

Abstract
Background
Mutations in either of two genes comprising the STSL locus, ATP-binding cassette (ABC)-transporters ABCG5 (encoding sterolin-1) and ABCG8 (encoding sterolin-2), result in sitosterolemia, a rare autosomal recessive disorder of sterol trafficking characterized by increased plasma plant sterol levels. Based upon the genetics of sitosterolemia, ABCG5/sterolin-1 and ABCG8/sterolin-2 are hypothesized to function as obligate heterodimers. No phenotypic difference has yet been described in humans with complete defects in either ABCG5 or ABCG8. These proteins, based upon the defects in humans, are responsible for regulating dietary sterol entry and biliary sterol secretion.
Methods
In order to mimic the human disease, we created, by a targeted disruption, a mouse model of sitosterolemia resulting in Abcg8/sterolin-2 deficiency alone. Homozygous knockout mice are viable and exhibit sitosterolemia.
Results
Mice deficient in Abcg8 have significantly increased plasma and tissue plant sterol levels (sitosterol and campesterol) consistent with sitosterolemia. Interestingly, Abcg5/sterolin-1 was expressed in both liver and intestine in Abcg8/sterolin-2 deficient mice and continued to show an apical expression. Remarkably, Abcg8 deficient mice had an impaired ability to secrete cholesterol into bile, but still maintained the ability to secrete sitosterol. We also report an intermediate phenotype in the heterozygous Abcg8+/- mice that are not sitosterolemic, but have a decreased level of biliary sterol secretion relative to wild-type mice.
Conclusion
These data indicate that Abcg8/sterolin-2 is necessary for biliary sterol secretion and that loss of Abcg8/sterolin-2 has a more profound effect upon biliary cholesterol secretion than sitosterol. Since biliary sitosterol secretion is preserved, although not elevated in the sitosterolemic mice, this observation suggests that mechanisms other than by Abcg8/sterolin-2 may be responsible for its secretion into bile.
Background
Absorption of dietary cholesterol from the intestine is an important part of cholesterol homeostasis and represents the initial step that allows dietary cholesterol to exert its metabolic effects. A typical western diet contains relatively equal amounts of cholesterol and non-cholesterol sterols, mainly plant sterols, of which about 55% of the dietary cholesterol is absorbed and retained compared to ~1% of the dietary non-cholesterol sterols [1-3]. Schoenheimer recognized more than 75 years ago that only cholesterol, not non-cholesterol sterols, is absorbed in the intestine, although the exact molecular mechanisms by which preferential cholesterol absorption occurs has not been fully elucidated [4]. Similarly, although the liver secretes free cholesterol into bile, it can preferentially excrete non-cholesterol sterols into bile and the mechanism(s) of this process has yet to be elucidated as well. Only recently, through the study of a rare human disease, have clues to this process been revealed.
Sitosterolemia (MIM 210250, also known as phytosterolemia) is a rare autosomal, recessively inherited disorder characterized by elevated blood and tissue plant sterol levels [5]. Affected individuals can develop tendon and/or tuberous xanthomas, hemolytic episodes, arthralgias, arthritis, and premature atherosclerosis [6,7]. Sitosterolemic patients have diagnostically elevated plasma plant sterol levels (for example, sitosterol, campesterol, stigmasterol, and avenosterol, etc.) and their 5-saturated stanol metabolites, particularly sitostanol, with normal or only moderately increased cholesterol levels. Clinical studies have shown that affected individuals hyperabsorb all dietary sterols (thus have lost the ability to restrict the sterol species absorbed) and have lost the ability to excrete sterols into bile [8,9]. Infusion of sitosterol and cholesterol into normal individuals leads to a rapid and preferential excretion of sitosterol into bile, but in sitosterolemic individuals biliary sterol secretion is almost absent [7,9]. Previous studies on sitosterolemia have suggested a defect involving a putative sterol 'transporter' expressed in the intestinal brush border and/or the hepatic canalicular membrane [10,11]. Genetic analyses of sitosterolemia pedigrees allowed the mapping of the STSL locus to human chromosome 2p21, between D2S2294 and D2S2298 [12,13]. By using positional cloning procedures or by screening for genes induced by exposure to LXR agonists, two groups identified not one but two genes, ABCG5 and ABCG8, which encode the proteins sterolin-1 and sterolin-2, mutations of which cause sitosterolemia [14-16]. Interestingly, complete mutation in either ABCG5 alone or ABCG8 alone is not only necessary, but sufficient to cause the disease [14]. To date, no patient with sitosterolemia has been identified with mutations in both genes. Based upon these data, and the similarity of the clinical and biochemical phenotype, sterolin-1 and sterolin-2 are hypothesized to function as obligate heterodimers. This is also supported by the fact that each of these proteins is a 'half-transporter', containing six transmembrane domains, and not the classical 12 transmembrane characteristic of functional ABC transporters. The exact function of these two proteins still remains unknown but recent studies have shown that overexpression of ABCG5/ABCG8 in a transgenic mouse model results in an increased biliary cholesterol concentration and a 50% reduction in the fractional absorption of dietary cholesterol [17]. It has been suggested that ABCG5 and ABCG8 act as mutual chaperones for both maturation and correct apical targeting. Co-expression of both was required for apical trafficking in a polarized cell line, as well in vivo using Abcg5/Abcg8 double-knockout mice [18,19]. However, no functional assay has yet been developed to demonstrate whether or not the two half-transporters function as homo- or heterodimers and whether they selectively pump sterols. Likewise, the cellular localization of these two proteins has yet to be shown in a model deficient in either Abcg5 or Abcg8 alone. A mouse model disrupting both Abcg5 and Abcg8 simultaneously results in a phenotype similar to that of the human disease [20]. However, it is important to point out that the human disease is caused by complete mutation in one or the other gene, but not both simultaneously.
We hypothesized that disruption of only one of the half-transporters would result in sitosterolemia and selective loss of either sterolin may show a differential effect on the biliary secretion of sterols or intestinal absorption of sterols. These studies are difficult to test in humans, as the target organs are relatively inaccessible. In order to test this hypothesis, we report a mouse model that has a selective genetic mutation of Abcg8. Mice homozygous for Abcg8 loss exhibit many of the characteristics of the human disease sitosterolemia. In addition, we report studies of biliary sterol secretion that show that despite forced secretion, Abcg8-deficient animals cannot secrete cholesterol into bile, although sitosterol secretion seemed unaffected.
Methods
Targeting vector construction and generation of knockout mice
A mouse genomic bacterial artificial chromosome (BAC) library (CitbCJ7, ES cell line/129Sv, Research Genetics, Inc., Huntsville, AL, USA) was screened by using primers designed from the sequences of mouse Abcg5 and Abcg8 cDNA as previously reported [21]. A positive BAC clone was used as a template to amplify genomic DNA fragments of Abcg8. Long-fragment polymerase chain reaction (PCR) was performed using Expanded Long Template PCR system kit (Roche Applied Science, Indianapolis, IA, USA). An approximately 4.5 kb 'long-arm' genomic fragment containing partial exon 1 to partial exon 3 was inserted into the Pml I restriction-cloning site A of OSDUPDEL vector. The 'short-arm' genomic fragment, containing partial exon 4 to partial exon 6, was cloned into the Not I-Kpn I of cloning site B. Homologous targeting would result in complete intron 3, partial exon 3 and partial exon 4 replacement by the neomycin-resistance cassette, resulting in the disruption of the ABC Walker A motif (Figure 1a).
Figure 1.
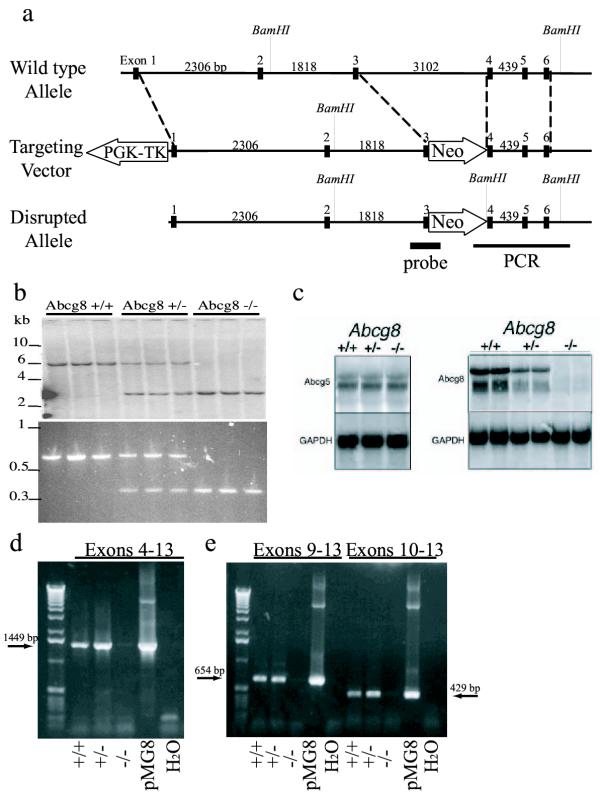
Generation of mice deficient in Abcg8/sterolin-2. The targeted disruption strategy of Abcg8 is as shown (panel a). Southern blot analysis of BamHI digested mouse genomic DNA, probed with [32P]-randomly labelled probe resulting in a 6.0 kb band for wild type, a 2.7 kb band for homozygous and two bands of 5.9 and 2.6 kb for the heterozygote (panel b). Northern blot analysis of hepatic RNA showed a loss of Abcg8/sterolin-2 mRNA in the homozygote and decreased Abcg8/sterolin-2 mRNA in the heterozygote, although Abcg5/sterolin-1 mRNA appeared relatively unaffected in the knockout mice (panel c). Probes were approximately 1.9 kb for Abcg5 and 2 kb for Abcg8 (see Methods for more detail). RT-PCR analyses of hepatic cDNA showed no Abcg8/sterolin-2 message, downstream of exon 4 in the Abcg8-/- mice, whether primers were located in exons 4 and 13 (panel d), exons 9 and 13 or exons 10 and 13 (panel e).
ES cells (129/SvEvTac-cell line) were electroporated with the linearized targeting vector DNA and cultured on sub-confluent embryonic fibroblasts. Transfected cell colonies were selected by culturing cells in medium containing 200 μg/mL G418 (Life Technologies, Rockville, MD, USA). The ES cells were screened for homologous recombination by PCR with the forward primer, neoF (5'-GGGTCGTTTGTTCGGATCAA-3') from neo cassette, and reverse primer intron 6R (5'-ACCAGTTGGTCCTAGCTCGA-3'), which is located outside the targeting construct in mouse Abcg8 intron 6. Positively targeted ES cell clones were confirmed by Southern blotting, using ES cell DNA digested by BamHI and a probe comprising of a 562 bp PCR fragment from partial intron 2 and exon 3 of Abcg8 gene.
