

Abstract
Glutathione (GSH) is a critical intracellular antioxidant that is active in free radical scavenging and as a reducing equivalent in biological reactions. Recent studies have suggested that GSH can affect cellular function at the level of gene transcription as well, in particular by affecting NF-κB activation. Additionally, increased or decreased GSH levels in vitro have been tied to increased or decreased hepatocyte proliferation, respectively. Here, we investigated the effect of GSH on the liver’s response to TNF-α injection and 2/3 partial hepatectomy (PH), using mice deficient for the modifier subunit of glutamate-cysteine ligase (GCLM), the rate-limiting enzyme in de novo GSH synthesis. We demonstrate that Gclm−/− mice have a delay in IκBα degradation after TNF-α injection, resulting in delayed NF-κB nuclear translocation. These mice display profound deficiencies in GSH levels both before and during regeneration, and after PH, Gclm−/− mice have an overall delay in cell cycle progression, with slower DNA synthesis, mitosis, and expression of cell cycle proteins. Moreover, there is a delay in expression of downstream targets of NF-κB in the regenerating liver in Gclm−/− mice. These data suggest that GSH may play a role in hepatic NF-κB activation in vivo, which is necessary for accurate timing of liver regeneration.
Keywords: Hepatocytes, Glutathione, Liver disease, Liver regeneration
Introduction
Glutathione (GSH) is a tripeptide of glutamate, cysteine, and glycine. It is the most abundant non-protein thiol in the cell, and is present at 5–10 mM in hepatocytes [1]. GSH scavenges reactive oxygen species (ROS) and acts as a cofactor in the metabolism of xenobiotics through reduction and conjugation reactions. Several methods have been used in vivo and in vitro to study the effects of short-term GSH depletion on the hepatocyte cell cycle. Previous experiments have demonstrated that GSH levels increase in proliferating hepatocytes [2], and short-term depletion of GSH by chemical inhibitors of glutamate cysteine ligase (GCL), the enzyme that catalyzes the rate limiting step in GSH biosynthesis, has been shown to delay hepatocyte proliferation in vitro [3]. GSH levels are also elevated in human hepatocellular carcinoma [3]. These data suggest a significant interaction between cellular GSH levels and cell proliferation in the liver.
In addition to long-appreciated effects on cellular redox balance, recent studies demonstrate that GSH directly modifies proteins to affect their function. Specifically, Reynaert et al. [4] found that post-translational S-glutathionylation of the inhibitory κB kinase (IKK), which normally functions to activate NF-κB, can directly affect its activity in vitro, and that oxidative stress directly leads to this modification. Glutathionylation of mitochondrial proteins such as ATP synthase appears to directly affect their activity [5]. Further, glutathionylation of the NF-κB subunit p65 in cultured hepatoma cells is dependent on redox status, such that oxidative stress leads to this modification, subsequent inhibition of NF-κB activation, and decreased cell survival [6]. In the liver, NF-κB functions in diverse processes, including regulating innate immunity, preventing apoptosis, and the development of cancer [7–9]. NF-κB has also been shown to have a key role in liver regeneration after partial hepatectomy (PH), both by preventing apoptosis and allowing cell cycle progression [10,11]. Thus, we were interested in studying the potential interplay between GSH levels, NF-κB activation in the liver, and hepatocyte proliferation after 2/3 PH.
Prior work investigating the role of GSH in hepatocyte proliferation used in vivo chemical inhibition of GCL by D,L-buthionine sulfoximine (BSO) to temporarily decrease hepatic GSH content, though levels eventually increased to normal despite repeated doses of BSO [3]. In order to achieve sustained GSH deficiency during liver regeneration, we took a genetic approach rather than a chemical approach. GCL is composed of catalytic (GCLC) and modifier (GCLM) subunits [12,13]. Constitutive knockout of Gclc in mice is embryonic lethal [14,15], so we employed mice that lack Gclm [16] in our studies. While GCLM does not have any catalytic activity itself, it increases the efficiency of GCLC by lowering the Km for glutamate and ATP, and by decreasing feedback inhibition of GCL activity by GSH. We have previously reported that Gclm−/− mice are particularly susceptible to acetaminophen-induced toxicity [16], but resistant to the development of diet-induced steatohepatitis [17]. Here, we demonstrate that this model effectively maintains low GSH levels throughout liver regeneration, and are thus able to describe the consequences of compromised de novo GSH synthesis on this complex physiologic process. We show that GSH depletion interferes with the liver response to TNF-α in terms of NF-κB activation, and with the priming of liver regeneration. GSH depletion also results in a significant delay in DNA replication after PH but does not cause a prolonged blockage of liver mass restitution.
