

Abstract
Haploinsufficiency of the single-minded homology 1 (SIM1) gene in humans and mice leads to severe obesity, suggesting that altered expression of SIM1, by way of regulatory elements such as enhancers, could predispose individuals to obesity. Here, we identified transcriptional enhancers that could regulate SIM1, using comparative genomics coupled with zebrafish and mouse transgenic enhancer assays. Owing to the dual role of Sim1 in hypothalamic development and in adult energy homeostasis, the enhancer activity of these sequences was annotated from embryonic to adult age. Of the seventeen tested sequences, two SIM1 candidate enhancers (SCE2 and SCE8) were found to have brain-enhancer activity in zebrafish. Both SCE2 and SCE8 also exhibited embryonic brain-enhancer expression in mice, and time course analysis of SCE2 activity showed overlapping expression with Sim1 from embryonic to adult age, notably in the hypothalamus in adult mice. Using a deletion series, we identified the critical region in SCE2 that is needed for enhancer activity in the developing brain. Sequencing this region in obese and lean cohorts revealed a higher prevalence of single nucleotide polymorphisms (SNPs) that were unique to obese individuals, with one variant reducing developmental-enhancer activity in zebrafish. In summary, we have characterized two brain enhancers in the SIM1 locus and identified a set of obesity-specific SNPs within one of them, which may predispose individuals to obesity.
INTRODUCTION
In the last decade, obesity has reached epidemic rates of prevalence in the USA. According to the National Center for Health Statistics, it is estimated that over one-third (35.7%) of the US population is currently obese compared with 27.5% in 1999–2000 (1). In addition, another third of the US population is considered overweight and global overweight/obesity rates are reaching similar prevalence (2,3). Obesity is a major public health concern due to its contribution to increasing risk for other diseases such as cancer, cardiovascular disease, type 2 diabetes, stroke, sleep apnea, asthma, osteoarthritis, organ diseases and a host of other co-morbidities (4–6). The cause of obesity has been attributed to a combination of reduced energy expenditure and physical activity, increased sedentary lifestyle, dietary excess and other environmental factors. Equally important is the underlying genetic predisposition that lays the foundation on which environmental and exogenous factors manifest their propensity to drive the obesity phenotype. The heritability of obesity has been well-documented, through family (7) and twin studies (8,9) and it is estimated to be between 40 and 70% (10), indicating the importance of understanding and elucidating the genes and pathways associated with obesity. To date, the leptin–melanocortin pathway is the most genetically implicated pathway in severe monogenic human obesity. Clinical reports have described individuals carrying mutations in this pathway to be the cause of a severe obesity phenotype (11).
One member of this pathway is the single-minded homology 1 (SIM1) gene. Originally discovered as a transcription factor essential for midline development in drosophila (12,13), it has also been shown to be important in the leptin–melanocortin pathway and is associated with monogenic and syndromic forms of obesity in humans and mice. In humans, chromosomal aberrations in the SIM1 locus result in hyperphagic obesity (14). A proportion of individuals who have Prader–Willi-like syndrome (PWS-like; OMIM #176270) carry interstitial deletions and rearrangements that include the SIM1 locus and are also obese (15–22). In addition, mutation analyses of SIM1 in obese and lean cohorts found an increase in unique rare [minor allele frequency (MAF) <1%] variants in the obese population, second only in prevalence to MC4R (23). A SIM1 haplotype of two common (MAF <1%) non-synonymous single nucleotide polymorphisms (SNPs) (P352T/A371V) in complete linkage disequilibrium was also found to associate with obesity [or higher body mass index (BMI)] in Caucasian males (24) and nominally associated in French Europeans (25). Furthermore, major SIM1 allele variants in the Pima Indian population were also found to be associated with BMI (26). Most recently, three groups identified and functionally characterized protein coding mutations in SIM1. Bonnefond et al. (27) identified rare variants in morbidly obese individuals with and without PWS-like syndrome features; most of these protein coding mutations altered SIM1 activity in functional assays. Ramachandrappa et al. (28) discovered rare variants, most of which reduced SIM1 function; probands carrying these rare variants segregated with hyperphagic behavior and increased BMI. Finally, Zeger et al. (29) also identified four rare variants in obese individuals, two of which reduced protein function. Collectively, these studies support SIM1’s role in the genetic etiology of obesity.