Positively targeted ES cells were microinjected into C57BL6 blastocysts and transplanted into pseudopregnant recipients. Five highly chimeric mice (agouti coat color, three males and two females) were isolated and bred with C57BL/6J mice to generate germ-line transmission heterozygous mice of Abcg8 gene disruption. Heterozygous offspring mice were back-crossed to C57BL/6J mice (n > 5) to produce disrupted line and inter-crossed to generate knockout mice.
Animals and diets
All mice were maintained on a standard rodent chow (Harlan Teklad mouse/rat diet LM-485) which contained 61 mg/kg cholesterol, 31 mg/kg campesterol, and 105 mg/kg sitosterol, given free access to water and maintained at 25°C with a 12-h light, 12-h dark cycle. All animals were housed in the facilities for laboratory animals provided by the Division of Laboratory Animal Resources ACLAM. The Institutional Animal Care and Research Advisory Committee at Medical University of South Carolina approved all experiments.
Genotyping by PCR
Mouse tail DNA was isolated and purified by cutting approximately 0.5 cm of mouse tail and digesting it in 500 μl lysis buffer (50 mM Tris-HCl, pH8.0, 100 mM EDTA, 125 mM NaCl, 1% SDS, 200 μg Proteinase K) with rocking overnight at 55°C. 200 μl saturated NaCl was added into digested solution with vigorous shaking for at least 60 seconds. Mixed solution was spun down at maximum speed in top micro-centrifuge for 20 min. The supernatant was transferred to a new tube with equal volume 100% EtOH and mixed by inversion. The DNA pellet was washed in 70% EtOH and resuspended in 100–150 μl TE buffer. UV spectrophotometry and electrophoresis were used to analyze the quality and quantity of the genomic DNA. Genotyping was performed by multiplex PCR reaction using three separate primers (two Abcg8 gene specific primers, mg8-in3F 5'-CCCAATGATGAATGAGACGCT-3', mg8-R9 5'-TTTGTCACGCTGGGCCTGG-3' and neoF for identification of Abcg8 target status). The PCR reaction consisted of 1x PCR buffer (1.5 mM MgCl2, 16 mM (NH4)2SO4, 0.1 mM dNTPs, 67 mM Tris-HCl (pH8.8)) 1 μM of each primer, water and 1.0 units of Taq polymerase. The reactions were cycled as follows: 94°C for 30 sec, 60°C for 30 sec, 72°C for 1 min for 35 cycles. The PCR products were identified by electrophoresis.
Southern blot analysis
To confirm PCR results, DNAs were digested with Bam HI, subjected to electrophoresis in a 0.6% agarose gel and transferred to nylon membrane. Southern blot filters were hybridized with a 562 bp PCR fragment from partial intron 2 and exon 3 of Abcg8 32P-randomly-labeled probe as previously described [22].
Northern blot analysis
Isolation of liver and intestinal total RNA from Abcg8+/+, Abcg8+/- and Abcg8-/- mice and Northern blot analyses were performed as previously described [23]. The probe for Abcg5 was 1964 bp (nt 136–2099, GeneBank™ accession no. AH011511) and the probe for Abcg8 was 2019 bp (nt 103–2126, GeneBank™ accession no. AH011518).
Reverse Transcription (RT)-PCR
To confirm that there was no alternative splicing of the disrupted allele, RT-PCR was performed on cDNAs of pooled mouse liver. Briefly, samples of total RNA (0.5 μg) from pooled mouse livers (n = 4) were reverse transcribed according to the SuperScript™ First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) using random hexamers in a final total reaction volume of 20 μl. RT-PCR was performed using primers located outside of the targeted region: F1 (CCTCAGCTGGTGAGGAGGTG) with R1 (GATGGAGAAGGTGAAGTTGCC), F2 (ATTTCCAATGACTTCCGGGAC) with R1 and F3 (CTGGAAGACGGGCTGTACACT) with R1, to produce expected product lengths of 1449, 654 and 429 bp respectively in wild-type cDNA.
Quantitative RT-PCR
Primers were based upon previously published primer sets [24-26] or designed using MacVector, which were designed from either mRNA or cDNA to avoid the amplification of potentially contaminating genomic DNA in the total RNA sample (Table 1). Samples of total RNA (0.5 μg) from pooled mouse livers (n = 4) were reverse transcribed according to the SuperScript™ First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) using random hexamers with a final total reaction volume of 20 μl. Quantitative RT-PCR was performed on a PE Biosystems GeneAmp® 5700 sequence Detection System (Forest City, CA, USA). Standard reaction volume was 10 μl containing 1 × QuantiTect SYBR Green PCR master mix (Qiagen, Valencia, CA, USA), 0.002 U AmpErase UNG enzyme (PE Biosystems, Forest City, CA, USA), 0.7 μl of cDNA template from reverse transcription reaction as above and 100–500 nM of oligonucleotide primers. Initial steps of RT-PCR were two minutes at 50°C for UNG activation, followed by a 15 minute hold at 95°C. Cycles (n = 40) consisted of a 15 second melt at 95°C, followed by a 1 minute annealing/extension at 60°C. The final step was a 60°C incubation for 1 minute. At the end of the run, samples were heated to 95°C with a ramp time of 20 minutes to construct dissociation curves to ensure that single PCR products were obtained. All reactions were performed in triplicate. Threshold cycle (CT) analysis of all samples was set at 0.5 relative fluorescence units. The relative quantities of message of genes of interest from the mouse liver samples used in real-time RT-PCR were normalized to cyclophilin to compensate for variations in input RNA amounts. The data were analyzed using the comparative threshold cycle method (CT). Briefly, the CT values are averaged, from the triplicates defining a Δ-CT value calculated by taking the average CT of the gene of interest and subtracting it from the average CT of cyclophilin. The ΔΔ-CT was calculated by subtracting the average Δ-CT(calibrator) values from the Δ-CT(sample). The relative quantification was then calculated by the expression 2-AverageΔΔ-CT. The mRNA quantity for the calibrator (wild type) was expressed as 1 and all other quantities were expressed as a fold difference relative to the calibrator (wild type).
Table 1.
Oligonucleotide primers used for quantitative RT-PCR
| Target gene | Primer sequence | Length of amplicon | GenBank accession no. |
| Abcg5 | Forward: 5'-AGGTCATGATGCTAGATGAGC-3' | 260 bp | AH011511 |
| Reverse: 5'-CAAAGGGATTGGAATGTTCAG-3' | |||
| Abcg8 | Forward: 5'-CTCCTCGGAAAGTGACAACAG-3' | 190 bp | AH011518 |
| Reverse: 5'-TAGATTTCGGATGCCCAGCTC-3' | |||
| Hmgr | Forward: 5'-CCGGCAACAACAAGATCTGTG-3' | 114 bp | BB664708 |
| Reverse: 5'-ATGTACAGGATGGCGATGCA-3' | |||
| Cyp7a1 | Forward: 5'-CAGGGAGATGCTCTGTGTTCA-3' | 121 bp | NM_007824 |
| Reverse: 5'-AGGCATACATCCCTTCCGTGA-3' | |||
| Abca1 | Forward: 5'-CCCAGAGCAAAAAGCGACTC-3' | 89 bp | NM_013454 |
| Reverse: 5'-GGTCATCATCACTTTGGTCCTTG-3' | |||
| Mdr2 (Abcb4) | Forward: 5'-GCAGCGAGAAACGGAACAG-3' | 64 bp | NM_008830 |
| Reverse: 5'-GGTTGCTGATGCTGCCTAGTT-3' | |||
| LXRα | Forward: 5'-GCTCTGCTCATTGCCATCAG-3' | 79 bp | AF085745 |
| Reverse: 5'-TGTTGCAGCCTCTCTACTTGGA-3' | |||
| Srebp-1c | Forward: 5'-GGAGCCATGGATTGCACATT-3' | 103 bp | BI656094 |
| Reverse: 5'-CCTGTCTCACCCCCAGCATA-3' | |||
| Srebp-2 | Forward: 5'-CTGCAGCCTCAAGTGCAAAG-3' | 119 bp | AF374267 |
| Reverse: 5'-CAGTGTGCCATTGGCTGTCT-3' | |||
| Cyclophilin | Forward: 5'-AAGTTCCATCGTGTCATCAAGGAC-3' | 173 bp | M60456 |
| Reverse: 5'-CCATTGGTGTCTTTGCCTGC-3' |
Plasma lipid analysis by fast protein liquid chromatography
Following 16 hrs of fasting, blood samples (n = 3) from each of the Abcg8+/+, Abcg8+/- and Abcg8-/- mice were collected from the retro-orbital venous plexus using heparinized capillary tubes under isoflurane anesthesia, and placed into precooled tubes containing 10 μl of 0.5 M EDTA. Equal volumes of plasma from each genotype were pooled and 100 μL of the pooled sample were injected and fractionated by fast protein liquid chromatography (FPLC) using a Superose 6 HR10/30 column (Pharmacia Biotech Inc., Piscataway, NJ, USA) and analyzed as described [27].
Sterol composition of plasma and tissues
Following a four-hour fast of 12-week-old mice fed on regular rodent chow diet; blood and tissues were collected under isofluorane anesthesia from each of the Abcg8+/+, Abcg8+/- and Abcg8-/- mice (n = 3 for each group). Blood was collected from the retro-orbital venous plexus by heparinized glass tube. The animals were sacrificed by cervical dislocation for tissue collection. Plasma (100 μl) was saponified in 1 N NaOH for one hour, 1.5 mL water added and sterols extracted with three sequential portions of 1.5 mL ethyl acetate, containing 70 μg 5α-cholestane as an internal standard, pooled and dried. Mouse tissues (liver, spleen and brain) were weighed, homogenized in 1 mL phosphate buffer saline (PBS) with a dounce homogenizer (15 strokes at 500 rpm) and a small aliquot of the whole homogenate assayed for protein concentration. Aliquots for sterol analysis from whole tissue homogenates were extracted as described above. Dried sterol samples were derivatized as trimethylsilylethers, redissolved in 10 μL hexane and 2 μL samples were injected into a capillary column (26 m length, 0.32 mm ID, 0.45 mm OD) coated with liquid CpWAX 57 CB (Chrompak, Bridgewater, NJ, USA) in a Hewlett-Packard Gas Chromatograph equipped with a flame ionization detector [28]. Concentrations of plasma and tissue sterols were reported as mg/dL and μg/g wet weight tissue, respectively.
Polyclonal antibody production
Three separate polyclonal anti-Abcg5/sterolin-1 antibodies have been raised as previously described [20,29,30]. In experiments, anti-Abcg5/sterolin-1 antibodies are labelled as follows:
1) SC – from the Patel group, 2) AMC – from the AMC Liver Center group, and 3) UTSW – from the Hobbs group.
