

Abstract
Substituted quinoline-2,4-dicarboxylates (QDCs) are conformationally-restricted mimics of glutamate that were previously reported to selectively block the glutamate vesicular transporters (VGLUTs). We find that expanding the QDC scaffold to benzoquinoline dicarboxylic acids (BQDC) and naphthoquinoline dicarboxylic acids (NQDCs) improves inhibitory activity with the NQDCs showing IC50 ~ 70 µM. Modeling overlay studies showed that the polycyclic QDCs resembled steroid structures and led to the identification and testing of estrone sulfate, pregnenolone sulfate and pregnanolone sulfate that blocked the uptake of l-Glu by 50%, 70% and 85% of control, respectively. Pregnanolone sulfate was further characterized by kinetic pharmacological determinations that demonstrated competitive inhibition and a Ki of ≈ 20 µM.
Keywords: glutamate; VGLUT; synaptic vesicle; quinoline 2,4-dicarboxylic acid; neuroactive steroids
Glutamate (l-Glu) is the major excitatory neurotransmitter in the CNS participating in fast synaptic transmission, plasticity and higher cognitive functions. The uptake of l-Glu into synaptic vesicles is mediated by the vesicular glutamate transporters (VGLUTs).1,2 Ca-dependent vesicle exocytosis releases l-Glu into the synaptic cleft where it activates ionotropic (iGluR) and/or metabotropic (mGluR) receptors. This signaling is terminated via the removal of l-Glu by high-affinity, Na-dependent excitatory amino acid transporters (EAATs) that move L-Glu into neurons and glia.
The vesicular l-Glu transporter isoforms (VGLUT1–3) belong to the solute carrier family SLC17 type I that are highly homologous proteins. VGLUT-mediated uptake of LGlu into vesicles is driven by a proton-dependent electrochemical gradient across the membrane created by a vacuolar-ATPase.1–3 In addition to differences in structure, mechanism, ionic preferences, and subcellular localization, VGLUT and EAAT (SLC 1 family) transport systems can be pharmacologically distinguished on the basis of substrate affinity with an l-Glu Km ~ 1–3 mM vs. 5–10 µM, respectively), and sensitivity to a structurally diverse group of competitive inhibitors. In contrast to the majority of EAAT blockers that are most often readily recognized as structural mimics of glutamate or aspartate,4–6 inhibitors of VGLUT range in structure from those with obvious similarities to LGlu (e.g., trans-ACPD, Ki ≈ 0.7 mM) to those that appear structurally unrelated to l-Glu including the ergot bromocryptine (Ki ≈ 22 µM)7 and the azo-dye Trypan Blue (Ki ≈ 50 nM) (Fig. 1).8 Such broad inhibitor structure diversity may be inherent to vesicular transporters, which appear to interact not only with excitatory neurotransmitters, but a variety of exogenous toxins, drugs and, perhaps, as yet unidentified endogenous molecules.
Figure 1.

Structures of glutamate and VGLUT inhibitors.
To better delineate the pharmacological specificity of VGLUT and further refine a ligand-based pharmacophore model, we have synthesized and evaluated a number of VGLUT inhibitors.8–14 In particular, the quinoline-2,4-dicarboxylates (2,4-QDCs; Fig. 1) have been shown to be a versatile, conformationally-constrained scaffold from which new compounds can be developed for SAR analyses.8,10,11 We previously showed that QDCs bearing lipophilic groups (phenyl, styryl, biphenyl, etc.) were more potent VGLUT inhibitors when compared to hydrophilic substituents10 suggesting the presence of one or more lipophilic domains associated with the VGLUT substrate binding site.
In this report, four novel QDC structures were prepared in which phenyl and naphthyl groups were fused onto the QDC scaffold rather than substituted. Aromatic groups were used in this study because they were found to improve inhibition as compared with aliphatic substituents.10,11 The resultant ligand panel was comprised of 5,6- and 7,8- benzo[h]quinoline dicarboxylic acids (BQDCs), and 5,6- and 7,8-naphtho[2,3- h]quinoline dicarboxylic acids (NQDCs) (Table 1).
Table 1.
