

Abstract
Background
Parasites incur periodic mutations which must ultimately be eliminated to maintain their genetic integrity.
Methods
It is hypothesised that these mutations are eliminated not by the conventional mechanisms of competition between parasites in different hosts but primarily by competition between parasites within the same infection.
Results
This process is enhanced by the production of a large number of parasites within individual infections, and this may significantly contribute to parasitic virulence.
Conclusions
Several features of the most virulent human malaria parasite Plasmodium falciparum can usefully be re-interpreted in this light and lend support to this interpretation. More generally, it constitutes a novel explanation for the evolution of virulence in a wider range of microparasites.
Background
Why are some parasites so virulent? This is one of the key questions in the extension of evolutionary thinking into medical sciences (for a recent access to the empirical and theoretical literature, see Refs [1,2]). There are several plausible explanations for virulence. One is that a certain degree of virulence is beneficial to the organism: a human rendered prone by a malaria fever is less likely to move and kill the mosquitoes which feed on him and spread the infection; the diarrhoea associated with cholera may result in more widespread transmission of the disease, and so on. Occasional host mortality is regrettable from all points of view because host mortality generally also kills the parasite, but is explicable as it occurs at one end of a spectrum of disease states. A second explanation for virulence is that a high parasitaemia causes host pathology but concomitantly results in more assured transmission of the parasite. According to this hypothesis the observed level of virulence is a "trade-off" between minimising host mortality and maximising transmission. [This argument is appropriate to P. falciparum despite the observation that some people with high parasitaemia show no symptoms while some exhibit severe disease with far fewer parasites, most probably as a consequence of differing levels of immunity; the key argument is that all other things being equal, higher parasitaemia tends to produce more severe disease, an assertion that seems reasonable for P. falciparum]. These two explanations both assume a degree of evolutionary fine-tuning to achieve an optimal level of virulence. There are two other explanations for virulence that rest not on evolutionary trade-offs but purely on bad luck. If an infection consists of millions or billions of individual parasites, then periodically a rogue "selfish" mutation may occur that grows rapidly, comes to dominate the infection and can cause a pathology which is detrimental for both host and parasite. Alternately, virulence can occur when a parasite finds itself in an exotic host species to which it is not adapted; the most familiar examples are human zoonosis such as Lassa fever or Ebola. Here we present a novel, fifth reason why virulence may occur. Like the first two hypotheses, it rests on an evolutionary, rather than bad-luck, argument. Moreover, it explains several puzzling features of the biology of Plasmodium falciparum, the organism responsible for the most severe form of human malaria.
Many severe human infections are characterised by a large number of individual parasites that acutely infect a host for a relatively short time before being eradicated by host immunity. Central to our thesis is a consideration of how deleterious mutations (for example, those disrupting protein structure and general cellular machinery) are eliminated during the life cycles of these organisms. In conventional evolutionary thinking, deleterious mutations are eliminated by direct competition with individuals carrying the normal non-mutated wildtype gene. Thus, for example, a mutation decreasing flight efficiency in a mosquito would ultimately be eliminated through being out-competed by mosquitoes with competent flight physiology. This type of mutation elimination is termed "purifying selection" and prevents mutations establishing themselves in the gene pool of the organisms. The problem is that populations of parasites such as malaria are highly sub-divided into numerous separate human hosts and face the overwhelming selective pressure of host immunity. Direct competition between separate clones of malaria in different hosts will be small and purifying selection correspondingly weak. ['Clones' is used here to denote the large number of parasites in a human descended from a single inoculated sporozoite. People in highly endemic areas may contain up to seven distinct clones, derived from separate infective bites, a feature quantified as their 'clonal multiplicity'. Critically, 'clones' does not imply that they are genetically stable over time, more realistically they are transitory and broken down during meiosis in the obligate sexual phase of the P. falciparum life cycle]. Our central argument is that mutations in this type of life history may be nullified by compensatory mutation rather than being eliminated by purifying selection. This occurs when a second "compensatory" mutation occurs within the gene which restores the metabolic activity of the gene product and hence nullifies the effect of the original mutation. There are two lines of evidence supporting this hypothesis. Firstly, the patterns of molecular evolution observed in P. falciparum and, in particular, the rapid rate of non-synonymous (i.e. protein altering) mutations are consistent with their being compensatory. Secondly, compensatory mutations are noted in laboratory populations of organisms kept in isolation (see later).