Membrane protein preparations
Total membranes were prepared by taking approximately 500 mg of liver tissue cut into small pieces and homogenized in ice cold lysis buffer (5 mM Tris-pH7.5, 250 mM sucrose with protease inhibitors) by a dounce homogenizer times 10 strokes. Samples were then subjected to centrifugation – 1300 g for 10 minutes at 4°C. The supernant was saved and the pellet was resuspended and again subjected to dounce homogenization times 10 strokes then centrifuged 1000 g for 10 minutes at 4°C. This step was repeated twice each time with the supernant being collected and kept on ice. The supernants were then pooled and subjected to ultracentrifugation at 100,000 g for 60 minutes at 4°C. The supernant was removed and the pellet was resuspended in lysis buffer. Plasma membranes were prepared as previously described [31]. Protein sample concentrations were then determined by Bio-Rad protein colormetric assay per manufacturer's protocol (BioRad, Hercules, CA, USA). 50 μg of membrane proteins were then resolved on 7.5% SDS acrylamide gel.
Immunoblotting
Proteins resolved by SDS-PAGE were transferred to nitrocellulose membranes. Membranes were then blocked for one hour in 5% dry milk in PBS-T (Phosphate Buffered saline and 0.1% Tween 20). Blots were then probed with primary antibody against Abcg5 in 5% milk in PBS-T overnight at 4°C. The blot was then washed three times for five minutes in TBS-T (Tris Buffered Saline/0.1% Tween-20) with 150 mM NaCl, and secondarily stained with goat-anti-rabbit conjugated HRP 1:10,000 dilution in 5% milk in PBS-T for one hour. The blot was again washed three times for five minutes in TBS-T with 150 mM NaCl then covered in Western Lightning® Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Inc. Boston, MA, USA), wrapped in plastic, exposed to film and developed.
Immunohistochemical analysis
Snap-frozen liver and intestine tissue were cut to produce 8 μm thick frozen sections, air-dried for 30 minutes onto glass slides and stored at -80°C until required. The slides were stained with hematoxylin, rinsed three times with PBS, fixed for 10 min with cooled methanol at -20°C and then rinsed three times with PBS. Slides for antibody staining were initially blocked in PBS/0.1 M glycine/10% goat serum for 60 min at room temperature then incubated with anti-sterolin-1 antibody (1:15 dilution) overnight at 4°C. The slides were washed with 1 × PBS and incubated with secondary antibody (goat-anti-rabbit IgG conjugated with rhodamine, 1:1000 dilution) for 30 min at room temperature, washed and then observed under an Olympus BX-5 confocal microscope with Fluoview. The sections stained at the AMC Liver Center were fixed in 100% acetone for 30 min at room temperature, blocked in a solution of PBS/0.05% Tween-20/5% goat serum (GS) for one hour, then incubated with Anti-sterolin-1 (1:40 dilution) and anti-Bsep (1:200 dilution) diluted in PBS/Tween/GS, for one hour. Thereafter, slides were washed 3 × 10 min in PBS/Tween and the secondary antibody (goat-anti-rabbit Alexa 488) was applied in 1:1000 dilution in PBST/GS for one hour at room temperature. After washing for 3 × 10 min the sections were mounted in Dapi/Dapco solution. Fluorescence microscopy was done with a Leica DM RA2 microscope equipped with a Leica DC350F photo camera. For Dapi staining, 359 nm (30.38 ms exposure) was used and for Abcg5 and Bsep staining, 514 nm (for both: 1654.34 ms exposure) was used and the pictures were analyzed and overlaid using FW4000 software (Leica).
Transfection and immunostaining of COS-1 cells
COS-1 cells were transiently transfected with pCMV-mouse Abcg5 and pCMV-mouse Abcg8 expression constructs using the Qiagen SuperFect reagent per protocol (Valencia, CA, USA). Briefly, COS-1 cells were seeded onto a six-well plate the day prior to transfection to generate 60% confluence when grown at 37°C and 5% CO2 in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 1 × glutamine. On the day of transfection, 2 μg of DNA was mixed with Qiagen SuperFect reagent and incubated for 10 minutes. This mixture was then added to serum-free media and placed on the cells to incubate for two hours. The cells were washed once with 1x PBS and fresh media was added. The transfected cells were then incubated for 48 hours to ensure expression and subsequently fixed with a 1:1 mixture of acetone:methanol. In preparation for immunostaining, cells were blocked with 2% goat serum in PBS for 30 minutes, then incubated with 1:50 dilution of primary antibody anti-Abcg5 in 2% goat serum-PBS for 1.5 hours, and subsequently incubated with secondary rhodamine anti-rabbit IgG antibody for 30 minutes. Cells were then observed under an Olympus BX-5 confocal microscope with Fluoview.
Dynamic biliary lipid collection and analysis
Mice were anaesthetized by intraperitoneal injection of 1 mL/kg fentanyl/fluanisone and 10 mg/kg Diazepam. The abdomen was opened and, after distal ligation of the bile duct, the gallbladder was cannulated. Bile sampling started directly after cannulation and was collected for 10 min. Bile flow was determined gravimetrically. Biliary bile salt, sterol and phospholipid concentrations were determined as described previously [32]. To determine maximal secretion rates of biliary lipid secretion, bile was diverted for 90 min to deplete the endogenous bile salt pool. Subsequently, tauroursodeoxycholate (Sigma Chemical Co., MO, USA) dissolved in PBS was infused into the jugular vein in stepwise increasing rates from 600–1,800 nmol/min/100 g body weight. Bile was collected in 10 min fractions and analyzed for bile salt and lipid content. In separate experiments, following distal ligation of the bile duct and cannulation of gallbladder, bile was diverted for 30 minutes while mice were infused with sterile PBS. Subsequently, the mice were infused continuously with 1,200 nmol/min/100 g body weight of tauroursodeoxycholate and two 30-minute fractions were collected. Sterol analyses of bile was performed by GC analysis as described above.
Assay of HMG-CoA and cholesterol 7α-hydroxylase enzyme activity
Mouse liver microsomes were prepared by differential centrifugation [33]. Briefly, 0.1–0.2 g liver was homogenized in a Potter-Elvjhem homogenizer with five volumes of buffer (0.25 M sucrose, 0.1 M Tris, 0.1 mM disodium EDTA, 0.1 mM DTT, pH 7.4). Microsomes were isolated by differential centrifugation (10,000 g to 100,000 g). The pellets were washed and resuspended in storage buffer (0.1 M dipotassium phosphate, 0.05 M KCl, 1 mM DTT, 5 mM disodium EDTA, 20% glycerol, pH 7.4) at 25–30 mg protein/ml. Protein concentrations were determined according to the method of Lowry et al. [34]. The activity of cholesterol 7α-hydroxylase (EC 1.14.12.17) was measured by an isotope incorporation method according to Shefer et al. [33] with some modifications. The reaction mixture (final volume 0.5 ml) consisted of potassium phosphate buffer (100 mM K2HPO4, 0.1 mM EDTA, 5 mM DTT, 30 mM nicotinamide, pH 7.4), [14C]-cholesterol (5 × 105 dpm) solubilized in 50 μl of 25% (wt/vol) β-cyclodextrin (final concentration 0.8%), and 50–200 μg of microsomal protein. The reaction was initiated by the addition of NADPH or an NADPH-generating system (3.4 mM NADP+, 30 mM glucose-6-phosphate, 0.3 U of glucose-6-phosphate dehydrogenase) and continued for 15 min at 37°C. The reaction was stopped with 0.5 ml of 1 N KOH, 5 μg of butylated hydroxytoluene, and 10 μl of ethanolic potassium hydroxide. [3H] 7α-hydroxycholesterol (1 × 104 dpm/5 μg) was added as an internal recovery standard. After saponification at 37°C for one hour, sterols were extracted twice with 3 ml of n-hexane and the extracts were evaporated to dryness under nitrogen. The residue was dissolved in 0.3 ml of n-hexane:2-propanol (97.3 vol/vol) and applied to a silica column (500 mg; sep-pak, by 4 ml of n-hexane:2-propanol (97.3 vol/vol)). 7α-hydroxycholesterol was eluted with 3 ml of n-hexane:2-propanol (80:20 vol:vol) and further isolated by TLC on silica gel plates (Silica gel 60, EM Science, Gibbestown, NJ, USA) with diethyl ether, and quantified by liquid scintillation counting using Ecolume (ICN Radiochemicals Irvine, CA, USA). Assays were carried out in duplicate with correction for zero-time controls. HMG-CoA reductase (EC 1.1.1.3.4) activity was determined according to the method of Xu et al. [35] with modifications as described [36].
Statistical analysis
Data are shown as means ± SD. The Student's t test was used to determine the statistical significance of differences between the groups of animals. Significance was set at P values <0.05.
Results
Isolation of Abcg8/sterolin-2 deficient mice
The targeting construct (Figure 1a) results in the potential disruption of normal splicing, as well as loss of some coding sequences involving exons 3 and 4. After ES cell electroporation with the linearized targeted plasmid DNA, two homologous recombinant ES clones (out of 58 ES clones screened) were identified. Blastocyst injection of these resulted in five highly chimeric mice (three males, two females). One male and one female, both from the same ES clone, showed germline transmission and the female line was used to establish a multi-generational colony bred onto a C57Bl/6J background.
Heterozygous mice were fertile and gave rise to normal litter sizes. Breeding heterozygous animals led to wild-type, heterozygous and homozygous knockout mice in the expected Mendelian distribution (data not shown). Although not quantitative, Northern analyses showed that while the knockout mice showed no detectable mRNA for Abcg8, expression of Abcg5 appeared unaltered (Figure 1c). To exclude the possibility of alternative splicing with the production of a non-functional but truncated Abcg8/sterolin-2 protein that may serve as a chaperone for Abcg5/sterolin-1, RT-PCR was performed on cDNA reverse transcribed from total liver RNA from Abcg8-deficient mice. No mRNA for Abcg8/sterolin-2 was detected containing any sequences downstream of exon 4 in the Abcg8-deficient mice, whether primers were located in exons 4 and 13, exons 9 and 13 or exons 10 and 13 (Figure 1d,1e). This demonstrates that if an alternatively spliced message could potentially code for a truncated protein, it is not detectable in the Abcg8-deficient mice. The membrane-spanning domains of Abcg8/sterolin-2 are encoded by exons 9–13.