Inhibition of VGLUT by QDCs at 250 µM.*
| Compound | Structure |
3H-L-Glu Uptake (% of control |
|---|---|---|
| 5,6-benz-quinoline-2,4-dicarboxylic acid: 5,6-BQDC (1) | 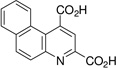 |
75 ± 4 |
| 7,8-benzo-quinoline-2,4-dicarboxylic acid: 7,8-BQDC (2) | 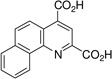 |
34 ± 1 |
| 5,6-naphtho-quinoline-2,4-dicarboxylic acids: 5,6-NQDC (3) | 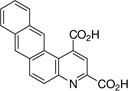 |
17 ± 3 |
| 7,8-naphtho-quinoline-2,4-dicarboxylic acid: 7,8-NQDC (4) | 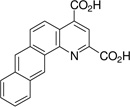 |
26 ± 1 |
| Estrone (Es) | 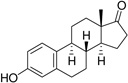 |
98 ± 8 |
| Estrone sulfate (Es-S) | 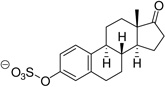 |
52 ± 8 |
| Estrone hemisuccinate (Es-Hs) | 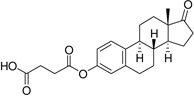 |
109 ± 20 |
| Pregnenolone (PGNE) | 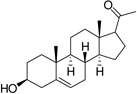 |
100 ± 4 |
| Pregnenolone acetate (PGNE-OAc) | 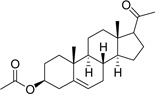 |
86 ± 5 |
| Pregnenolone sulfate (PGNE-S) | 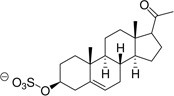 |
31 ± 3 |
| Pregnanolone (PGNA) | 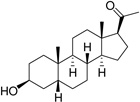 |
100 ± 6 |
| Pregnanolone sulfate (PGNA-S) | 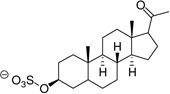 |
15 ± 4 |
Inhibitory activity is reported as mean % of control (uptake of 250 µM 3H-L-Glu in the absence of QDCs) ± SEM, n ≥ 3. Lower uptake values represent a greater degree of inhibition.
The 5,6- and 7,8-BQDC dimethyl esters were synthesized by a condensation reaction between dimethyl-2-ketoglutaconate (DMKG)15 and α- or β-aminonaphthalene, respectively, per our prior report (Scheme 1).10 Likewise, 5,6- and 7,8-NQDC dimethyl esters were synthesized from 1- or 2- aminoanthracene, respectively (Scheme 1). A single QDC regioisomer was isolated in each case. Following flash chromatography, each of the polycyclic BQDC/NQDC dimethyl esters were hydrolyzed to the corresponding BQDC or NQDC diacids (1–4; Table 1) using NaOH in THF/H2O (1:1), acidified with HCl to precipitate the product, and purified by recrystallization.16
Scheme 1.

Syntheses of fused QDCs from aminonaphthalenes and aminoanthracenes.10
The compounds were assessed as VGLUT inhibitors by quantifying their ability to reduce the uptake of 3H-L-Glu into synaptic vesicles prepared from rat brain.10,17 This preparation would be expected to contain all three VGLUT subtypes.18 As reported in Table 1, the benzoquinoline dicarboxylic acids (BQDCs) showed different binding activities: 5,6-BQDC 1 reduced L-Glu uptake by only 25%, whereas the 7,8-BQDC 2 isomer was far more effective, blocking 66% of uptake. The naphthyl derivatives were nearly equal in potency. The tetracyclic 5,6-NQDC 3 compound reduced 3H-L-Glu (250 µM) uptake over 80% and the angular analog 7,8-NQDC 4 was similarly effective. The NQDC analogs were better inhibitors as compared to the smaller, but positionally-equivalent BQDCs, whereas the 7,8-substituted analogs 2 and 4 did not differ greatly as inhibitors. The 5,6-NQDC 3 was the most potent of those tested, which is consistent with other aromatic-linked substituents on QDCs that are effective as VGLUT inhibitors, including 5- and 8-α-naphthyl, 6-[4,4’]biphenyl and 6-styryl.8,10
Taken together, the most effective QDC inhibitors of VGLUT were substituted with highly lipophilic, polycyclic groups.10 On inspection, these structures resemble steroids, and when combined with the observation that sulfonate-substituted azo-dyes like Trypan Blue (Fig 1) exhibit nanomolar Ki's at VGLUT, the neuroactive steroid hormones and their sulfated metabolites emerged as likely candidate structures to screen. Since QDCs and azo-dyes contain aromatic rings, the steroid panel included estrone, pregnenolone (PGNE) and pregnanolone (PGNA) and/or their sulfated metabolites to assay for inhibitory activity (Table 1). Estrone sulfate (Es-S) has been shown to inhibit the uptake of other aromatic anion dyes in CNS tissue, presumably mediated by an organic anion transporter.19
Estrone, PGNE and PGNA did not block L-Glu uptake to any appreciable level (Table 1). Assuming ligand binding to VGLUT requires the presence of one or more ionizable groups to mimic L-Glu, this result is not surprising. PGNE-acetate showed a modest reduction in L-Glu uptake while estrone hemisuccinate (Es-Hs) was inactive even though it is ionized at physiologic pH. It is possible the oxyanion may be too distant from the ring scaffold to bind transporter residues. Conversely, each of the sulfated neuroactive steroids showed markedly greater inhibition of VGLUT. For example, Es-S reduced L-Glu uptake by 50%, PGNE-S by 70% and PGNA-S by 85%, which is comparable to VGLUT inhibition by the BQDCs and NQDCs (Table 1).