Methods
The efficacy of compensatory mutation depends on two factors, the size of the parasite population within the human host (which determines whether the rare compensatory mutation will actually occur within an individual host) and the number of replication cycles in the life cycle (which determines the extent to which the compensatory mutation spreads within the host prior to its transmission). The stochastic element in the process means that it is best investigated by computer simulation. The simulations are used to calculate the probability that (i) a fully-functional "wildtype" malaria infection will suffer a deleterious mutation and be transmitted by a mosquito as a mutated "dysfunctional" form, and (ii) that a compensatory mutation will occur in a "dysfunctional" infection and spread within this infection to the extent that it will be transmitted by a mosquito as the compensated, metabolically functional, wildtype form. The relative magnitudes of the two processes determine the equilibrium frequency of wildtype and dysfunctional malaria parasites within the human population.
The principle is best illustrated with the simplest possible case where the parasites consist of only two types: the fully functional "wildtype" and a mutated "dysfunctional" type. The latter suffers a growth disadvantage compared to the wildtype, so are gradually eliminated if both types are simultaneously present in the same human host. Parasites can mutate from one type to the other. Mutations occur in wildtype parasites converting them to the dysfunctional type, and compensatory mutations occur in the dysfunctional types restoring the gene function and thereby converting them back to the wildtype form. These are referred to as "dysfunctional" and "beneficial" mutations respectively and are quantified as the rate per Plasmodium per erythrocyte cycle. Note that dysfunctional mutations can occur in a large number of genes, all of which periodically mutate to the dysfunctional form. Conversely, beneficial mutations can occur only in those few codons where a compensatory mutation can compensate for the original deleterious mutation. Thus, dysfunctional mutation rates will be several orders of magnitude higher than the beneficial rate. In the example shown on figure 1, these rate are 10-5 and 10-8 per erythrocytic cycle respectively, which means that, on average, one in 100,000 wildtype plasmodia mutates to a deleterious form in each erythrocyte cycle (and similarly that 1 in 10,000,000 mutants have their function restored each erythrocyte cycle). The key phrase in the preceding sentence is "on average". In particular, if the number of parasites within a host is small then zero mutations may occur. The variation in the dynamics between individual infections can be large, so the results are best presented as the equilibrium frequency of the wildtype forms in the population. The exact shape of figure 1 depends on the values of mutation rates and the selective disadvantage of the deleterious mutation (10% per erythrocytic cycle in this example), but the general results are robust.
Figure 1.

Equilibrium frequency of the fully functional wildtype form of a parasite as a consequence of the number of parasites within the human host (its clonal population size) and number of parasite erythrocyte cycles (each of which lasts two days) which elapse prior to transmission. Computational details are as described in the main text i.e. dysfunctional mutation rates = 10-5, beneficial mutation rates = 10-8, and the selective disadvantage of the mutation is 10% per erythrocytic cycle.
Technical aspects of the simulations are as follows. The infection starts out as either entirely wildtype or entirely dysfunctional type, depending on the type of sporozoite that initiated the infection. The mean number of mutations expected per cycle is calculated and the actual number randomly drawn from a Poisson distribution around this mean; for example, if the clonal population size is 107 and the mutation rate is 10-8, then the program will pick zero mutations per cycle in about 90% of simulations, one in about 10% of simulations and two or more in a small proportion of simulations. Once the number of new mutations has been simulated, these are added to the number that already exist and the next erythrocytic cycle is simulated. The number of mutations in the next cycle is also drawn from a Poisson distribution but taking into account selective differences. For example, if 20 wildtype parasites already exist, one is added by mutation, and wildtypes have a 10% growth advantage then the number next generation is drawn from a Poisson distribution with mean (1+20) × 1.1 = 23.1, and so on for the required number of erythrocytic cycles. The simulations are run 10,000 times for each value of clonal population size, so that each combination of population size and number of erythrocytic cycles produces two parameters: 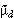 , the probability that an infection, originally wildtype, is subsequently transmitted as the dysfunctional type, and
, the probability that an infection, originally wildtype, is subsequently transmitted as the dysfunctional type, and 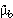 , the probability that an infection originally dysfunctional is subsequently transmitted as wildtype. The equilibrium frequency of the wildtype expected under this type of mutation elimination is
, the probability that an infection originally dysfunctional is subsequently transmitted as wildtype. The equilibrium frequency of the wildtype expected under this type of mutation elimination is 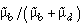 . This simple case assumes there are only two types of parasites, wildtype and dysfunctional. The calculations can be easily extended to a case where dysfunctional types may contain 1,2,3,4.... deleterious mutations. The same qualitative results arise but at the cost of greatly increased model complexity.