Sterol levels in Abcg8/sterolin-2 deficient animals
Elevated plant sterols in tissue and plasma are diagnostic of sitosterolemia. Sterol levels, as determined by gas chromatography (GC) analysis, in plasma and tissues of the Abcg8-/- mice are shown in Table 2. There were no differences in tissue weights between wild types, heterozygotes and knockouts. Plasma cholesterol levels of the homozygous and heterozygous mice were decreased by 52% and 26%, respectively, compared to those of wild-type mice (n = 3 all groups, Table 2). Plant sterols were almost undetectable in wild-type and heterozygous mice, but were significantly elevated in Abcg8-/- mice. Plasma campesterol and sitosterol levels were eight- and 36-fold higher in Abcg8-/- mice compared to wild-type mice (8.76 ± 2.10 and 18.52 ± 5.03 mg/dL, respectively, versus 1.14 ± 0.69 and 0.50 ± 0.55 mg/dL). There was no significant difference in the plasma plant sterol levels between heterozygous and wild-type mice. Liver cholesterol content of the Abcg8-/- mice was reduced by ~50% relative to wild-type liver (1202 ± 264 μg/g wet weight tissue, versus 2280 ± 310 μg/g wet weight tissue, respectively). The reduced cholesterol content was offset by a five-fold increase in campesterol and a 22-fold increase in sitosterol (233.9 ± 48.1 and 376.3 ± 96.1 μg/g wet weight tissue, respectively) in Abcg8-/- knockout mice compared to wild-type mice (52.2 ± 15.0 and 16.7 ± 8.6 μg/g wet weight tissue, respectively, Table 2). There were no significant differences of liver cholesterol or plant sterol contents between Abcg8+/+ and Abcg8+/- mice. Spleen sterol contents reflected the liver profiles (Table 2). No significant differences in brain sterol contents were observed, as would be expected, since the blood brain barrier is intact and prevents entry of these sterols in sitosterolemia (Table 2). There were only traces of plant sterols in the brains from knockout animals, reflecting blood contamination during tissue harvesting.
Table 2.
Mouse plasma and tissue sterol analyses
| Plasma (mg/dL) | ||||
| Group | Cholesterol | Campesterol | Stigmasterol | β-Sitosterol |
| Abcg8+/+ | 73.0 ± 14 | 1.1 ± 0.7 | ND | 0.5 ± 0.5 |
| Abcg8+/- | 54.0 ± 10* | 1.8 ± 0.3* | ND | 0.8 ± 0.2* |
| Abcg8-/- | 35.0 ± 6.0* | 8.8 ± 2.1* | ND | 18.5 ± 5.0* |
| Liver (μg/g wet tissue) | ||||
| Group | Cholesterol | Campesterol | Stigmasterol | β-Sitosterol |
| Abcg8+/+ | 2280 ± 310 | 52.2 ± 15.0 | ND | 16.7 ± 8.6 |
| Abcg8+/- | 2460 ± 588 | 76.4 ± 12.3* | 1.6 ± 3.3* | 26.7 ± 6.4* |
| Abcg8-/- | 1202 ± 264* | 233.9 ± 48.1* | 22.7 ± 6.3* | 376.3 ± 91.6* |
| Spleen (μg/g wet tissue) | ||||
| Group | Cholesterol | Campesterol | Stigmasterol | β-Sitosterol |
| Abcg8+/+ | 2702 ± 310 | 41.7 ± 7.6 | 15.1 ± 14.1 | 21.9 ± 10.9 |
| Abcg8+/- | 2578 ± 320 | 66.8 ± 18.8* | 14.5 ± 16.9* | 32.2 ± 6.1* |
| Abcg8-/- | 1779 ± 195* | 353.8 ± 97.5* | ND* | 436.6 ± 148.5* |
| Brain (μg/g wet tissue) | ||||
| Group | Cholesterol | Plant sterols | Desmosterol | Lathosterol |
| Abcg8+/+ | 11301 ± 156 | 10.2 ± 3.9 | 102.0 ± 13.3 | 52.0 ± 0.3 |
| Abcg8+/- | 11846 ± 814 | 24.5 ± 5.5 | 113.7 ± 25.5 | 49.1 ± 4.2 |
| Abcg8-/- | 11846 ± 1061 | 63.0 ± 6.5 | 123 ± 18.3 | 56.9 ± 9.5 |
Animals used in these studies were 12 weeks of age, a mixture of male and female and fed a regular chow diet. *P < 0.05 for -/- versus +/+ and -/- versus +/-. ND, not detectable.
Interestingly, the majority of the elevated tissue plant sterols are unesterified. The livers of Abcg8+/+, +/- and -/- mice have relatively similar levels of esterified cholesterol (Figure 2a). However, there is little if any esterification of sitosterol or campesterol (Figure 2b,2c) consistent with previous findings [37,38]. Thus all of the expansion of tissue sterol pools are as free sterols.
Figure 2.
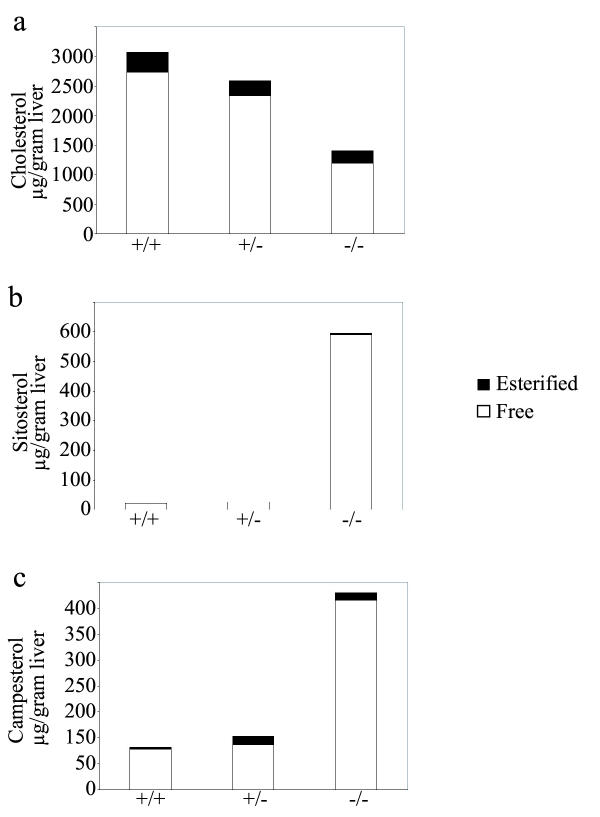
Free versus esterified sterol levels in the livers of Abcg8+/+, Abcg8+/- and Abcg8-/- mice. Sterol contents of liver extracts of 12-week-old female mice fed a regular chow diet, determined by GC analysis show that the majority of sterols in all genotypes are unesterified. Esterified cholesterol (panel a) remains relatively constant for each genotype. However, no esterified sitosterol in the livers from Abcg8+/+ and Abcg8+/- mice and very little from Abcg8-/- mice was detected (panel b). Small amounts of esterified campesterol were detected in each of the genotypes (panel c).
FPLC analyses for sterols (measured enzymatically and thus reflecting total sterols) and triglycerides were performed on plasma samples from fasted animals fed a regular chow diet. No significant differences were observed for the sterol profiles (Figure 3a), but surprisingly, the Abcg8-/- knockout mice had a significantly higher triglyceride level in the LDL lipoprotein fractions compared to wild-type and heterozygous mice (Figure 3b). The size of this peak was variable between different littermate analyses, but is always increased in the knockout animals. Total plasma triglyceride levels of the Abcg8-/- mice were slightly higher (79.2 ± 14 mg/dL compared to the wild type 63.4 ± 13 mg/dL and heterozygotes 46.6 ± 12 mg/dL, n = 3 for all genotypes). Preliminary analyses of proteins in the isolated FPLC fractions did not show any qualitative changes in apolipoproteins B, E, AI, or AII. The significance of this triglyceride rich peak remains unclear at present.
Figure 3.
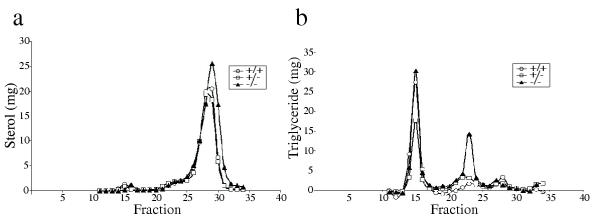
Lipid profiles of the plasma of Abcg8+/+, Abcg8+/- and Abcg8-/- mice. Lipoproteins were separated from pooled mouse plasma samples by FPLC. Total sterols (panel a) and triglyceride (panel b) profiles of the fractions are shown. There was no difference of cholesterol profiles in the groups, but there was a small triglyceride peak in Abcg8-/- mice in fractions 21–25, corresponding to the LDL-size range, the significance of which is not known at present.
Liver gene expression and enzyme activity change in Abcg8/sterolin-2 deficient mice
To investigate the effects of a deficiency of Abcg8/sterolin-2 on the genes that regulate sterol metabolism, quantitative RT-PCR was performed looking at the expression levels of Abcg5, Abcg8, Hmgr, Cyp7a1, Abca1, Mdr2, Lxr, Srebp-1c, and Srebp-2 mRNA in the livers of mice fed a regular chow diet (Figure 4a). As expected, Abcg8 mRNA expression levels were undetectable in the Abcg8-/- mice and were reduced by ~50% in the heterozygous mice, relative to wild-type mice. Interestingly, by quantitative RT-PCR, the mRNA expression of Abcg5 in the knock-out mice was also reduced by more than 60% compared to the wild-type mice, although no changes were noted in the heterozygous mice. Expression of HMG-CoA reductase mRNA was decreased by ~50% and ~80% in the heterozygote and knockout mice respectively, in keeping with limited observations in human patients with this disorder [7].
Figure 4.
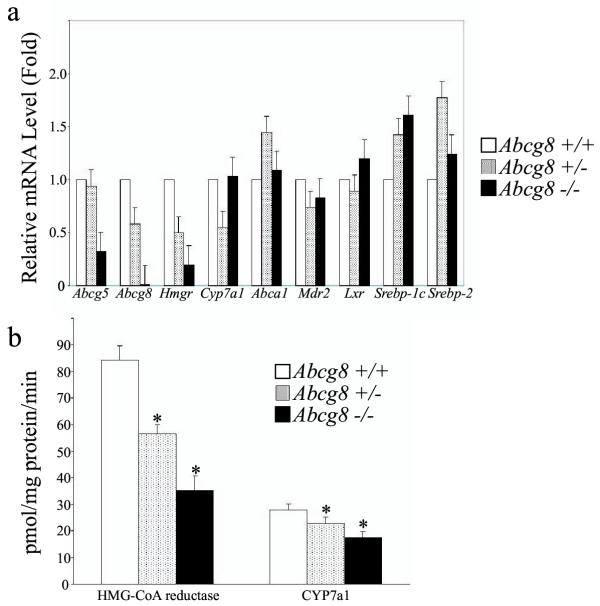
Analyses of mRNA expression and enzyme activity in mouse livers. Panel (a) shows RT-PCR quantitation of mRNA levels for Abcg5, Abcg8, Hmgr, Cyp7a1, Abca1, Mdr2, Lxr, Srebp-1c and Srebp-2 in mouse livers from Abcg8+/+ (open bars), Abcg8+/- (hatched bars) and Abcg8-/- (filled bars). Knockout mice showed an ~60% reduction in mRNA for Abcg5/sterolin-1 and an 80% reduction in the message for HMG CoA reductase. Relatively no message for Abcg8/sterolin-2 was detected in the knockout mice. Panel (b) shows the enzyme activities for HMG-CoA reductase and CYP7a1 in livers from Abcg8+/+, Abcg8+/- and Abcg8-/- mice. Activities of HMG-CoA reductase and CYP7a1 were significantly reduced in the Abcg8-/- mouse liver (*P < 0.05, see text for discussion).