Materials and Methods
Animal studies/ethics statement
All animal procedures were in accordance with the NIH Guide for the Use and Care of Laboratory Animals and were approved by the University of Washington Institutional Animal Care and Use Committee (protocol 2877-01). Gclm+/− mice have been previously described [16], and we used a breeding scheme in which heterozygous Gclm females were mated with heterozygous Gclm males, but only Gclm−/− and Gclm+/+ (wild type) mice were used for our experiments. 3 to 6 mice per genotype per time point or condition were used for each experiment. Murine TNF-α (25 μg/kg body weight, R&D Systems, Minneapolis, MN, USA) was injected intraperitoneally (IP) into 8 to 10 week old male Gclm−/− and wild type (wt) mice, and animals were sacrificed at the indicated time points after injection. In a separate cohort of animals, 2/3 PH was performed on 8–10 week old male mice under isoflurane anesthesia following an overnight fast as described [18]. Mice were injected IP with bromodeoxyuridine (BrdU) (50 μg/g body weight, Roche Diagnostics, Indianapolis, IN, USA) 2 hours prior to sacrifice. Livers were harvested after CO2 euthanasia and cardiac puncture, and weighed prior to sectioning and storage.
Immunoblotting
Whole liver homogenates were prepared using 1% Triton X-100 lysis buffer containing protease inhibitors, quantified, and 40 μg of total protein were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes as described [18]. Immunoblotting was performed using standard procedures with the following antibodies: IκBα (Cell Signaling, Danvers, MA, USA), cyclin E (Upstate Biotechnologies, Billerica, MA, USA), or β-actin (Sigma, St. Louis, MO, USA).
Immunohistochemistry
Harvested livers were immediately fixed either in 10% neutral buffered formalin or methacarn (60% methanol, 30% chloroform, 10% glacial acetic acid) and prepared for histological analysis. Immunohistochemistry (IHC) for p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), BrdU (Dako, Carpinteria, CA, USA), and activated caspase 3 (Cell Signaling) was performed using standard techniques. To quantify hepatocyte proliferation, BrdU positive nuclei and mitotic figures were counted on slides cut from methacarn or formalin-fixed livers as described [19].
RNA isolation and real-time RT-PCR analysis
Total liver RNA was prepared using TRIzol (Invitrogen, Carlsbad, CA, USA), quantified using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE), and 1 μg was reverse transcribed using the Retroscript kit (Ambion, Carlsbad, CA, USA) as described [18]. Real time RT-PCR was then performed for Tnfα or Il6 using off the shelf FAM-labeled primers and reagents (Life Technologies, Carlsbad, CA, USA).
Determination of GSH levels & measurement of GCL activity
Total hepatic GSH levels were determined in protein lysates in TES/SB buffer with protease inhibititors from Gclm−/− and wt mice before and after PH using a fluorogenic assay as described previously [16]. Hepatic GCL activity was measured in liver lysates from Gclm−/− and wt mice before and after PH using a fluorogenic 96-well microtiter plate assay as described [20].
Enzyme Linked Immunosorbent Assay (ELISA)
Serum was obtained by cardiac puncture at the time of animal sacrifice, and IL-6 concentration therein was measured using a specific ELISA kit (R&D Systems) per the manufacturer’s instructions.
Fluorogenic caspase assay
Caspase 3 activity was measured in 100 μg of protein lysates in Triton X-100 buffer with protease inhibitors using DEVD-7-amino-4-methylcoumarin (Enzo Life Sciences, Inc, Farmingdale, NY, USA) as a substrate [21]. Enzymatic assays and standard curves were generated in duplicate using a fluorescent plate reader (Packard Instruments, Palo Alto, CA, USA), with AML12 cells [22] treated with Actinomycin D (Sigma-Aldrich) and TNF-α serving as positive controls [21].
Statistical analysis
Statistical analysis was done by non-parametric analysis using ANOVA with GraphPad Prism software (GraphPad for Science Inc, San Diego CA). Data are presented as average +/− S.E.M., with p<0.05 considered statistically significant.