Mouse studies have also determined that Sim1 plays a role in energy homeostasis. Initial investigations determined that the gene is involved in the terminal differentiation of neurons in the diencephalon/hypothalamus (30–32). Germline knockout of Sim1 in mice by two separate groups resulted in perinatal lethality, due to an underdeveloped hypothalamus in homozygous mice (33,34). Sim1 heterozygous mice are obese, with hyperphagic behavior, increased adipose tissue, have accelerated linear growth (nose to tail length), hyperinsulinemia and hyperleptinemia (35). This phenotype is remarkably similar to melancortin 4 receptor (Mc4r) knockout mice in a dose-dependent manner (36,37), further implicating Sim1 in the leptin–melanocortin pathway. Whether the obesity phenotype is due to the germline perturbation of the gene during development or due to improper activation of the leptin–melanocortin pathway later on in adult time points was addressed with the generation of a conditional knockout of postnatal hypothalamic Sim1. Both heterozygotes and full inactivation of the Sim1 gene at the postnatal day P21 time point led to dose-dependent diet-induced obesity, demonstrating that this gene indeed plays a distinct role in energy homeostasis that did not depend on the proper differentiation of the developing hypothalamus (38). Overexpression of Sim1 in the paraventricular nucleus (PVN) of wild-type mice also led to reduced food intake, whereas knockdown by a short-hairpin RNA brought about increased food intake, further supporting the hypothesis that expression and function of postnatal Sim1 has a distinct function in the PVN to regulate energy consumption (39,40).
As inferred by human and mouse SIM1 deletions, altered gene dosage of SIM1 can cause obesity, suggesting that variation in SIM1 regulatory elements could predispose to this phenotype. In this study, we utilized a zebrafish transgenic assay (41) to screen and annotate enhancers in the SIM1 locus from embryonic to adult stages. We found two brain SIM1 candidate enhancers (SCE2 and SCE8) that showed similar enhancer activity in mice and overlapped Sim1 expression. Using a deletion series, we determined the region critical for neuronal-enhancer activity of SCE2 and sequencing this region in obese and lean cohorts identified obesity-associated SNPs. Functional analyses of these SNPs revealed differential-enhancer activity for one obesity-associated variant. These findings suggest that regulatory elements in this region may control SIM1 during developmental and post-developmental time points and nucleotide variants in them could alter the expression of SIM1 and contribute to obesity susceptibility.
RESULTS
Comparative genomic analysis of the SIM1 locus
To identify potential SIM1 enhancers, we carried out a comparative genomic analysis on the SIM1 locus. We searched for conserved non-coding regions in this locus, defined as one gene upstream [activating signal cointegrator 1 complex subunit 3 (ASCC3)] and downstream [melanin-concentrating hormone receptor 2 (MCHR2)] of SIM1, for a total genomic distance of ∼1 mb (Fig. 1). Analysis of the SIM1 locus revealed a human–mouse synteny block that ends upstream of MCHR2 and separates/eliminates any non-coding conservation between human and mouse ∼93 kb upstream of MCHR2 (Fig. 1). This suggests that enhancers found within this synteny block likely regulate Sim1 in mice (42). Furthermore, while MCHR2 is expressed in the hypothalamus in humans and is modestly associated with polygenic obesity (43), MCHR2 and its ligand, MCH, are not present in mice.
Figure 1.

SIM1 locus and SCEs tested for enhancer activity. UCSC Genome Browser snapshot of the SIM1 locus. SCEs tested for enhancer activity in zebrafish are indicated by dashes above the genes. These SCEs are within one gene upstream (ASCC3) and one gene downstream (MCHR2) of SIM1. Genes are depicted using the UCSC gene track and the black arrow indicates the end of the human-mouse synteny block (∼93 kb upstream of MCHR2).