To verify whether the mRNA changes resulted in alteration of the enzyme activity changes, liver samples were analyzed for HMG-CoA reductase activity and Cyp7a1 activity (Figure 4b). HMG-CoA reductase activity was reduced by 30% and 60% in the Abcg8+/- and Abcg8-/- mice, respectively (Figure 4b), and thus reflected the changes in mRNA expression. In contrast, although the Cyp7a1 mRNA expression levels were essentially unchanged in the knockout mouse, Cyp7a1 activity was significantly decreased by 37% (P < 0.01). In the heterozygous mice, both the mRNA and activity of Cyp7a1 were decreased. Sitosterol is known to be a direct competitive inhibitor of Cyp7a1 and it is likely that the elevated plant sterols in the liver are responsible for the inhibition in the knockout mouse [39].
Localization of Abcg5 protein in Abcg8-/- knockout mice
Does the loss of Abcg8/sterolin-2 result in loss of Abcg5/sterolin-1 expression, as might be predicted from the genetic studies and more recently from the in vitro and in vivo expression studies? A robust antibody to mouse Abcg8/sterolin-2 is not currently available. However, three separate groups have developed rabbit polyclonal antibody to mouse Abcg5/sterolin-1. These three antibodies were used in Western blotting and immunohistochemistry experiments on the liver and intestine of Abcg8-/- mice. Western blotting of liver total membrane preparations using the three separately-developed anti-Abcg5/sterolin-1 antibodies showed different results. As has been previously published, Abcg5/sterolin-1 exists in two separate forms, the 'immature' 75 kDa protein and a fully glycosylated 'mature' 93 kDa protein [18,30]. Using the SC anti-Abcg5/sterolin-1 peptide antibody, a 75 kDa band is detected (Figure 5a). This antibody does not detect a 'mature' 93 kDa band in either wild-type or knockout animals nor is the 75 kDa band sensitive to either Endo-H or PNGaseF (lower panel shows same aliquots probed with anti-transferrin). The AMC antibody detects the 'mature' form of Abcg5/sterolin-1 in wild-type animals but not the knockouts. Interestingly, the 'immature' 75 kDa band is detected by this antibody but the band shifts with Endo-H and PNGase F treatment (Figure 5b). The UTSW antibody detects the 'mature' and 'immature' forms of Abcg5/sterolin-1 in wild-type animals but detects no forms in the knockout mouse (Figure 5c). What does hold true for all antibodies used against Abcg5/sterolin-1 is the detection of a 75 kDa band.
Figure 5.
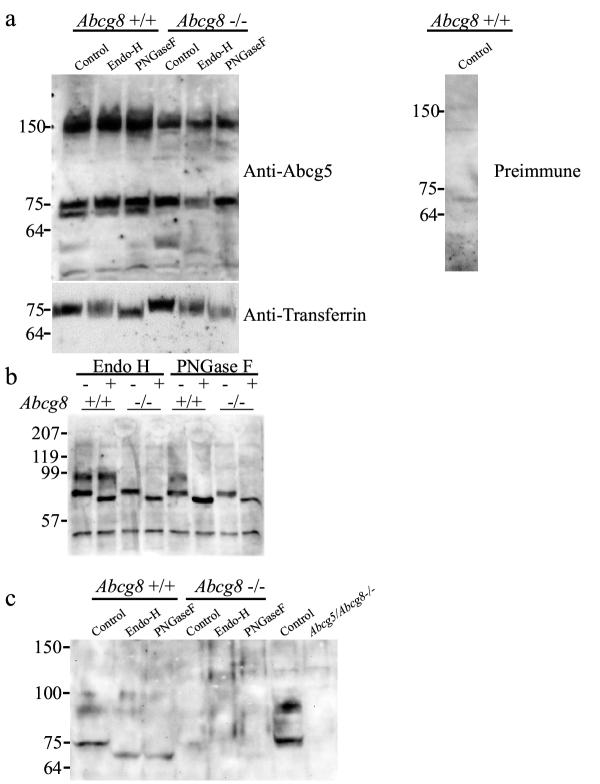
N-glycosylation of Abcg5/sterolin-1 analyses in Abcg8-/- mouse liver. Mouse liver homogenate stained with SC anti-Abcg5/sterolin-1 after treatment with Endo-H or PNGaseF shows a 75 kDa band present in both genotypes, which is resistant to deglycosylation (panel a). Staining of wild-type liver homogenate with preimmune serum showed no detectable bands. Lower portion of panel (a) shows the same aliquots stained for anti-transferrin as a control for deglycosylation. AMC anti-Abcg5/sterolin-1 staining of mouse liver homogenate shows a 'mature' ~90 kDa band in the wild-type mice but not in the Abcg8-/- mice (panel b). A 75 kDa form is present which is sensitive to deglycosylation. Mouse liver homogenate stained with UTSW anti-Abcg5/sterolin-1 shows a 'mature' ~90 kDa band and an 'immature' 75 kDa band in the wild-type mice but no signal is detected in the Abcg8-/- mice. Treatment with Endo-H or PNGaseF results in a lower molecular weight protein in the wild-type mice. Abcg5/Abcg8-/- liver homogenate used for negative control.
Immunohistochemistry experiments were carried out using all three antibodies. In acute transfection studies, the SC antibody recognized over-expressed mouse Abcg5/sterolin-1 in COS-1 cells, but not mouse Abcg8/sterolin-2 (Figure 6b,6c). Co-transfection of COS-1 cells with both Abcg5 and Abcg8 cDNAs did not alter the pattern of immunofluorescence, although we are not able to confirm simultaneous expression of Abcg8 in cells expressing Abcg5/sterolin-1, as we have no suitable antibody for Abcg8/sterolin-2 (Figure 6d). Nevertheless, this experiment indicates that the SC antibody can recognize Abcg5/sterolin-1 but does not cross-react with Abcg8/sterolin-2.
Figure 6.
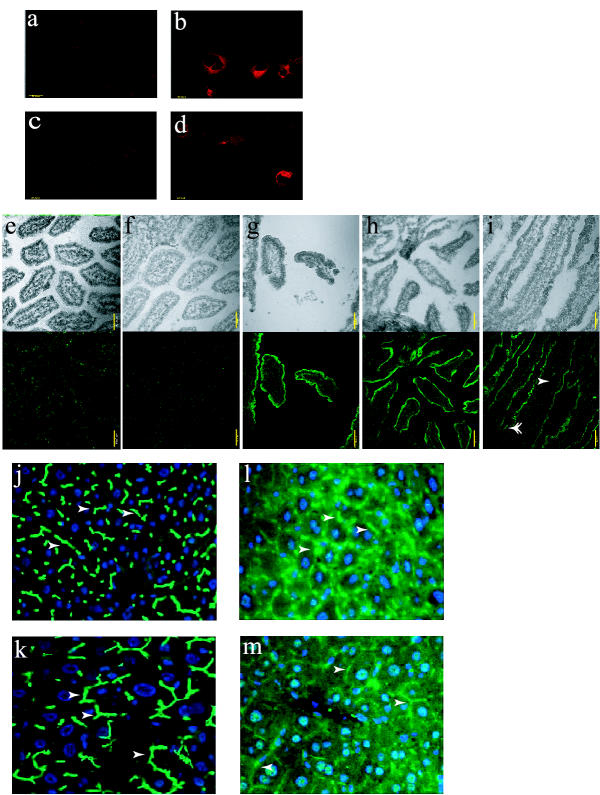
Immunohistochemical evaluation of mouse Abcg5/sterolin-1 expression in liver and intestine. COS-1 cells were transiently transfected with pCMV-mouse Abcg5 or pCMV-mouse Abcg8 constructs, allowed to express for 48 hours then fixed and incubated with antibody. pCMV empty vector transfected COS-1 cells incubated with SC anti-Abcg5/sterolin-1 showed no significant fluorescence (panel a). Abcg5 transfected COS-1 cells incubated with SC anti-Abcg5/sterolin-1 antibody showed a membrane distribution (panel b), where as Abcg8 transfected cells incubated with SC anti-Abcg5 antibody showed no significant fluorescence (panel c). Abcg5 and Abcg8 co-transfected COS-1 cells incubated with SC anti-Abcg5/sterolin-1 antibody resulted in a fluorescence pattern similar to Abcg5 alone (panel d). The yellow bar represents 20 μm. Wild-type intestine incubated with SC pre-immune serum (panel e), or SC anti-Abcg5/sterolin-1 antibody pre-incubated with the blocking peptide (panel f) showed no specific signals. Wild-type, Abcg8+/- and Abcg8-/- intestine (panels g, h and i respectively) incubated with SC anti-Abcg5/sterolin-1 antibody, showed no difference in expression patterns, despite the loss of Abcg8/sterolin-2 in the knockout mice. Single arrowhead shows intestinal villus and double arrowhead shows crypt. The yellow bar represents 50 μm. Antibody staining of liver sections was also performed at the AMC Liver Center. As a control, an antibody to Bsep/Abcb11 was used and showed a clear apical distribution in both wild-type (panel j) and Abcg8 knockout mice (panel k). Using AMC antibody against Abcg5/sterolin-1 (see text), in both wild-type (panel l) and Abcg8 knockout liver (panel m), the pattern of expression was also apical and unchanged although the signal is fainter compared to that for Bsep/Abcb11 (see text for discussion). Arrows indicate bile canaliculi.
Serial sections of intestinal tissue were incubated with anti-Abcg5/sterolin-1 to determine the cellular location of Abcg5/sterolin-1. The pattern of staining of Abcg5/sterolin-1 in the intestinal sections is clearly apical and localized to the villi of the enterocytes (Figure 6g,6h,6i). Loss of Abcg8/sterolin-2 does not seem to affect this pattern of staining. To further confirm that the antibody used recognizes Abcg5/sterolin-1; this pattern is blocked if the antibody is pre-incubated with the peptide to which it was raised (Figure 6f). To further confirm that Abcg5/sterolin-1 expression is preserved and may be apical, immunohistochemistry was performed in Amsterdam using the AMC antibody to stain liver sections from wild-type and knockout mice [30]. As a control, the expression of Abcb11, an ABC transporter known to be responsible for the export of bile salts, was compared to that of Abcg5/sterolin-1 (Figure 6j,6k). Again, both proteins are apically localized in liver sections from wild-type mice and this pattern is essentially unperturbed in liver from knockout mice, although the expression level of Abcg5/sterolin-1 seems to be less robust qualitatively (Figure 6l,6m). To further confirm the apical expression of Abcg5/sterolin-1 in the knockout mice, the UTSW antibody was used to stain liver sections of wild-type and knockout mice. Again, there is apical expression of Abcg5/sterolin-1 in both wild-type and knockout livers (Figure 7a,7b).