. This simple case assumes there are only two types of parasites, wildtype and dysfunctional. The calculations can be easily extended to a case where dysfunctional types may contain 1,2,3,4.... deleterious mutations. The same qualitative results arise but at the cost of greatly increased model complexity.
Results
A typical result is shown in figure 1 where the equilibrium frequency of the wildtype form is shown as a function of the parasite population size within the human host (clonal population size) and the number of erythrocyte cycles before transmission. Increasing the clonal population size increases the rate at which deleterious mutations are nullified by compensatory mutations, thereby increasing the equilibrium frequency of fully functional wildtype infections. As the clonal population size increases, a law of diminishing returns applies and the evolutionary optimal size occurs when the (short-term) risk of host mortality balances the (long-term) advantages of mutation elimination.
Discussion
This effect can be seen in controlled laboratory experiments. Burch and Chao [3] showed that increasing the clonal population size of bacteriophages increased the speed at which beneficial mutations accumulate. Miralles et al. [4] presented similar data for an RNA virus. Compensatory mutations may be a direct reversal of the original mutation but, more typically, they restore gene function through another compensatory mutation at another site within the same gene. Presumably this second mutation restores, partially or completely, the tertiary structure of the molecule and hence its biological activity. Several studies [3,5-9] have demonstrated this process in E. coli and bacteriophages. P. falciparum appears to be the only example where the process of an advantageous mutation occurring and spreading to dominate the infection can be directly and easily observed. If an infection is drug sensitive and the human host is treated with a drug, then the symptoms go into remission as the majority of the parasites are eliminated. However, a small number of parasites may contain a spontaneous mutation conferring drug resistance. These survive and flourish even in the presence of drugs, so that the infection reappears shortly afterwards ("recrudesces") as a drug-resistant strain. This process appears to be remarkably effective: 33% of infections treated with the antimalarial drug atovaquone subsequently recrudesce as a drug-resistant infection [10]. The effect is extremely dependent on the size of infection. As an example, if a human is inoculated with a drug-sensitive clone and there are 1011 parasites in the host and the mutation rate to the drug resistant form is 10-8, then there will be a sub-population of 103 resistant parasites. Following drug treatment, the sensitive forms will be eliminated and the sub-population of 103 resistant forms will expand to dominate the infection. Needless to say, if the mutation rate to resistance is 10-8, then infection sizes of less than ten thousand would rarely contain the appropriate mutation. Current antimalarial drug deployment strategies utilise this effect by using drugs in combination. If mutation rates to resistance are 10-8 for each drug in a two-drug mixture, then even an infection of 1012 individual malaria parasites is highly unlikely (a probability of 0.0001) to simultaneous contain a spontaneous drug-resistant mutation in each gene.
P. falciparum, in common with several other parasites, appears to try and evade host immunity by continually changing its immune characteristics. It does so, at least in part, by expressing a sequence of var genes [11,12]. An interesting consequence of the above argument is that var switching in P. falciparum may select for increased infection load (because increased parasite numbers are required to increase effective population size) and hence to increased virulence. The population size illustrated in figure 1 is best interpreted as an effective population size [13,14]. This is a key concept in population genetic theory and is defined as being equivalent to an idealised population whose size does not fluctuate. Effective population size decreases rapidly if the population goes through a series of "bottlenecks" of small population sizes. As P. falciparum switches through its antigenic repertoire, it appears likely that only a small portion of the population will actually survive. As human immunity to the current var product expressed in the infection builds up and starts to eliminate the parasites, a small proportion will have switched to another var gene in the repertoire to escape immunity and expand to dominate the infection [15-17]. This lowers the effective population size considerably below the crudely observed population size of 109 to 1011 parasites in a typical human malarial infection. For example, if the population size is 1010 but every 10 erythrocyte cycles goes through a bottleneck of 1000 individuals which switch var expression, the effective size decreases to 10,000. Intuitively, it is possible to see how this impedes the spread of a compensatory mutation through an infection: even if a compensatory mutation has occurred and is spreading through an infection, the chances are that it has occurred in a cell lineage that will ultimately be destroyed by human immunity acting against its var gene expression [18].