Using the ECR Browser (44), 488 evolutionary conserved sequences (ECRs) between human and mouse were found that were at least 70% conserved for at least 100 bp within the defined SIM1 locus (Supplementary Material, Table S1). ECRs were analyzed manually for repetitive sequences and any RNA coding evidence using the UCSC Genome Browser (45), removing any that contained either. The remaining 360 non-coding ECRs were then ranked by species conservation to prioritize for enhancer assays. Seventeen ECRs, conserved between human and frog, were chosen for subsequent enhancer assays (Supplementary Material, Table S2) and were termed SCEs.
Zebrafish-enhancer screen
The human sequences of the seventeen SCEs were cloned into a vector containing the E1b-minimal promoter followed by the Green Fluorescent Protein (GFP) reporter gene (46). These constructs were microinjected into Casper zebrafish embryos at the one-cell stage using the Tol2 transposase system for germline integration of the transgene (41). Casper zebrafish were utilized due to their transparency, allowing GFP expression to be easily visualized at both developmental and post-developmental stages (47). Enhancer activity was annotated at three developmental time points: 24, 48 and 72 h post fertilization (hpf) (Supplementary Material, Table S3) and three post-developmental time points: 1, 2 and 3 months post fertilization (mpf) (Supplementary Material, Table S4).
From the seventeen sequences, several SCEs demonstrated enhancer activity in the vicinity of the hypothalamus at developmental and post-developmental time points as defined by ≥20% GFP expression over background: SCE2 and SCE8 during developmental time points (Supplementary Material, Table S3) and SCE2, SCE4, SCE11 and SCE13 during post-developmental time points (Supplementary Material, Table S4). In stable zebrafish lines of these six constructs, only SCE2 and SCE8 consistently showed the observed expression patterns across developmental and adult time points. SCE2 and SCE8 exhibited specific GFP expression in the diencephalon/hypothalamus region during both developmental and post-developmental time points (Fig. 2; Supplementary Material, Tables S3 and S4). The expression of sim1 at 24–48 hpf as characterized by whole-mount in situ hybridization (48,49) overlaps the GFP expression of SCE2 and SCE8.
Figure 2.

SCE2 and SCE8 zebrafish-enhancer expression across developmental and post-developmental time points. (A–D) SCE2 drives expression of GFP in the forebrain (f), hypothalamus (hy), hindbrain (hb), somites (sm) and spinal cord (sp) during development and localizes to the hypothalamic region following development. (E–H) SCE8 is a hypothalamus and epidermis (ep) enhancer during development and maintains GFP expression in the hypothalamus at later stages.
Mouse-enhancer assays
To test whether our zebrafish-enhancers have comparable expression patterns in mammals, we tested SCE2 and SCE8 using a mouse transgenic enhancer assay (50). SCE2 and SCE8 were cloned into the Hsp68-LacZ reporter plasmid (51) and were used to generate transgenic mouse embryos using standard techniques (52). SCE8 showed enhancer activity in the developing hypothalamus and overlapped Sim1 expression at embryonic day (E) 12.5 (Supplementary Material, Fig. S2), a time point during hypothalamic development at which Sim1 expression has been documented at high levels (30,53,54). SCE2 was previously found to be an enhancer in the developing mesencephalon and cranial nerve at E11.5 (55).
Since SCE2 showed the strongest consistent expression in the developing brain both in zebrafish and mice, we further investigated its mouse expression across developmental and adult time points. A stable SCE2 transgenic mouse line was generated and expression was assayed at different time points. In addition to repeating the previous finding at E11.5, we found that the activity of SCE2, as reflected by LacZ staining, is evident at E9.5, maintained during adulthood and is similar to that of Sim1 (Fig. 3). Enhancer activity is localized primarily throughout the developing forebrain, specifically in the zona limitans intrathalamica (ZLI) and diencephalon, and the midbrain, notably the tegmentum, and then transitions to the hippocampus from E13.5 to E15.5 (Supplementary Material, Fig. S1). Postnatally, enhancer activity is observed in the PVN of the hypothalamus and the basal amygdala and remains in the hippocampus at P56 (Fig. 3). Other than the hippocampus expression, the adult expression of SCE2 is similar to Sim1 expression as determined by in situ hybridization on mouse brain sections (Fig. 3) (56).
Figure 3.