Results
Gclm−/− mice as a model of severe and consistent GSH depletion before and after PH
Previous reports have demonstrated an increase in hepatic GSH levels in regenerating rat liver after PH [2]. As Gclm−/− mice lack the modulatory subunit of GCL, they lack the capacity to enzymatically regulate the catalytic subunit (GCLC) in response to changing cellular levels of the enzyme’s substrates, ATP and glutamate [16]. With these feed-forward mechanisms disrupted and the subsequent non-inducible nature of GSH production, we hypothesized that Gclm−/− mice [16] would not show an increase in GSH after 2/3 PH. Consistent with the initial characterization of Gclm−/− mice, we observed that hepatic GSH levels in non-operated (non-op) mice are roughly 15% of wt levels (Figure 1A). Similarly, GCL activity in Gclm−/− mice is less than 25% of that of wt mice (Figure 1B). We then measured hepatic GSH levels and GCL activity in wt and Gclm−/− mice after PH, and found that Gclm−/− mice maintained these low baseline GSH levels and GCL activity during liver regeneration. Conversely, wt mice have a two-fold increase in whole liver GSH levels during the early phase of regeneration. These data validate our use of Gclm−/− mice as a robust model of the effects of GSH deficiency on the regenerating liver.
Figure 1.
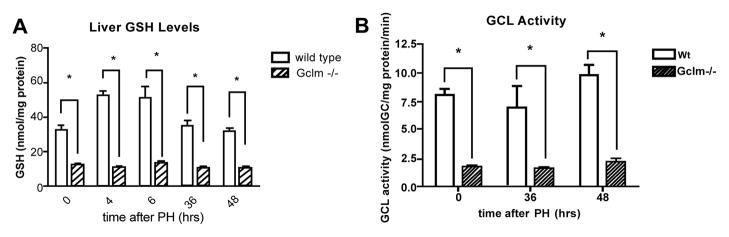
Gclm−/− mice are an adequate model of GSH depletion.
A: GSH content in the livers of wt and Gclm−/− mice before and after PH. *= p<0.05 by one-way ANOVA. GSH: glutathione; PH: partial hepatectomy; wt: wild type; Gclm: glutamate cysteine ligase modifier subunit.
B: GCL activity in the livers of Gclm−/− and wt mice before and after PH. *=p<0.05 by one-way ANOVA. GCL: glutamate cysteine ligase; n=3–6 mice per genotype per time point.
Abnormal NF-κB activation after TNFα injection into Gclm−/− mice
Recent studies have demonstrated that GSH can alter protein function directly [4]. One protein that is altered in this way is IKKβ, which normally functions to phosphorylate IκBα, thus allowing NF-κB to translocate to the nucleus to effect gene transcription [7]. We thus hypothesized that Gclm−/− mice would have abnormal NF-κB activation in the liver. We first chose to use TNF-α injection, a commonly used experimental method for activating NF-κB dependent transcription [23]. Gclm−/− mice and wt littermates received IP injections of TNF-α and were sacrificed either 15 or 30 minutes after injection. We first examined the kinetics of IκBα degradation, as this is an early step allowing subsequent NF-κB nuclear translocation and DNA binding. Immunoblot analysis demonstrated similar levels of IκBα protein in non-injected wt and Gclm−/− mice. Fifteen minutes after TNF-α injection, IκBα levels were equivalent in livers of wt and Gclm−/− mice. In wt mice, we observed that IκBα was almost completely absent from liver lysates by 30 minutes after TNF-α injection, in agreement with previously published data (Figure 2A). In contrast, Gclm−/− mice had persistent hepatic IκBα at 30 minutes after TNF-α injection, indicating that its proteasomal degradation is delayed in mice with low levels of GSH.
Figure 2.