Figure 7.
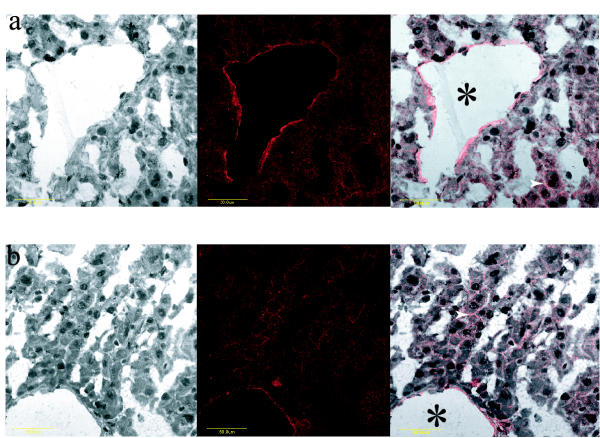
Immunohistochemical evaluation of mouse Abcg5/sterolin-1 expression in liver. Wild-type liver incubated with UTSW anti-Abcg5/sterolin-1 resulted in apical expression (panel a). Merged image clearly shows the apical distribution. Likewise Abcg8-/- liver incubated with the same UTSW anti-Abcg5/sterolin-1 antibody resulted in a similar apical expression pattern relative to the wild type (panel b). White arrows indicate bile canaliculi and asterisk indicates a bile duct. The yellow bar represents 50 μm.
Biliary secretion of bile salts, sterol and phospholipids in Abcg8/sterolin-2 deficient mice
Sitosterolemic individuals have an impaired ability to secrete cholesterol and plant sterols into bile [40,41]. We analyzed the knockout mice for any alterations in biliary sterol handling. Analyses of the initial basal bile secretion showed that the sterol and phospholipid contents in Abcg8-/- mice were reduced as compared to wild-type mice, (Figure 8a,8b,8c, bile salt secretion not statistically significant, for sterol secretion P = 0.01 and for phospholipid secretion P = 0.03).
Figure 8.
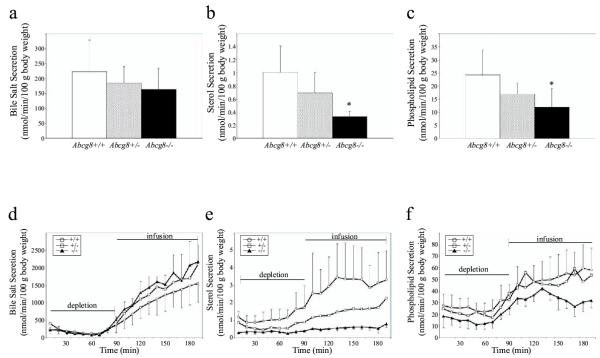
Biliary sterol, phospholipid and bile salt analyses. Bile salt, sterol and phospholipid contents from male Abcg8+/+ (n = 8), Abcg8+/- (n = 7), and Abcg8-/- (n = 5) mice were examined as described in Methods. Bile salt secretion following a 10 minute collection was lower in Abcg8+/- and Abcg8-/- mice compared to wild type (panel a), but these differences were not statistically significant. Biliary sterol and phospholipid were significantly reduced in the Abcg8-/- mice compared to the wild-type (panels b and c). *P < 0.05. The lower panel shows biliary bile salt, sterol and phospholipid secretion rates from female Abcg8+/+ (n = 5), Abcg8+/- (n = 4), and Abcg8-/- (n = 3) mice measured following 90-minute bile salt depletion followed by stepwise TUDC infusion as described in the text. Bars represent phase TUDC infusion rates. No differences were observed in the ability of the Abcg8-/- mice to secrete bile salts (panel d), but there is a marked inability of the knockout mice to secrete sterols (panel e) and a trend towards a reduced ability to secrete phospholipid (panel f) compared to wild type.
To investigate whether the defect in biliary sterol secretion could be (partly) restored by forced biliary sterol secretion, we infused mice with stepwise increasing doses of tauroursodeoxycholic acid (TUDC). Mice were first depleted of their endogenous bile salt pools for 90 min and subsequently infused via the jugular vein with increasing doses of TUDC. As shown in Figure 8d, bile salt secretion was no different between the different genotypes during the depletion or the TUDC infusion phases of the experiment. However, a different pattern emerged for the secretion of phospholipid (Figure 8f). There was no difference between wild-type and heterozygous mice, but knockout mice showed a trend of lower phospholipid secretion during both the depletion and infusion phase. Even more dramatic was the effect on biliary sterol secretion although the sterol contents were determined enzymatically and no distinction is made as to whether this is sitosterol or cholesterol (Figure 8e, but see below). In the knockout mice, almost no stimulated sterol secretion was noted, with an intermediate phenotype in the heterozygous mice. In the heterozygous mice, sterol secretion increased to reach a maximal level of about 50% of the wild type suggesting that Abcg8/sterolin-2 is a rate-limiting step for biliary 'cholesterol' secretion. In the knockout mice, sterol secretion remained at a constant level during the depletion phase and increased minimally upon infusion of TUDC.
Since the Abcg8-/- mice still secreted some sterols we were interested in which biliary sterol species were being secreted. Therefore bile was collected from mice during a constant infusion of TUDC and analyzed by GC. Mice were first depleted of their endogenous bile acid pools for 30 minutes then infused with a continuous dose of TUDC (1,200 nmol/min/100 g body weight). The knockout mice showed a significantly diminished ability to secrete cholesterol, yet still maintained the ability to secrete sitosterol and campesterol compared to wild-type mice (Figure 9). Surprisingly, the Abcg8+/- mice tended to show an increased ability to secrete all sterols above the levels seen in the wild-type mice, although this was not statistically significant.
Figure 9.
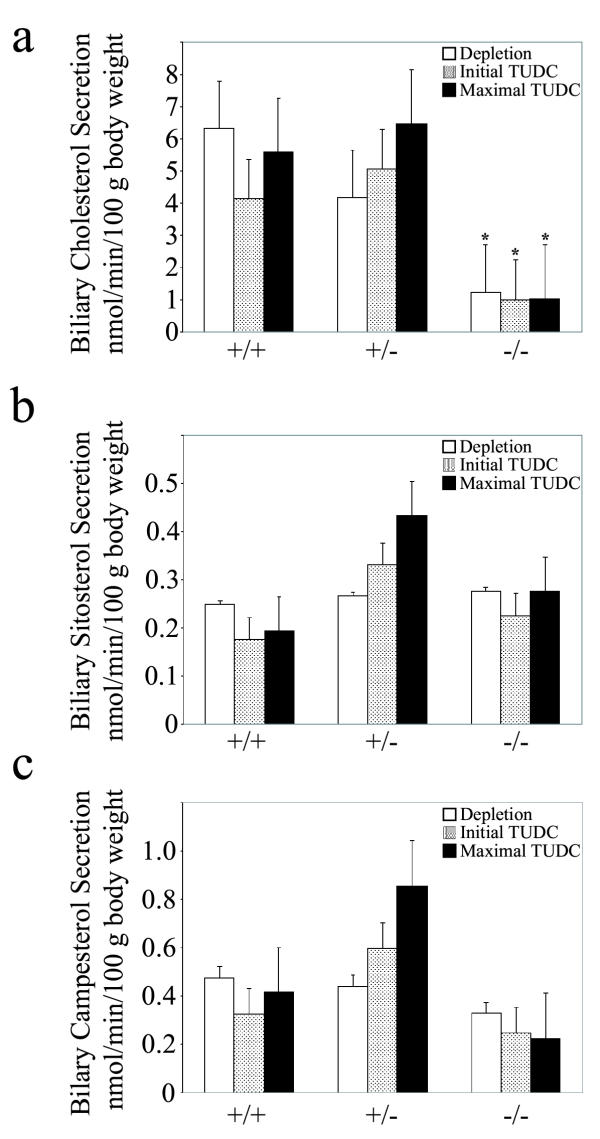
Biliary cholesterol and sitosterol secretion. Biliary sterols were analyzed by GC analyses in Abcg8+/+ (n = 4), Abcg8+/- (n = 3) and Abcg8-/- (n = 4) mice following 30-minute bile salt depletion followed by a continuous TUDC infusion as described in Methods. Abcg8-/- mice were unable to secrete cholesterol into bile with forced TUDC administration relative to the wild-type mice (panel a, *P < 0.05). Interestingly, the Abcg8-/- mice were able to still secrete sitosterol (panel b) and campesterol (panel c). Heterozygous mice showed an increased ability to secrete all sterols with forced TUDC administration, although the results were not statistically significant.
Discussion
The mechanism by which dietary cholesterol is specifically absorbed and dietary non-cholesterol sterols primarily excluded or the mechanism(s) by which the liver can selectively secrete sterols has not been elucidated. Identification of the genetic defect(s) in a rare human disorder, sitosterolemia, where these processes are specifically disrupted, may finally have led to the identification of the 'transporters' responsible for these processes. Complete defects in one of two genes (but not both), organized in a head-to-head configuration at the STSL locus, causes sitosterolemia. We report a mouse model of sitosterolemia, with a selective, but complete, defect in one of these genes, Abcg8. This mouse reflects the known defects described in human sitosterolemia [5,7,42]. Plasma and all tissues, apart from the brain, have significantly elevated sitosterol levels. Homozygous knockout mice are viable, fertile and sitosterolemic. However, fertility seems to be reduced when homozygous mice are bred together (S Patel and J Oatis, unpublished observation).
This model reflects many other observed changes described in limited studies in humans. For example, in humans, the activity of HMG-CoA reductase and CYP7a1 have been reported to be low [7,39]. In this study, we show that the mRNA and enzyme activities in the livers from knockout animals are also significantly reduced. Additionally, the activity of the rate-limiting enzyme for bile acid synthesis, CYP7a1, is reduced, although no changes at the mRNA level are noted. This is also in keeping with previous studies that show that sitosterol is a direct inhibitor of this enzyme in vitro [39].
Biliary sterols of Abcg8-deficient mice were dramatically different compared to wild-type mice. Measurement of biliary sterol secretion rates in Abcg8-/- mice demonstrated a failure to increase sterol secretion into bile despite exogenous infusion of bile acids. Furthermore, complete loss of Abcg8/sterolin-2 results in an inability to secrete cholesterol into bile, although secretion of sitosterol seemed to be preserved. Arguably, since the body pools and plasma sitosterol levels in the knockout mice are so considerably elevated, perhaps the biliary sitosterol levels could be considered to be inappropriately low. Despite this reservation, the finding that sitosterol is present in the bile suggests that plant sterols may be secreted independent of Abcg8/sterolin-2. The ability of Abcg8-/- mice to secrete bile salt and phospholipid into bile as compared to wild-type mice was not significantly impaired. Collectively these data suggest that Abcg8/sterolin-2 is necessary for hepatobiliary cholesterol secretion in mice.