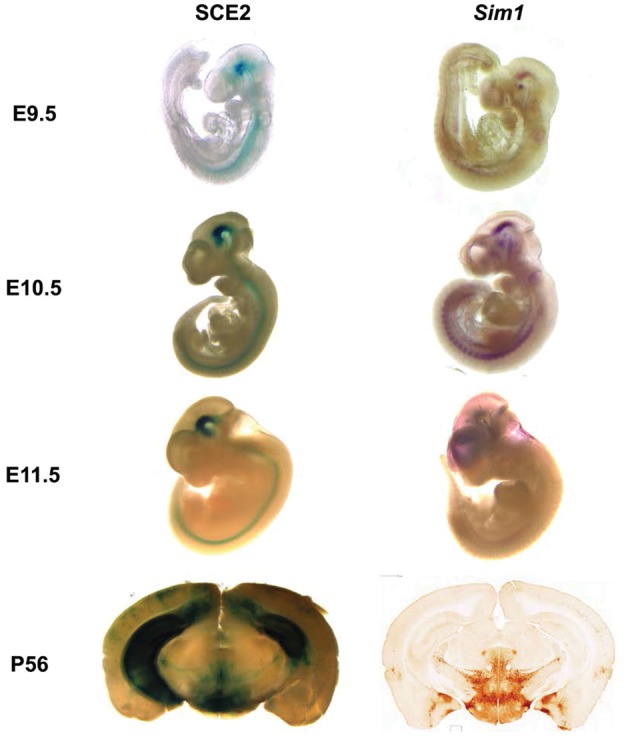
SCE2-enhancer activity and Sim1 expression in mice. SCE2 is active in the developing forebrain (diencephalon, ZLI), developing midbrain (mesencephalon, tegmentum) and the neural tube across various time points as reflected by LacZ staining. SCE2-enhancer activity overlaps Sim1 expression from E9.5 to E12.5 and in the adult hypothalamus. Adult hypothalamus in situ expression was obtained from GENSAT (56).
SCE2 deletion series identifies a functional hypothalamus-enhancer core
To further refine the domain driving brain expression in SCE2, we carried out a deletion series in zebrafish. SCE2 is 87% conserved between human and mouse across the length of the enhancer (Fig. 4A). Further genomic comparisons reveal ∼700 bp sequence, named here as the core element (CE), that is conserved between human and fish. In addition, examination of a mouse E11.5 forebrain E1A-binding protein p300 (EP300 or p300; a protein that co-localizes with enhancers) chromatin immunoprecipitation followed by sequencing (ChIP-seq) data set (57) identified a peak that overlaps the CE. Combined, the evolutionary conservation and p300 ChIP-seq peak suggest that this 700 bp CE within SCE2 is the major functional domain of this enhancer and that it may be sufficient to drive the enhancer expression observed in our original screen.
Figure 4.

SCE2-enhancer deletion series. (A) A UCSC Genome Browser snapshot showing that SCE2 is 87% conserved between human and mouse and has a 700 bp core sequence that is conserved between human and fish. The location of the mouse E11.5 forebrain p300 ChIP-seq peak (57) is also shown. (B) The 3′ region of SCE2 drives GFP expression in the spinal cord (sp). (C) The core element (CE) drives enhancer activity in the forebrain (fb), hypothalamus (hy), midbrain (mb), hindbrain (hb) and somites (sm). (D) The 5′ SCE2 sequence was negative for enhancer activity.
The CE and the flanking sequences (5′ and 3′ elements relative to SIM1) that comprise SCE2 were individually cloned into the E1b-GFP zebrafish-enhancer vector and microinjected into zebrafish as previously described. The CE was sufficient to drive the majority of SCE2 GFP expression in the forebrain, midbrain, hindbrain and somites at 24–72 hpf (Fig. 4C; Supplementary Material, Table S5). The 3′ element appeared to drive the spinal cord expression pattern, the only pattern that was not observed in the enhancer assay for SCE2-CE (Fig. 4B). The 5′ element was negative for GFP expression (Fig. 4D). These results suggest that the CE of SCE2 is the functional domain that is important for the brain activity of this enhancer.