Conclusions
It is argued that virulence occurs as a consequence of the large parasite density (quantified as the clonal population size within a human) required to effectively eliminate mutations or nullify them through compensatory mutations, the evolutionary optimal level of virulence being determined by the conflicting necessity of removing mutations balanced against the need to minimise host mortality. This constitutes a novel hypothesis for the evolution of virulence and is a simple consequence of an inevitable fact of life: that deleterious mutations occur and must be effectively eliminated. It is difficult to see how this can be done effectively solely by the actions of the purifying selection envisaged in conventional population genetic theory, and it appears that compensatory mutations play a major role. P. falciparum life history traits may, therefore, have evolved to facilitate the effective incorporation of compensatory mutations, the most notable consequence being that they reach very high numbers within individual hosts. Virulence may therefore be simply an unfortunate side effect of large infection sizes, parasites evolving to be as benign as possible while still maintaining an infection density (clonal population size within the human) sufficient to allow the effective removal of deleterious mutations. The trade off between these conflicting interests determines the optimal level of virulence (see figure 1). The alternative hypotheses for explaining the evolution of virulence cannot be resolved, all being entirely plausible post-hoc explanations. However, the mutation/selection hypothesis explains three interesting facets of the P. falciparum lifecycle. Firstly, that the correlation between parasite load and infectivity in the field is weak [19], suggesting that increased infectivity is a relatively weak selective force for increased parasite load. Secondly, that despite a large asexual parasite load, the number of infective stages is surprisingly small [20] and usually consists of around 5% of the total parasite number. There are several plausible explanations for this. For example, that they are kept at low levels by host immunity, or are produced at low frequencies to minimise their stimulation of host immunity, or that low levels avoid damaging the mosquito vector.
These explanations are immaterial to our argument which addresses the issue of trying to understand why 95% of the parasites which, moreover, are the ones responsible for clinical disease and host death, are necessary to produce the remaining 5% of infective stages. The explanation proposed here is that this 95% constitutes a large selective environment ensuring that the subsequent infective stages harbour a minimal number of deleterious mutations. Thirdly, the conventional hypothesis that increasing parasite load increases transmissibility does not explain why the turnover of parasites is so high (especially because, as noted above, gametocytes are present at a low level and are relatively long-lived), whereas our analysis of mutation elimination demonstrates that increasing turnover of parasites increases the rate at which compensatory mutations spread (figure 1).
These arguments have been developed with specific reference to the human malaria parasite P. falciparum. The same line of argument applies to many other infective agents. Many parasitic protozoa, bacteria and viruses also build up to huge infection levels. Conventionally this has been interpreted as a strategy to increase infectivity, but it can just as validly be regarded as a strategy to increase the efficacy of eliminating harmful mutations. The argument here is not that increasing parasite load does not increase transmissibility (it seems plausible that it does to some extent), but that the primary function of a large, rapidly dividing parasite population is to eliminate mutations rather than increase transmission and that it is rewarding to re-examine several facets of P. falciparum biology, and presumably the biology of similar organisms, in the light of this hypothesis.
Authors' contribution
All authors contributed to the conceptual development of this approach and the interpretation of published data. IH wrote, ran and collated the simulations which were subsequently reviewed by S. P-M and A.S.
Acknowledgments
Acknowledgements
This work was partly financed by the DFID-funded Malaria Knowledge Programme of the Liverpool School of Tropical Medicine. However, the Department for International Development can accept no responsibility for any information or views expressed.
Contributor Information
Ian M Hastings, Email: hastings@liverpool.ac.uk.
S Paget-McNicol, Email: smcnicol@optusnet.com.au.