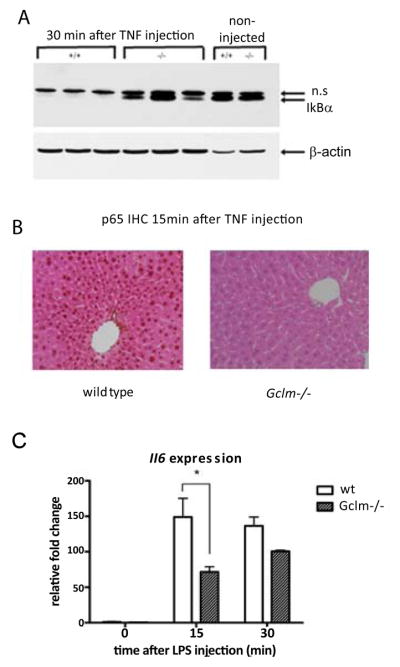
Gclm−/− mice have delayed NFκB activation after TNF-α injection.
A: Western blot demonstrating IκBα degradation 30 min after TNF-α injection. TNF-α: tumor necrosis factor-α; n.s.: non-specific band.
B: IHC for p65 in wild type and Gclm−/− mice injected with TNF-α. Images shown are at 40x magnification and are representative of 4–6 mice per group. Scale bar is 100 microns.
C: Real-time PCR for Il6 in whole liver RNA after TNF-α injection. Wt: wild type; Il6: interleukin-6; n=3–6 mice per time point per genotype. *=p, 0.05 by one-way ANOVA.
The continued presence of IκBα, an inhibitor of NF-κB, in Gclm−/− mice suggested a delay or attenuation of NF-κB signaling after TNF-α injection. To determine whether this delay altered TNF-α induced NF-κB nuclear translocation, we examined the cellular distribution of NF-κB in hepatocytes by IHC. In wt mice, we found strong nuclear staining for the NF-κB p65 subunit 15 minutes after TNF-α injection (Figure 2B), whereas Gclm−/− mice had an absence of nuclear staining at this time point, suggesting a defect in nuclear translocation of NF-κB in the setting of low GSH levels. By 30 minutes after TNF-α injection, we found nuclear p65 localization in both genotypes (data not shown).
Following IκBα degradation, NF-κB activates gene transcription to affect a broad range of cellular functions. To determine whether the delay in NF-κB nuclear translocation leads to a delay in NF-κB mediated gene transcription in Gclm−/− mice, we examined the expression of a known NF-κB target gene, Il6, after TNF-α injection by real-time PCR. In wt mice, there is an increase in Il6 expression at 15 and 30 minutes after injection (Figure 2C). In Gclm−/− mice however, induction of Il6 expression 15 minutes after TNF-α injection is significantly blunted. Our findings of decreased NF-κB activation after TNF-α injection in Gclm−/− mice lead us to hypothesize that liver regeneration, an NF-κB dependent process, would be defective in these mice.
Delayed DNA replication and hepatocyte proliferation in Gclm−/− mice after 2/3 PH
Two thirds PH in rodents stimulates compensatory hyperplasia of the remaining hepatocytes wherein they exit G0, synchronously re-enter the cell cycle, divide once or twice and return to quiescence [24]. In mice, the peak of the first round of DNA replication has been reported to occur around 36 hours after PH. To determine whether hepatocyte proliferation is delayed in Gclm−/− mice following PH, we injected the thymidine analog BrdU IP 2 hours prior to sacrifice, and analyzed newly synthesized hepatocyte DNA by BrdU staining. Consistent with previous data, wt mice have a peak of hepatocyte DNA replication at 36 hours that declines by 48 hours. In Gclm−/− mice, however, DNA replication is relatively low at 36 hours after PH, but increases to peak at 48 hours after PH (Figure 3A), demonstrating an overall delay in DNA synthesis after PH when hepatic levels of GSH are reduced.
Figure 3.

Delayed hepatocyte proliferation after PH in Gclm−/− mice.
A: BrdU incorporation in hepatocytes after PH, presented as the percentage of positively staining hepatocytes in 3000 cells examined for each mouse. *=p<0.05. BrdU: bromodeoxyuridine; PH: partial hepatectomy; wt: wild type. B: Hepatocyte mitotic counts after PH, presented as number of mitoses per 3000 hepatocytes examined. *=p<0.05. C: Western blot demonstrating cyclin E expression 36 hours after PH in wild type (+/+) and Gclm−/− (−/−) mice.
D: Liver weights expressed as a percentage of body weight 1, 2, and 6 days after PH in wt (wild type) and Gclm−/− mice; n=3–6 mice per genotype per time point.