A more detailed investigation of the concentration-dependence with which select inhibitors blocked 3H-L-Glu (250 µM) uptake demonstrated that 5,6-NQDC and 7,8-NQDC derivatives are equipotent, exhibiting IC50 values of about 70 µM (Fig. 2). Using the Cheng-Prusoff equation (Ki = IC50/(1 + [S]/Km)20 where [L-Glu] =250 µM and Km = 2.5 mM, affords Ki's of ~ 60 µM placing these NQDCs among the more potent QDC-based inhibitors yet identified.
Figure 2.

The inhibitory dose-response of 5,6-NQDC, 7,8-NQDC, Es-S and PGNA-S on the uptake of 3H-L-Glu (250 µM) into synaptic vesicles. Values are reported as % of control with each curve representing the average of at least three determinations. Error bars represent SEM. Control rates in these assays for L-Glu uptake were 1589 ± 127 pmol/min/mg protein.
The dose-response analyses for Es-S and PGNA-S yielded IC50 values of 121 µM and 42 µM, respectively (Fig. 2). These results further support the notion that select sulfate groups present on the azo-dye VGLUT inhibitors act as carboxylate bioisosteres.8,21,22 The most potent of the steroids tested, PGNA-S, was investigated in greater detail to elucidate the mechanism of inhibition. The resulting Lineweaver-Burk plot (Fig. 3) yielded a pattern of competitive inhibition showing an increase in Km,app with no change in Vmax. A replot of the slopes from the double reciprocal plots vs [I] yielded a Ki of 22 µM for PGNA-S that is approximately 100-fold more active than the endogenous substrate l-Glu (Km ~ 1–3 mM).
Figure 3.

Lineweaver-Burk plot demonstrating competitive inhibition by PGNA-S. Rates represent triplicates averaged from four different assays. The above plot yielded a Km of 2.5 mM, while a slope replot (inset) yielded a Ki for the inhibitor of 22 µM.
To better understand structure-activity relationships (SARs) governing Table 1 data, our previously described VGLUT pharmacophore model (GASP; Tripos Inc., St. Louis)8,14 was used to appraise energy minimized ligand overlays. The original VGLUT pharmacophore (Fig. 4, Panel A) was constructed from the inhibitors bromocryptine (BCP; orange), Chicago Sky Blue (CSB; yellow), and, 6-(4’-biphenyl)-2,4-QDC (QDC; gray) and used here to assess bioisostere group equivalencies of key moieties found in 7,8-NQDC (7,8-fused-naphthyl-2,4-QDC), 7,8-BQDC (7,8-fusedphenyl-2,4-QDC) and PGNA-S (Panel B). The overlays of the Panel B QDC's are found consistent with three previously described pharmacophore regions that contribute to enhanced binding to VGLUT: i) a carboxylic acid moiety, ii) an H-bond acceptor, and iii) a lipophilic domain; together that are consistent with other models.23
Figure 4.

Correlated VGLUT inhibitor ligand structural features; A: VGLUT pharmacophore8,14 overlay composed of one half of Chicago Sky Blue (CBS, yellow), bromocryptine (BCP, orange) and 6-(4’- biphenyl)-quinoline-2,4dicarboxylate (QDC, gray) - select regions described; and B: overlay of energy minimized 2 (7,8-BQDC, purple), 4 (7,8-NQDC, green) and PGNA-S (pink), where Panel B is correlated to the VGLUT pharmacophore Panel A regions.