A Saul, Email: ASAUL@niaid.nih.gov.
References
- Ebert D. The evolution and expression of parasite virulence. In: Stearns SC, editor. In Evolution in Health and Disease. Oxford: Oxford University Press; 1999. pp. 161–172. [Google Scholar]
- Weiss RA. Virulence and pathogenesis. Trends Microbiol. 2002;10:314–317. doi: 10.1016/S0966-842X(02)02391-0. [DOI] [PubMed] [Google Scholar]
- Burch CL, Chao L. Evolution by small steps and rugged landscapes in the RNA virus theta6. Genetics. 1999;151:921–927. doi: 10.1093/genetics/151.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles R, Moya A, Elena SF. Diminishing returns of population size in the rate of RNA virus adaptation. J Virol. 2000;74:3566–3571. doi: 10.1128/JVI.74.8.3566-3571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL. The physiology of weak selection. Genome. 1989;31:183–189. doi: 10.1139/g89-032. [DOI] [PubMed] [Google Scholar]
- Lenski RE. Experimental studies of pleiotropy and epistasis in Escherichia coli. II. Compensation for maladaptive effects associated with resistance to virus T4. Evolution. 1988;42:433–440. doi: 10.1111/j.1558-5646.1988.tb04150.x. [DOI] [PubMed] [Google Scholar]
- Levin BR, Perrot V, Walker N. Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics. 2000;154:985–997. doi: 10.1093/genetics/154.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore FBG, Rozen DE, Lenski RE. Pervasive compensatory adaptation in Escherichia coli. Proc R Soc Lond B Biol Sci. 2000;267:515–522. doi: 10.1098/rspb.2000.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag SJ, Perrot V, Levin BR. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc Lond B Biol Sci. 1997;264:1287–1291. doi: 10.1098/rspb.1997.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looareesuwan S, Viravan C, Webster HK, Kyle DE, Canfield CJ. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg. 1996;54:62–66. doi: 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- Baruch DI, Pasloske BL, Singh HB, Bi XH, Ma XC, Feldman M, Taraschi TF, Howard RJ. Cloning the Plasmodium falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE. The large diverse gene family Var encodes proteins involved in cytoadherence and antigenic varaiation of Plasmodium-falciparum-infected erythrocytes. Cell. 1995;82:89–100. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Harlow: Longman; 1996. [Google Scholar]
- Hartl DL, Clark AG. Principles of Population Genetics. 3. Sunderland, Ma, USA: Sinauer Associates; 1997. [Google Scholar]
- Brannan LR, Turner CMR, Phillips RS. Malaria parasites undergo antigenic variation at high rates in vivo. Proc R Soc Lond B Biol Sci. 1994;256:71–75. doi: 10.1098/rspb.1994.0051. [DOI] [PubMed] [Google Scholar]
- Molineaux L, Diebner HH, Eichner M, Collins WE, Jeffery GM, Dietz K. Plasmodium falciparum parasitaemia described by a new mathematical model. Parasitology. 2001;122:379–391. doi: 10.1017/S0031182001007533. [DOI] [PubMed] [Google Scholar]
- Paget-McNicol S, Gatton M, Hastings IM, Saul A. The Plasmodium falciparum var gene switching rate, switching mechanism and patterns of parasite recrudescence described by mathematical modelling. Parasitology. 2002;124:225–235. doi: 10.1017/S0031182001001160. [DOI] [PubMed] [Google Scholar]
- Gatton ML, Hogarth W, Saul A. Time of treatment influences the appearance of drug-resistant parasites in Plasmodium falciparum infections. Parasitology. 2001;123:537–546. doi: 10.1017/S0031182001008824. [DOI] [PubMed] [Google Scholar]
- Carter R, Graves PM. Gametocytes. In: Wernsdorfer WH, McGregor I, editor. In Malaria: principles and practice of malariology. Edinburgh: Churchill Livingston; 1988. pp. 253–305. [Google Scholar]
- Taylor LH, Read AF. Why so few transmission stages? Reproductive restraint by malaria parasites. Parasitol Today. 1997;13:135–140. doi: 10.1016/S0169-4758(97)89810-9. [DOI] [PubMed] [Google Scholar]