To determine whether the delay in hepatocyte proliferation in Gclm−/− mice extended beyond the S phase of the cell cycle, we assessed mitotic figures in hematoxylin and eosin stained liver sections after PH. We observed the highest number of hepatocytes with mitotic nuclei at 40 hours after PH in wt mice. Consistent with the delay in BrdU incorporation into proliferating hepatocytes in Gclm−/− mice, there were significantly fewer mitotic hepatocytes at this time point in Gclm−/− mice than in wt mice (Figure 3B), though the number of mitoses in Gclm−/− livers appeared to be increasing at 48 hours. Similar to the decreased peak of BrdU labeling, Gclm−/− mice show a lower peak number of mitotic hepatocytes when compared to wt animals.
To determine whether the regenerative delay in Gclm−/− mice was present at the level of cell cycle control, we examined the expression of a late G1/S phase cell cycle protein, cyclin E, by immunoblot. We found that its expression is decreased in Gclm−/− mice compared to wt mice at 36 hours after PH (Figure 3C). Our data outlined above suggest that liver regeneration is delayed in Gclm−/− mice. To determine whether a delay in the hepatocyte cell cycle also altered the liver to body weight ratios in these mice, we measured this parameter in Gclm−/− mice up to 6 days after PH and compared them to wt mice. We did not observe a significant difference in liver to body weight ratios, suggesting that although liver regeneration is initially delayed in Gclm−/− mice, it does continue to completion (Figure 3D).
Increased apoptosis in Gclm−/− mice after PH
With their inability to up-regulate GSH levels in response to cellular stress, one might expect Gclm−/− mice to have more cellular injury after PH than do wt mice. While apoptosis is not typically noted in the remnant wt liver after 2/3 PH [25], several studies have linked inadequate antioxidant defenses to apoptosis in other systems [26]. We thus used a fluorogenic assay to measure the activity of caspase 3, the final apoptosis-executing enzyme, to assess whether there is apoptosis in the livers of Gclm−/− mice after PH. We confirmed the absence of detectable caspase 3 activity in wt liver lysates at 6, 24, and 48 hours after PH (Figure 4A). In Gclm−/− mice, however, there was a small but consistent amount of caspase activity at 24 and 48 hours after PH, suggesting that without sufficient GSH there is an apoptotic response to PH that is not seen in other animals.
Figure 4.

Elevated caspase activity and delayed cytokine induction in Gclm−/− mice after PH.
A: Fluorogenic assay for activated caspase 3 performed on liver lysates at the indicated times after PH. *=p<0.05 by one-way ANOVA.
B: Representative section demonstrating IHC for cleaved caspase 3, 24 hours after PH in a Gclm−/− liver at 20x. Inset is the hepatocyte designated by arrowhead, at 40x.
C: Real-time PCR for Tnfα in whole liver RNA after PH; *=p<0.05.
D: Circulating IL-6 levels after PH as measured by ELISA; *=p<0.05. wt: wild type; n=3–6 mice per genotype per time point.
In order to determine in which Gclm−/− cell types this modest apoptotic activity after PH was occurring, we performed IHC for cleaved caspase 3. We did not see any staining for cleaved caspase 3 in wt livers at 0, 24, or 36 hours after PH, but did see patchy cytosolic staining of hepatocytes in Gclm−/− livers 24 hours after PH (representative section shown in figure 4B), with no significant staining in the non-parenchymal cells of any livers examined. It should be noted that we did not see any evidence of liver necrosis in Gclm−/− or wt mice at any time point after PH. These data suggest that in the absence of adequate GSH, there is a small but consistent amount of hepatocyte apoptosis after PH, while apoptosis does not occur if GSH levels are normal.
Cytokine induction is delayed in Gclm−/− mice following PH
In order to determine whether the delay in liver regeneration seen in Gclm−/− mice could be related to defective NF-κB activation, we evaluated events known to be downstream of NF-κB after PH. In the regenerating liver, NF-κB activation in Kupffer cells leads to expression of Tnfα, transactivation of the type 1 TNF receptor, and the release of IL-6, which has been shown to play several roles early in liver regeneration [24]. We first measured expression of Tnfα after PH, and found that the induction of this gene at 2 hours after PH in wt mice is deficient in Gclm−/− mice (Figure 4C), confirming lack of induction of an NF-κB target gene in the GSH-deficient regenerating liver. Circulating levels of IL-6 typically peak within a few hours after PH, and consistent with previous findings, we found a marked increase in circulating IL-6 levels in wt mice at 4 hours after PH that decreases by 6 hours after surgery (Figure 4D). In contrast, serum levels of IL-6 in Gclm−/− mice are lower than those of wt littermates at 4 hours after PH. Six hours after PH IL-6 levels increase only slightly in Gclm−/− mice and are similar to the relatively low levels seen in wt mice at 6 hours after PH, suggesting that the release of IL-6 is attenuated in Gclm−/− mice, possibly related to deficient NF-κB activation.
