenization of the visual cortex and NL-1 immunoprecipitations were performed as described in the section on immunoblotting.
Statistical analyses
The statistical significance of the immunoblots, immunocytochemistries, mEPSCs and dual whole-cell recordings was tested using a Mann-Whitney U test. The data distribution was assumed to not be normal. All experiments were done at least three independent times. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in the literature. All data were collected and processed randomly.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to A. Sanz-Clemente and A. Scimemi for technical assistance and for discussions on the project and manuscript. We thank the NINDS sequencing facility and light imaging facility for their expertise. This research was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (M.A.B., T.H., J.D.B., Y.L., J.S.D. and K.W.R.) and the National Institute of Mental Health grant number 5 R37 MH038256 (S.L.S., B.E.H. and R.A.N.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in theonline version of the paper.
AUTHOR CONTRIBUTIONS
M.A.B. designed experiments, performed all biochemical and imaging experiments, conducted electrophysiology experiments in disassociated hippocampal cultures and executed data analysis. M.A.B. and K.W.R. wrote the manuscript. S.L.S. and B.E.H. designed and conducted all electrophysiology experiments in slice cultures. T.H. designed constructs and aided in biochemistry and imaging experiments. Y.L. performed and analyzed all mass spectrometry data. J.D.B. aided in animal and biochemical experiments. K.W.R., J.S.D. and R.A.N. helped design experiments and supervised the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Yamagata M, Sanes JR, Weiner JA. Synaptic adhesion molecules. Curr. Opin. Cell Biol. 2003;15:621–632. doi: 10.1016/s0955-0674(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 2.Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- 7.Dean C, et al. Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levinson JN, et al. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1ß in neuroligin-induced synaptic specificity. J. Biol. Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- 10.Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl. Acad. Sci. USA. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wittenmayer N, et al. Postsynaptic Neuroligin1 regulates presynaptic maturation. Proc. Natl. Acad. Sci. USA. 2009;106:13564–13569. doi: 10.1073/pnas.0905819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichtchenko K, et al. Neuroligin 1: a splice site-specific ligand for b-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 13.Irie M, et al. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- 14.Iida J, Hirabayashi S, Sato Y, Hata Y. Synaptic scaffolding molecule is involved in the synaptic clustering of neuroligin. Mol. Cell. Neurosci. 2004;27:497–508. doi: 10.1016/j.mcn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Poulopoulos A, et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Ichtchenko K, Nguyen T, Südhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J. Biol. Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 17.Song JY, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- 19.Budreck EC, Scheiffele P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur. J. Neurosci. 2007;26:1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x. [DOI] [PubMed] [Google Scholar]
- 20.Hoon M, et al. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc. Natl. Acad. Sci. USA. 2011;108:3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chubykin AA, et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko J, et al. Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 2009;28:3244–3255. doi: 10.1038/emboj.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shipman SL, et al. Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat. Neurosci. 2011;14:718–726. doi: 10.1038/nn.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon HB, et al. Neuroligin-1–dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci. 2012;15:1667–1674. doi: 10.1038/nn.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc. Natl. Acad. Sci. USA. 2008;105:9087–9092. doi: 10.1073/pnas.0803448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blundell J, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010;30:2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlhaus R, El-Husseini A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav. Brain Res. 2010;208:96–105. doi: 10.1016/j.bbr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 28.Jung SY, et al. Input-specific synaptic plasticity in the amygdala is regulated by neuroligin-1 via postsynaptic NMDA receptors. Proc. Natl. Acad. Sci. USA. 2010;107:4710–4715. doi: 10.1073/pnas.1001084107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YB, et al. Neurexin-neuroligin transsynaptic interaction mediates learning-related synaptic remodeling and long-term facilitation in aplysia. Neuron. 2011;70:468–481. doi: 10.1016/j.neuron.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shipman SL, Nicoll RA. A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron. 2012;76:309–316. doi: 10.1016/j.neuron.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dresbach T, Neeb A, Meyer G, Gundelfinger ED, Brose N. Synaptic targeting of neuroligin is independent of neurexin and SAP90/PSD95 binding. Mol. Cell. Neurosci. 2004;27:227–235. doi: 10.1016/j.mcn.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Schapitz IU, et al. Neuroligin 1 is dynamically exchanged at postsynaptic sites. J. Neurosci. 2010;30:12733–12744. doi: 10.1523/JNEUROSCI.0896-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutiérrez RC, et al. Activity-driven mobilization of post-synaptic proteins. Eur. J. Neurosci. 2009;30:2042–2052. doi: 10.1111/j.1460-9568.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- 34.Kim JH, Huganir RL. Organization and regulation of proteins at synapses. Curr. Opin. Cell Biol. 1999;11:248–254. doi: 10.1016/s0955-0674(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 35.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 36.Bolliger MF, et al. Unusually rapid evolution of Neuroligin-4 in mice. Proc. Natl. Acad. Sci. USA. 2008;105:6421–6426. doi: 10.1073/pnas.0801383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peixoto RT, et al. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron. 2012;76:396–409. doi: 10.1016/j.neuron.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki K, et al. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron. 2012;76:410–422. doi: 10.1016/j.neuron.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. USA. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Futai K, et al. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95–neuroligin. Nat. Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shipman SL, Nicoll RA. Dimerization of postsynaptic neuroligin drives synaptic assembly via transsynaptic clustering of neurexin. Proc. Natl. Acad. Sci. USA. 2012;109:19432–19437. doi: 10.1073/pnas.1217633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- 43.Tropea D, Majewska AK, Garcia R, Sur M. Structural dynamics of synapses in vivo correlate with functional changes during experience-dependent plasticity in visual cortex. J. Neurosci. 2010;30:11086–11095. doi: 10.1523/JNEUROSCI.1661-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giannone G, et al. Neurexin-1b binding to Neuroligin-1 triggers the preferential recruitment of PSD-95 versus gephyrin through tyrosine phosphorylation of Neuroligin-1. Cell Rep. 2013;3:1996–2007. doi: 10.1016/j.celrep.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 45.Esteban JA, et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 46.Yan J, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry. 2005;10:329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 47.Jamain S, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Etherton MR, Tabuchi K, Sharma M, Ko J, Südhof TC. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor–mediated synaptic transmission in hippocampus. EMBO J. 2011;30:2908–2919. doi: 10.1038/emboj.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comoletti D, et al. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. 2004;24:4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linhoff MW, et al. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.