Abcg8-/- mice appeared normal and healthy maintained on a regular rodent chow diet. Their phenotypic features are very similar to those recently reported for a mouse deficient in both sterolins simultaneously [7,20]. Plasma and hepatic plant sterol levels in Abcg8-/- and Abcg5/Abcg8 knockout are increased similarly with marked decreases in cholesterol levels. Interestingly, plasma triglyceride levels of the Abcg8-/- mice were slightly higher than wild-type animals and this increased triglyceride is carried in the LDL fraction range, as measured by FPLC. The significance of this is not clear, although preliminary SDS-PAGE analyses of all the fractions failed to reveal any differences between wild-type and knockout samples.
Hepatic levels of GC-measured cholesterol were greatly reduced in Abcg8-/- mice compared to wild-type mice, yet mRNA levels of HMG-CoA reductase and enzyme activity are also reduced without any significant change in mRNA of the Srebps. The basis for this is not clear at present, unless the increase in non-plant sterols leads to a suppression of SREBP activation. Sitosterol has been shown to directly inhibit Cyp7a1 activity in vitro and we presume this may account for the reduced enzyme activity [39]. In preliminary studies, when knockout mice are placed on a low sitosterol diet, the activity of this enzyme, as well as the mRNA levels are increased (E Klett and S Patel, unpublished observations). Another prediction, based upon the enzyme and mRNA levels in knockout mice, is that if these mice are placed on a high cholesterol/high sitosterol diet, they may show significant accumulation of both sterols in the body. This may also be relevant to the human disorder where some, although not all affected individuals, manifest premature coronary artery disease.
Abcg8/sterolin-2 deficient mice also allow us to examine the role of this protein in biliary sterol secretion. The bile is sterol poor and upon stimulation by increasing bile salt excretion, Abcg8/sterolin-2 deficient mice cannot respond, in contrast to the wild-type mice. Thus, Abcg8/sterolin-2 is necessary for cholesterol secretion but not necessary for plant sterol secretion. It could be argued that although the biliary sterol output is comparable to that seen in wild-type mice that this is inappropriately low, since the tissue and plasma pools of sitosterol are so elevated in the knockout mice. Despite this caveat, the findings of sitosterol in significant quantities in the bile suggests that mechanisms other than via the sterolins can 'export' some of these sterols. Interestingly, although humans who are heterozygous for genetic defects in either ABCG5 or ABCG8 seem to be phenotypically normal, mice heterozygous for Abcg8/sterolin-2 deficiency show that sterol secretion is impaired, but not absent. Note that under steady-state conditions on a rodent chow diet, heterozygous animals show no significant elevations in plasma or tissue sitosterol levels, suggesting that the activity of these proteins may not be rate limiting. Furthermore, the heterozygous Abcg8+/- mice showed higher levels of biliary sitosterol relative to the wild-type mice. While the rate of total biliary sterol secretion is reduced compared to wild-type mice, since the heterozygous mice are not sitosterolemic, either the activity of these proteins is not rate limiting or other mechanisms can compensate for a 50% loss of activity. On the other hand, since feeding is intermittent, a slow, but continuous secretion during post-prandial periods could easily negate any mild temporary increase in tissue and plasma plant sterol levels in heterozygous animals. This may be amenable to testing by placing these animals on a high plant sterol diet and measuring the plasma sterol levels.
Recently, Kosters et al. reported that Abcg5/Abcg8 mRNA expression in a variety of different mouse strains correlated with biliary cholesterol secretion rates [30]. In their studies, although diosgenin-fed mice showed a marked increase in biliary cholesterol output, mRNA levels of Abcg5 and Abcg8 were not altered. Using Western blot analyses, the protein level of Abcg5 was also not altered by diosgenin, although Abcg8 was not measured. They concluded that a parallel route for biliary cholesterol secretion might be operational, independent of the sterolins. While biliary cholesterol secretion is not completely absent in the Abcg8-deficient mice it would appear that sterolin-2 plays a major role in this process. In a genetic screen of plasma plant sterol levels, Sehayek et al. identified three loci that may be responsible for controlling plasma plant sterol levels and not one of these loci mapped to the murine STSL region [43]. Thus, there is support for at least four loci that may be involved in regulating plasma plant sterol levels. To date the STSL locus is the only one proven to be involved in dietary sterol trafficking and the identity of the others remains to be elucidated.
We, as well as others, have proposed Abcg5/sterolin-1 and Abcg8/sterolin-2 to function as obligate heterodimers. Thus knocking out Abcg8/sterolin-2 alone is equivalent to a functional loss of both sterolins. It has been shown that Abcg8/sterolin-2 expression is required for correct apical trafficking of Abcg5/sterolin-1 to the apical surface in a polarized cell line and more recently in vivo by over-expression experiments [18,19]. In experiments shown here, using three separately developed anti-Abcg5/sterolin-1 antibodies, we have obtained inconsistent data regarding the trafficking of Abcg5/sterolin-1 in Abcg8-/- mice. Based upon classical N-glycosylation maturation it would appear that Abcg5/sterolin-1, in the Abcg8-/- mice, does not exit the endoplasmic reticulum. However, by immunohistochemistry it appears that Abcg5/sterolin-1 is apically expressed in the Abcg8-deficient mice regardless of the anti-Abcg5/sterolin-1 antibody used. Interestingly, in a report of Abcg5/Abcg8 localization in canine gallbladder epithelial cells, these two proteins were found intracellularly under baseline conditions [44]. But when given liver X receptor alpha (LXRα) agonist, Abcg5/sterolin-1 and Abcg8/sterolin-2 appeared to be expressed at the apical surface. In these studies the UTSW anti-Abcg5/sterolin-1 antibody was used. It is apparent with these conflicting data that the trafficking of these transporters is not as clear-cut. In a recent paper, Mangrivite et al. showed that the apical sorting of rat SPNT in polarized renal epithelial cells was independent of N-glycosylation [45]. Therefore, at this time, we can neither exclude the possibility that Abcg5/sterolin-1 expression is functional in our Abcg8/sterolin-2 deficient mice nor can we rule it out. We have no functional assay for these proteins at present. More extensive fractionation experiments are underway to better determine the exact compartmental location of Abcg5/sterolin-1 in the Abcg8-/- mice. In this context, Plösch et al. have described an Abcg5/sterolin-1 deficient mouse that maintains the ability to secrete biliary cholesterol to the same extent as the wild-type mice and, when fed a LXR agonist, had higher Abcg8 mRNA expression and tended to secrete more biliary cholesterol than wild-type mice [46]. It has been argued that Abcg5/sterolin-1 and Abcg8/sterolin-2 are dependent upon each other for secretion of hepatic sterols into bile [19]. Given the data presented here and from the Abcg5-deficient mouse this does not appear to be the case. Perhaps in the previously published model a non-physiologic state has been made that generates these data. Taken together, these animal models suggest that Abcg5/sterolin-1 and Abcg8/sterolin-2 have independent function in vivo or that there are proteins other than the sterolins that can secrete biliary sterols.
Conclusions
The major findings in this study are: (1) disruption of only one of the two genes comprising the STSL locus is necessary and sufficient to cause sitosterolemia, (2) that the loss of Abcg8/sterolin-2 leads to the loss of biliary cholesterol secretion and (3) Abcg5/sterolin-1 appears to be still apically-expressed despite the absence of Abcg8/sterolin-2. These data strongly support the direct role of Abcg8/sterolin-2 as a key player in biliary cholesterol secretion, although biliary sitosterol secretion was apparently preserved. These data suggest that there are mechanisms other than Abcg8/sterolin-2 that allow for the secretion of sitosterol into bile.
Competing interests
None declared.
Authors' contributions
ELK carried out molecular genetics studies, quantitative RT-PCR, and dynamic bile collection for Figure 9. KL carried out molecular genetic studies and made the targeting construct. The knockout mice were isolated in the NM laboratory. HY performed Northern blotting. ELK, AK, EV and MHL performed Western blotting and immunohistochemistry experiments. AK and NL performed bile collection experiments and bile characterization for Figure 8. MA performed FPLC analysis. SS, AK and RK performed GC sterol analyses. JC assisted with dynamic bile collection and mouse surgery. ROE, AG, NM, GS and SBP were responsible for supervision, data analyses of experiments and for providing funding of these experiments. ELK and SBP wrote the paper.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We would like to thank Dr B Stieger for kindly providing anti-Bsep antibody and Dr H Hobbs for providing the UTSW anti-Abcg5/sterolin-1 antibody. We would also like to thank John Oatis, III for his excellent animal husbandry. Grants from the American Heart Association, Beginning Grant-In-Aid Mid-Atlantic Affiliate (KL), NIH Postdoctoral Training Grant T32 HL07260 (ELK), the Netherlands Organization for Scientific Research (NOW) 902-23-193 (AK) as well as the National Institutes of Health Grant HL60613 (SBP) and DK56830 (GS) supported this work.
Contributor Information
Eric L Klett, Email: klettel@musc.edu.
Kangmo Lu, Email: Kangmo-Lu@ouhsc.edu.
Astrid Kosters, Email: A.Kosters@amc.uva.nl.
Edwin Vink, Email: E.Vink@amc.uva.nl.
Mi-Hye Lee, Email: leemih@musc.edu.
Michael Altenburg, Email: mikelly@email.unc.edu.
Sarah Shefer, Email: salenge@umdnj.edu.
Ashok K Batta, Email: ashkbatta@hotmail.com.
Hongwei Yu, Email: yuh@musc.edu.
Jianliang Chen, Email: chenjl@musc.edu.
Richard Klein, Email: kleinrl@musc.edu.
Norbert Looije, Email: N.Looije@amc.uva.nl.
Ronald Oude-Elferink, Email: r.p.oude-elferink@amc.uva.nl.
Albert K Groen, Email: a.k.groen@amc.uva.nl.
Nobuyo Maeda, Email: nobuyo.maeda@pathology.unc.edu.
Gerald Salen, Email: salenge@umdnj.edu.
Shailendra B Patel, Email: patelsb@musc.edu.