Discussion
One of the early events in mammalian liver regeneration is the rapid activation of the NF-κB transcription factor [24]. This process is initiated by signaling through the type 1 TNF receptor and activation of IKK, which phosphorylates the inhibitor of NF-κB, known as IκBα [7]. The phosphorylation of IκBα leads to its ubiquitination and proteasomal degradation, which allows NF-κB to translocate to the nucleus and activate gene transcription. We show that in contrast to wt mice, GSH depleted mouse livers have persistent hepatic IκBα at 30 minutes after TNF-α injection, indicating that proteasomal degradation of IκBα is delayed and is possibly responsible for the inhibition of NFκB activation in these animals. Interestingly, TNFα signaling has previously been shown to induce survival or apoptosis depending upon the redox status of the cell [21,27,28].
Additionally, several recent reports have demonstrated that under certain conditions, GSH can form mixed disulfides with redox sensitive cysteine residues in proteins [4,29,30]. This process, which is called S-glutathionylation, has been shown to alter protein function. Several proteins in the NF-κB transcription factor pathway, including IKK, receptor-interacting protein (RIP), and the transcription factor components themselves, p50 and p65 [6] are among those that can be reversibly modified by GSH, thereby affecting their functions. Another possibility is that lack of GSH increases oxidative stress and alters protein function directly.
Given the decreased activation of NF-κB after TNF-α injection in Gclm−/− mice and the importance of NF-κB in liver regeneration, we expected that GSH depletion would lead to alterations in liver regeneration. Indeed, other investigators showed that temporary chemical depletion of GSH caused a delay in DNA replication after PH in rats [3]. The effect of oxidative stress after PH has also been studied in Nrf2 KO mice [31]. These animals have a deficiency in detoxifying enzymes and transient insulin/IGF resistance after PH. Oxidative stress in Nrf2 deficient animals causes steatosis and liver tumors, further demonstrating the crucial role of redox regulations in the liver. We were interested in the effects of prolonged and severe GSH depletion on the sequence of events that lead to hepatocyte proliferation. Gclm−/− mice lack the gene encoding the modifier subunit of GCL, and in the absence of GCLM, GCL enzyme function is compromised because of feedback inhibition of the enzyme by relatively low levels of GSH [13,16]. GCLM deficient mice are highly susceptible to the effects of acetaminophen, for instance, and conversely, animals that over-express GCLM are less sensitive to acetaminophen toxicity [16,32]. Here, we show that GSH is required for optimal release of IL-6 and activation of cell cycle components after PH, leading to a delay in DNA replication of approximately 8 hours in Gclm−/− mice. Interestingly, after this delay, even with a persistent GSH deficit, completion of regeneration in Gclm−/− livers is similar to that of wt livers.
The question of whether Gclm−/− mice have increased oxidative stress given their lack of GSH is an interesting one. The initial expectation was that Gclm−/− would have increased oxidative stress, but we have learned that these mice have significant compensation for their chronic lack of GSH by up-regulating several antioxidant genes, including thioredoxin reductase and heme oxigenase [17]. These data, in conjunction with evidence of decreased lipid peroxidation and catalase activity at baseline in Gclm−/− mice, lead us to believe that despite their inability to up-regulate GSH in response to oxidative stress, other compensatory pathways actually give Gclm−/− mice enhanced anti-oxidant capabilities [16,17]. We, therefore, do not believe that oxidative stress underlies the delay in liver regeneration in Gclm−/− mice.