The Panel B overlays suggest that either of the two carboxyl groups can be aligned to occupy the carboxyl region while occupying the Panel A lipophilic domain. This suggests that the QDCs may bind VGLUT in more than one orientation. The similar VGLUT inhibitor potencies of the 7,8-NQDC (green), 7,8-BQDC (purple) and 5,6-NQDC (not shown) suggests that their two COOH groups (and ring nitrogen atom) might be interchangeable with respect to binding the H-bonding and carboxylate bioisosteres (Fig. 4). The overlay further reveals that the 5,6-NQDC and 7,8-NQDC naphthyl groups assume similar spatial positions to produce better inhibitors, perhaps due to interactions between the naphthyl groups and a lipophilic region in VGLUT. The 7,8-BQDC analog may also be able to achieve a lipophilic interaction to a lesser extent, resulting in weaker VGLUT inhibition. Similarly, the reduced potency of 5,6-BQDC is considered a result of its less optimal lipophilic group position relative to the other moieties for VGLUT binding.
The low energy conformation of PGNA-S (Fig. 4, pink) aligns well with the VGLUT pharmacophore, including: i) the equivalency of A-C rings of the steroid with the QDCs and the steroid D ring relative to the pharmacophore lipophilic groups, ii) the alignment of the sulfate moiety with one of the QDC carboxylate groups, and iii) the element of steroid planarity relative carbon scaffold moieties of the other Panel A-B ligands. Thus, these sulfated steroid features are consistent with the pharmacophore model and provide a rationale for the observed inhibitory activity.
While inhibition by steroids would not likely have been obvious based upon a structural comparison with L-Glu, the sulfated neuroactive steroids do have structural similarities (e.g., carboxylate bioisostere, heteroatom, and a lipophilic domain) with other VGLUT inhibitors such as the aromatic azo-dyes and BCP (Fig. 1). Vesicular transporters, in general, exhibit a remarkably wide binding specificity as illustrated by the sensitivity of the monoamine transporter (VMAT 1 and 2) to inhibition by MPP+ (N-methyl-4-phenylpyridinium) and PCB's (penta- and hexa-polychlorinated biphenyls). Likewise, the inhibitory action of endogenous polyunsaturated fatty acids arachidonate, icosapentaneoate acid and linoleate at VGLUT and the VGAT (vesicular GABA transporter) support this.24 Since the constitution of VGLUT (and other neurotransmitter transporters e.g., EAATs) are with regions of lipophilic residues that are thought to be accessible to ligands, these regions could influence the binding of L-Glu during translocation guiding substrate permeation and/or regulate transporter activity via the binding of competitive endogenous inhibitors, such as neurosteroids.
This study demonstrates that VGLUT-mediated uptake of [3H]L-Glu into synaptic vesicles is inhibited to various extents by QDC ligands composed with discrete aromatic positional lipophilic groups and, for the first time sensitivity to steroid sulfates has been demonstrated. Sulfated steroids are produced by sulfotransferases localized to the cytoplasm of neurons.25,26 Sulfated neurosteroids have also have also been identified as modulators of ionic glutamate receptors and GABA receptors.27,28 In particular, pregnenolone sulfate has been reported to potentiate NMDA receptor signaling while pregnanolone sulfate inhibits NMDA, AMPA and KA receptors. The finding that neurosteroid sulfates inhibit VGLUT raises a number of interesting questions about their specificity and regulation of VGLUT, in addition to their plausible influences on other vesicular and related CNS transporters. Our efforts to further define the subtle aspects of QDC SARs at VGLUT are underway as are studies to delineate sulfate steroid activities at other CNS transporters.
Acknowledgments
This research was supported in part by NIH R21-NS42077 (RJB), RO1-NS38248 (CMT), and NCRR COBRE award P20-RR15583. We are also grateful to Kim Cylbuski for technical help and to the Core Laboratory for Neuromolecular Production P30 NS-055022 (CMT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Edwards RH. Neuron. 2007;55:835. doi: 10.1016/j.neuron.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Sudhof TC. Annu Rev Neurosci. 2004;27:509. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 3.Takamori S. Neurosci Res. 2006;55:343. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 4.Esslinger CS, Agarwal S, Gerdes J, Wilson PA, Davis ES, Awes AN, O'Brien E, Mavencamp T, Koch HP, Poulsen DJ, Rhoderick JF, Chamberlin AR, Kavanaugh MP, Bridges RJ. Neuropharm. 2005;49:850. doi: 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Esslinger CS, Koch HP, Kavanaugh MP, Philips DP, Chamberlin AR, Thompson CM, Bridges RJ. Bioorg Med Chem Lett. 1998;8:3101. doi: 10.1016/s0960-894x(98)00560-5. [DOI] [PubMed] [Google Scholar]