References
- Weihrauch JL, Gardner JM. Sterol content of foods of plant origin. J Am Diet Assoc. 1978;73:39–47. [PubMed] [Google Scholar]
- Nair PP, Turjman N, Kessie G, Calkins B, Goodman GT, Davidovitz H, Nimmagadda G. Diet, nutrition intake, and metabolism in populations at high and low risk for colon cancer. Dietary cholesterol, beta-sitosterol, and stigmasterol. Am J Clin Nutr. 1984;40:927–930. doi: 10.1093/ajcn/40.4.927. [DOI] [PubMed] [Google Scholar]
- Wilson MD, Rudel LL. Review of cholesterol absorption with emphasis on dietary and biliary cholesterol. J Lipid Res. 1994;35:943–955. [PubMed] [Google Scholar]
- Schoenheimer Rudolph. Uber die Bedeutung der Pflanzensterine fur den tierischen Organismus. Z für physiol Chem. 1929;180:1–5. [Google Scholar]
- Bhattacharyya AK, Connor WE. Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. Journal of Clinical Investigation. 1974;53:1033–1043. doi: 10.1172/JCI107640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RS, Bhattacharyya AK, Connor WE, Fredrickson DS. Beta-sitosterolemia and xanthomatosis. New England Journal of Medicine. 1976;294:482–483. doi: 10.1056/NEJM197602262940907. [DOI] [PubMed] [Google Scholar]
- Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Shore V. Sitosterolemia. Journal of Lipid Research. 1992;33:945–955. [PubMed] [Google Scholar]
- Salen G, Shore V, Tint GS, Forte T, Shefer S, Horak I, Horak E, Dayal B, Nguyen L, Batta AK, et al. Increased sitosterol absorption, decreased removal, and expanded body pools compensate for reduced cholesterol synthesis in sitosterolemia with xanthomatosis. Journal of Lipid Research. 1989;30:1319–1330. [PubMed] [Google Scholar]
- Bhattacharyya AK, Connor WE, Lin DS, McMurry MM, Shulman RS. Sluggish sitosterol turnover and hepatic failure to excrete sitosterol into bile cause expansion of body pool of sitosterol in patients with sitosterolemia and xanthomatosis. Arterioscler Thromb. 1991;11:1287–1294. doi: 10.1161/01.atv.11.5.1287. [DOI] [PubMed] [Google Scholar]
- Patel SB, Honda A, Salen G. Sitosterolemia: exclusion of genes involved in reduced cholesterol biosynthesis. Journal of Lipid Research. 1998;39:1055–1061. [PubMed] [Google Scholar]
- Patel SB, Salen G, Hidaka H, Kwiterovich PO, Stalenhoef AF, Miettinen TA, Grundy SM, Lee MH, Rubenstein JS, Polymeropoulos MH, Brownstein MJ. Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest. 1998;102:1041–1044. doi: 10.1172/JCI3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Gordon D, Ott J, Lu K, Ose L, Miettinen T, Gylling H, Stalenhoef AF, Pandya A, Hidaka H, Brewer Jr., B., Kojima H, Sakuma N, Pegoraro R, Salen G, Patel SB. Fine mapping of a gene responsible for regulating dietary cholesterol absorption; founder effects underlie cases of phytosterolemia in multiple communities. European Journal of Human Genetics. 2001;9:375–384. doi: 10.1038/sj.ejhg.5200628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Lee M-H, Carpten JD, Sekhon M, Patel SB. High-Resolution Physical and Transcript Map of Human Chromosome 2p21 Containing the Sitosterolemia Locus. European Journal of Human Genetics. 2001;9:364–374. doi: 10.1038/sj.ejhg.5200627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- Lu K, Lee M-H, Hazard S, Brooks-Wilson A, Hidaka H, Kojima H, Ose L, Stanlenhoef AFH, Mietinnen T, Bjorkhem I, Brukert E, A. Pandya., Brewer HB, Salen G, Dean M, Srivastava A, Patel SB. Two genes that map to the STSL locus cause sitosterolemia: Genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8 respectively. American Journal of Human Genetics. 2001;69:278–290. doi: 10.1086/321294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R, Srivastava AK, Salen G, Dean M, Patel SB. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nature Genetics. 2001;27:79–83. doi: 10.1038/87170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–680. doi: 10.1172/JCI200216001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf GA, Li WP, Gerard R, Gelissen I, White A, Cohen JC, Hobbs HH. Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J Clin Invest. 2002;110:659–669. doi: 10.1172/JCI200216000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf GA, Yu L, Li WP, Gerard RD, Tuma PL, Cohen JC, Hobbs HH. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. Epub 2003 Sep 22. [DOI] [PubMed] [Google Scholar]
- Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Lee MH, Yu H, Zhou Y, Sandell SA, Salen G, Patel SB. Molecular cloning, genomic organization, genetic variations, and characterization of murine sterolin genes Abcg5 and Abcg8. J Lipid Res. 2002;43:565–578. [PMC free article] [PubMed] [Google Scholar]
- Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhu YH, Patel SB. Cyclosporin-induced dyslipoproteinemia is associated with selective activation of SREBP-2. Am J Physiol. 1999;277:E1087–94. doi: 10.1152/ajpendo.1999.277.6.E1087. [DOI] [PubMed] [Google Scholar]
- Plosch T, Kok T, Bloks VW, Smit MJ, Havinga R, Chimini G, Groen AK, Kuipers F. Increased hepatobiliary and fecal cholesterol excretion upon activation of the liver X receptor is independent of ABCA1. J Biol Chem. 2002;277:33870–33877. doi: 10.1074/jbc.M206522200. [DOI] [PubMed] [Google Scholar]
- Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, Horton JD. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci U S A. 2001;98:13607–13612. doi: 10.1073/pnas.201524598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest. 1999;103:1579–1586. doi: 10.1172/JCI6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LB, Shefer S, Salen G, Horak I, Tint GS, McNamara DJ. The effect of abnormal plasma and cellular sterol content and composition on low density lipoprotein uptake and degradation by monocytes and lymphocytes in sitosterolemia with xanthomatosis. Metabolism. 1988;37:346–351. doi: 10.1016/0026-0495(88)90134-5. [DOI] [PubMed] [Google Scholar]
- Yu H, Pandit B, Klett E, Lee MH, Lu K, Helou K, Ikeda I, Egashira N, Sato M, Klein R, Batta A, Salen G, Patel SB. The rat STSL locus: characterization, chromosomal assignment, and genetic variations in sitosterolemic hypertensive rats. BMC Cardiovasc Disord. 2003;3:4. doi: 10.1186/1471-2261-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters A, Frijters RJ, Schaap FG, Vink E, Plosch T, Ottenhoff R, Jirsa M, De Cuyper IM, Kuipers F, Groen AK. Relation between hepatic expression of ATP-binding cassette transporters G5 and G8 and biliary cholesterol secretion in mice. J Hepatol. 2003;38:710–716. doi: 10.1016/S0168-8278(03)00093-X. [DOI] [PubMed] [Google Scholar]
- Meier PJ, Sztul ES, Reuben A, Boyer JL. Structural and functional polarity of canalicular and basolateral plasma membrane vesicles isolated in high yield from rat liver. J Cell Biol. 1984;98:991–1000. doi: 10.1083/jcb.98.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijters CM, Ottenhoff R, Van Wijland MJ, Van Nieuwkerk C, Groen AK, Oude Elferink RP. Influence of bile salts on hepatic mdr2 P-glycoprotein expression. Adv Enzyme Regul. 1996;36:351–363. doi: 10.1016/0065-2571(95)00019-4. [DOI] [PubMed] [Google Scholar]
- Shefer S, Salen, G and Batta, AK. Cholesterol 7a- hydroxylase (7a-monooxygenase EC 1.14.12.17) In: Fears R and Sabine JR, editor. Methods of Assay. Boca Raton, CRC; 1986. [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randell RJ. Protein measurements with the Folin phenol reagent. Journal of Biological Chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- Xu G, Salen G, Shefer S, Ness GC, Nguyen LB, Parker TS, Chen TS, Zhao Z, Donnelly TM, Tint GS. Unexpected inhibition of cholesterol 7 alpha-hydroxylase by cholesterol in New Zealand white and Watanabe heritable hyperlipidemic rabbits. J Clin Invest. 1995;95:1497–1504. doi: 10.1172/JCI117821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau G, Shefer S, Salen G, Mosbach EH. Determination of hepatic 3-hydroxy-3-methylglutaryl CoA reductase activity in man. J Lipid Res. 1974;15:94–98. [PubMed] [Google Scholar]
- Field FJ, Mathur SN. beta-sitosterol: esterification by intestinal acylcoenzyme A: cholesterol acyltransferase (ACAT) and its effect on cholesterol esterification. J Lipid Res. 1983;24:409–417. [PubMed] [Google Scholar]
- Tavani DM, Nes WR, Billheimer JT. The sterol substrate specificity of acyl CoA: :cholesterol acyltransferase from rat liver. J Lipid Res. 1982;23:774–781. [PubMed] [Google Scholar]
- Shefer S, Salen G, Nguyen L, Batta AK, Packin V, Tint GS, Hauser S. Competitive inhibition of bile acid synthesis by endogenous cholestanol and sitosterol in sitosterolemia with xanthomatosis. Effect on cholesterol 7 alpha-hydroxylase. Journal of Clinical Investigation. 1988;82:1833–1839. doi: 10.1172/JCI113799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salen G, Ahrens E. H., Jr., Grundy SM. Metabolism of beta-sitosterol in man. J Clin Invest. 1970;49:952–967. doi: 10.1172/JCI106315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen TA. Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: a case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur J Clin Invest. 1980;10:27–35. doi: 10.1111/j.1365-2362.1980.tb00006.x. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Boberg KM. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: Scriver CR, Beaudet AL, Sly WS and Valle D, editor. The Metabolic Basis of Inherited Disease. 7th. Vol. 2. New York, McGraw-Hill Inc.; 1995. pp. 2073–2102. [Google Scholar]
- Sehayek E, Duncan EM, Lutjohann D, Von Bergmann K, Ono JG, Batta AK, Salen G, Breslow JL. Loci on chromosomes 14 and 2, distinct from ABCG5/ABCG8, regulate plasma plant sterol levels in a C57BL/6J x CASA/Rk intercross. Proc Natl Acad Sci U S A. 2002;99:16215–16219. doi: 10.1073/pnas.212640599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauscher A, Kuver R. ABCG5 and ABCG8 are expressed in gallbladder epithelial cells. Biochem Biophys Res Commun. 2003;307:1021–1028. doi: 10.1016/S0006-291X(03)01296-8. [DOI] [PubMed] [Google Scholar]
- Mangravite LM, Giacomini KM. Sorting of rat SPNT in renal epithelium is independent of N-glycosylation. Pharm Res. 2003;20:319–323. doi: 10.1023/A:1022247826750. [DOI] [PubMed] [Google Scholar]
- Plosch T, Bloks VW, Terasawa Y, Berdy S, Siegler K, Van Der Sluijs F, Kema IP, Groen AK, Shan B, Kuipers F, Schwartz M. Sitosterolemia in ABC-Transporter G5-deficient mice is aggravated on activation of the liver-X receptor. Gastroenterology. 2004;126:290–300. doi: 10.1053/j.gastro.2003.10.074. [DOI] [PubMed] [Google Scholar]