An interesting, albeit subtle, finding in the regenerating livers of Gclm−/− mice was a small amount of hepatocyte apoptosis beginning 24 hours after PH. PH does not induce apoptosis in wt mice, unlike other models of regeneration, such as carbon tetrachloride injection [33]. This finding is in accordance with other studies demonstrating that redox stress can induce apoptosis in hepatocytes [34], but we do not believe that this low level of cell death could account for the delay in hepatocyte proliferation in Gclm−/− mice. Considering the profound GSH deficit in Gclm−/− mice, the phenotypes of delayed p65 activation after TNF-α injection and 8 hour delay in hepatocyte proliferation after PH may seem unimpressive. In our experience, any such delay in regeneration is significant, as liver regeneration is simultaneously driven by dozens of pathways, which easily compensate for one another in most settings [24,35]. It should be noted that although Gclm−/− mice have compromised de novo synthesis of GSH, they do show up-regulation of other genes that may partially compensate for the low levels of GSH in these mice [17,36]. These include altered expression of pathways important for maintaining thiol redox status (including glutathione disulfide reductase, thioredoxin reductase, sulfiredoxin, and ribonucleotide reductase), which are known to be important in nucleotide synthesis.
The effects of GSH depletion in the regenerating liver suggest that NF-κB activation is subject to regulation by GSH in this setting. Our results agree with those which demonstrate the redox regulation of NF-κB in mouse alveolar type II epithelial cells in culture [4]. Moreover, they showed that cys-179 of IKK is a target for activation by oxidative stress through S-glutathionylation. Another group demonstrated that in cultured hepatoma cells, oxidative stress leads to glutathionylation of p65, and subsequent decrease in NF-κB activation [6]. If NF-κB is similarly regulated in the liver in vivo, the increase in GSH reported to occur in the regenerating liver would modulate IKK activity and/or other NF-κB components, leading to activation of the transcription factor. We did not directly measure NF-κB activation itself in this setting, as it primarily occurs in Kupffer cells [10], which comprise a small portion of the total liver cells. Thus, in the evaluation of whole liver homogenates or RNA, signaling events in Kupffer cells may be overwhelmed by the lack of such signals in hepatocytes.
In summary, in the present work we demonstrate an in vivo dependence on intact GSH levels for normal NF-κB activation after TNF-α injection. Additionally, mice deficient in GSH synthesis have impaired priming, delayed DNA synthesis, and low level apoptosis after PH. This work may have implications for liver disease in humans, as relatively common genetic polymorphisms exist in both human GCLM and GCLC genes, and these have been shown to impact GCL expression, GSH synthesis, and risk for several diseases [12], including non-alcoholic steatohepatitis [37].
Acknowledgments
The authors thank Thomas Montine, Angela Wilson, Dianne Botta, Collin White, and B.J. Thompson for their input and technical assistance. This work was supported by the Herbert Coe Foundation, the American College of Surgeons Foundation, the American Surgical Association Foundation (all to KJR), and NIH grants CA-23226 (to NF), CA-127228 (to NF), and CA-174131 (to JSC).
References
- 1.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang ZZ, Li H, Cai J, Kuhlenkamp J, Kaplowitz N, et al. Changes in glutathione homeostasis during liver regeneration in the rat. Hepatology. 1998;27:147–153. doi: 10.1002/hep.510270123. [DOI] [PubMed] [Google Scholar]
- 3.Huang ZZ, Chen C, Zeng Z, Yang H, Oh J, et al. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001;15:19–21. doi: 10.1096/fj.00-0445fje. [DOI] [PubMed] [Google Scholar]
- 4.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, et al. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alisi A, Piemonte F, Pastore A, Panera N, Passarelli C, et al. Glutathionylation of p65NF-kappaB correlates with proliferating/apoptotic hepatoma cells exposed to pro- and anti-oxidants. Int J Mol Med. 2009;24:319–326. doi: 10.3892/ijmm_00000235. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty JB, Mann DA. NF-kappaB signalling: embracing complexity to achieve translation. J Hepatol. 2010;52:285–291. doi: 10.1016/j.jhep.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 8.He G, Karin M. NF-kappaB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Chaisson ML, Brooling JT, Ladiges W, Tsai S, Fausto N. Hepatocyte-specific inhibition of NF-kappaB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J Clin Invest. 2002;110:193–202. doi: 10.1172/JCI15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plumpe J, Malek NP, Bock CT, Rakemann T, Manns MP, et al. NF-kappaB determines between apoptosis and proliferation in hepatocytes during liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2000;278:G173–183. doi: 10.1152/ajpgi.2000.278.1.G173. [DOI] [PubMed] [Google Scholar]