- 6.Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR. J Med Chem. 1991;34:717. doi: 10.1021/jm00106a037. [DOI] [PubMed] [Google Scholar]
- 7.Carlson MD, Kish PE, Ueda T. J Neurochem. 1989;53:1889. doi: 10.1111/j.1471-4159.1989.tb09258.x. [DOI] [PubMed] [Google Scholar]
- 8.Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM. Curr Med Chem. 2005;12:2041. doi: 10.2174/0929867054637635. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed SK, Etoga JL, Patel SA, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 2011;21:4358. doi: 10.1016/j.bmcl.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM. J Med Chem. 2002;45:2260. doi: 10.1021/jm010261z. [DOI] [PubMed] [Google Scholar]
- 11.Carrigan CN, Esslinger CS, Bartlett RD, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 1999;9:2607. doi: 10.1016/s0960-894x(99)00444-8. [DOI] [PubMed] [Google Scholar]
- 12.Denton T, Seib T, Bridges R, Thompson C. Bioorg Med Chem Lett. 2002;12:3209. doi: 10.1016/s0960-894x(02)00520-6. [DOI] [PubMed] [Google Scholar]
- 13.Etoga JL, Ahmed SK, Patel S, Bridges RJ, Thompson CM. Bioorg Med Chem Lett. 2010;20:2680. doi: 10.1016/j.bmcl.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel SA, Nagy JO, Bolstad ED, Gerdes JM, Thompson CM. Bioorg Med Chem Lett. 2007;17:5125. doi: 10.1016/j.bmcl.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corey EJ, Tramontano A. J. Am. Chem. Soc. 1981;103:5599. [Google Scholar]
- 16.All compounds showed elemental and spectral characteristics consistent with the structures. Naphtho[2,3-f]quinoline-1,3-dicarboxylic acid (3) - 17 mg, 89 % yield: 1H NMR (DMSO-d6) δ 9.09 (s, 1H), 8.73 (s, 1H), 8.31 (d, J = 8.8 Hz, 1H), 8.21 (d, J = 8.8 Hz, 1H), 8.19 (s, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.71 (p, J = 9.0 Hz, 2H); 13C NMR (DMSO-d6) δ 172.6, 167.1, 150.7, 149.0, 142.3, 134.5, 133.6, 132.6, 131.8, 130.3, 129.9, 129.3, 129.2, 128.7, 127.7, 126.6, 123.9, 122.3, 121.5; HRMS (ESI) m/z calcd for C19H12NO4 (M+H) 318.0766, found 318.0763. Naphtho[2,3-h]quinoline-2,4-dicarboxylic acid (4) - 15 mg, 79 % yield: 1H NMR (DMSO-d6) δ 9.91 (s, 1H), 8.67 (s, 1H), 8.59 (s, 1H), 8.54 (d, J = 9.6 Hz, 1H), 8.37-8.35 (m, 1H), 8.22 (d, J = 9.6 Hz, 2H), 7.71-7.69 (m, 2H); 13C NMR (DMSO-d6) δ 168.7, 167.3, 149.0, 147.8, 138.8, 134.4, 133.4, 132.1, 130.5, 130.1, 129.5, 128.7, 128.5, 128.3, 127.1, 126.1, 124.1, 123.4; HRMS ESI m/z calcd for C19H12NO4 (M+) 318.0766 found 318.0769.
- 17.Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ. Neuropharm. 1998;37:839. doi: 10.1016/s0028-3908(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 18.Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. Proc Natl Acad Sci U S A. 2004;101:7158. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichel V, Miller DS, Fricker G. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1311. doi: 10.1152/ajpregu.90373.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 21.Fykse EM, Fonnum F. Neurochem Res. 1996;21:1053. doi: 10.1007/BF02532415. [DOI] [PubMed] [Google Scholar]
- 22.Roseth S, Fykse EM, Fonnum F. Biochem Pharmacol. 1998;56:1243. doi: 10.1016/s0006-2952(98)00200-7. [DOI] [PubMed] [Google Scholar]
- 23.Almqvist J, Huang Y, Laaksonen A, Wang DN, Hovmöller S. Prot Sci. 2007;16:1819. doi: 10.1110/ps.072944707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhry FA, Edwards RH, Fonnum F. Annu Rev Pharmacol Toxicol. 2008;48:277. doi: 10.1146/annurev.pharmtox.46.120604.141146. [DOI] [PubMed] [Google Scholar]
- 25.Falany CN. FASEB J. 1997;11:206. doi: 10.1096/fasebj.11.4.9068609. [DOI] [PubMed] [Google Scholar]
- 26.Falany CN. FASEB J. 1997;11:1. doi: 10.1096/fasebj.11.1.9034159. [DOI] [PubMed] [Google Scholar]
- 27.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Pharmacol Rev. 2010;62:405. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S. Neurosci Biobehav Rev. 2013;37:109. doi: 10.1016/j.neubiorev.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]