- 12.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, et al. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med. 2009;30:86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, et al. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm (−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 16.McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, et al. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99:628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- 17.Haque JA, McMahan RS, Campbell JS, Shimizu-Albergine M, Wilson AM, et al. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab Invest. 2010;90:1704–1717. doi: 10.1038/labinvest.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, et al. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, et al. Proinflammatory cytokine production in liver regeneration is Myd88- dependent, but independent of Cd14, Tlr2, and Tlr4. J Immunol. 2006;176:2522–2528. doi: 10.4049/jimmunol.176.4.2522. [DOI] [PubMed] [Google Scholar]
- 20.White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ. Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal Biochem. 2003;318:175–180. doi: 10.1016/s0003-2697(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 21.Pierce RH, Campbell JS, Stephenson AB, Franklin CC, Chaisson M, et al. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am J Pathol. 2000;157:221–236. doi: 10.1016/S0002-9440(10)64533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu JC, Merlino G, Fausto N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci USA. 1994;91:674–678. doi: 10.1073/pnas.91.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Streetz KL, Wustefeld T, Klein C, Manns MP, Trautwein C. Mediators of inflammation and acute phase response in the liver. Cell Mol Biol (Noisyle- grand) 2001;47:661–673. [PubMed] [Google Scholar]
- 24.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 25.Zorde-Khvalevsky E, Abramovitch R, Barash H, Spivak-Pohis I, Rivkin L, et al. Toll-like receptor 3 signaling attenuates liver regeneration. Hepatology. 2009;50:198–206. doi: 10.1002/hep.22973. [DOI] [PubMed] [Google Scholar]
- 26.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, et al. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184:241–252. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal. 2009;11:2245–2263. doi: 10.1089/ars.2009.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Botta D, Franklin CC, White CC, Krejsa CM, Dabrowski MJ, et al. Glutamate-cysteine ligase attenuates TNF-induced mitochondrial injury and apoptosis. Free Radic Biol Med. 2004;37:632–642. doi: 10.1016/j.freeradbiomed.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 29.Aesif SW, Anathy V, Havermans M, Guala AS, Ckless K, et al. In situ analysis of protein S-glutathionylation in lung tissue using glutaredoxin-1- catalyzed cysteine derivatization. Am J Pathol. 2009;175:36–45. doi: 10.2353/ajpath.2009.080736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, et al. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol. 2011;44:491–499. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beyer TA, Xu W, Teupser D, auf dem Keller U, Bugnon P, et al. Impaired liver regeneration in Nrf2 knockout mice: role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008;27:212–223. doi: 10.1038/sj.emboj.7601950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botta D, Shi S, White CC, Dabrowski MJ, Keener CL, et al. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J Biol Chem. 2006;281:28865–28875. doi: 10.1074/jbc.M605143200. [DOI] [PubMed] [Google Scholar]
- 33.Rio A, Gassull MA, Aldeguer X, Ojanguren I, Cabre E, et al. Reduced liver injury in the interleukin-6 knockout mice by chronic carbon tetrachloride administration. Eur J Clin Invest. 2008;38:306–316. doi: 10.1111/j.1365-2362.2008.01939.x. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Ruiz C, Fernandez-Checa JC. Redox regulation of hepatocyte apoptosis. J Gastroenterol Hepatol. 2007;22:S38–S42. doi: 10.1111/j.1440-1746.2006.04644.x. [DOI] [PubMed] [Google Scholar]
- 35.Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. J Gastroenterol Hepatol. 2011;26:203–212. doi: 10.1111/j.1440-1746.2010.06539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Krishan M, Nebert DW, Shertzer HG. Glutathione-deficient mice are susceptible to TCDD-Induced hepatocellular toxicity but resistant to steatosis. Chem Res Toxicol. 2012;25:94–100. doi: 10.1021/tx200242a. [DOI] [PubMed] [Google Scholar]
- 37.Oliveira CP, Stefano JT, Cavaleiro AM, Zanella Fortes MA, Vieira SM, et al. Association of polymorphisms of glutamate-cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2010;25:357–361. doi: 10.1111/j.1440-1746.2009.06001.x. [DOI] [PubMed] [Google Scholar]