Sequencing analysis of SCE2-CE in obese and lean cohorts
To determine whether nucleotide variants in SCE2 could be associated with predisposition to obesity, we sequenced a large obese cohort (n = 510) and compared our results to a matched lean cohort (n = 554) (58). We focused on the SCE2-CE region, since our zebrafish deletion series analysis demonstrated that this sequence is responsible for hypothalamus-enhancer activity. Two common SNPs, rs187302227 and rs192532320, which flank the portion of SCE2-CE that is conserved to fish, were present in both cohorts and did not show any significant MAF frequency differences (Table 1). However, four novel SNPs that were unique to the obese cohort were found as singletons, one of which, chr6:100658719 G>A (hg19), was found in a homozygous form versus one unique SNP (rs182500930) in the lean cohort (Table 1).
Table 1.
Sequencing summary of SCE2-CE
| SCE2-CE variant chromosomal position (hg19) | dbSNP # | Base change | Conservation | MAF (obese) | MAF (lean) |
|---|---|---|---|---|---|
| chr6:100658489 | rs187302277 | T/C | Mixed | 0.0114 | 0.0049 |
| chr6:100658548 | NA | C/T | Mammals | 0.0014 | |
| chr6:100658624 | NA | C/T | Stickleback | 0.0014 | |
| chr6:100658893 | rs182500930 | C/T | Stickleback | 0.0012 | |
| chr6:100658719 | NA | G/A | Stickleback | 0.0014 | |
| chr6:100658735 | NA | G/A | Stickleback | 0.0028 | |
| chr6:100659108 | rs192532320 | C/A | Opossum | 0.0055 | 0.0049 |
Sequencing of an obese cohort identified four rare SNPs that are not present in the lean controls cohort. One SNP, rs182500930, unique to the lean group is not present in the obese cohort and was identified in dbSNP. Two SNPs, rs18730227 and rs192532320, flanking the conserved SCE2 core element have also been previously reported and are present in both obese and lean cohorts without significant MAF differences.
To provide further support that the SNPs present in the obese individuals are unique, we analyzed the prevalence of SNPs in SCE2-CE in the 1000 Genomes Project (59). Other than the two flanking common SNPs (rs187302227 and rs192532320) previously found in the obese and lean cohorts, five other SNPs were identified in SCE2-CE (Supplementary Material, Table S7). Once weighted according to the race/ethnicity percentages of our obese cohort (85% White, 10% Black and 5% Latino race/ethnicity), two SNPs, rs150264974 and rs188023826, were present with weighted MAFs of 0.0008 and 0.0002, respectively. These SNPs were not shared by our case or control groups, suggesting that the SNPs identified in our obese cohort are unique to the obese cohort. In summary, these results show a potentially higher prevalence of rare variants in the obese versus the lean cohort and the 1000 Genomes Project.
Functional characterization of obese-specific SCE2-CE variants
The four obese-associated variants were functionally analyzed for differential-enhancer activity during development using the zebrafish-enhancer assay. Using site-directed mutagenesis, we cloned all four variants into SCE2-CE. The variants were microinjected into zebrafish embryos as previously described and annotated at developmental time points (24–72 hpf). We observed that, in general, the original pattern of SCE2-CE was maintained during developmental time points, regardless of the variant (data not shown). However, GFP-expressing fish showing a similar enhancer pattern differed between the variants compared with the reference sequence. The most notable difference was a significant reduction in brain-enhancer activity for SCE2-CE-100658719 G>A, the variant appearing in an individual in a homozygous form, across all three time points (Fig. 5; Supplementary Material, Table S6). Combined, these results suggest that this variant could alter the enhancer activity of SCE2.
Figure 5.

Developing hypothalamus-enhancer activity of obese-associated SCE2-CE variants in zebrafish. Variant 100658719 G>A exhibited statistically significant reduction of hypothalamus-enhancer activity (P < 0.05) at 24 hpf (A), 48 hpf (B) and 72 hpf (C) (one-way ANOVA, followed by Dunnett's multiple comparison test).
DISCUSSION
Using comparative genomics coupled with in vivo enhancer assays, our study identified two novel hypothalamus enhancers in the SIM1 locus. Both enhancers showed expression in zebrafish and mice at comparable developmental time points, further demonstrating the validity of using zebrafish as a rapid and cost-efficient filter to detect mammalian enhancers in this tissue. Furthermore, we were able to use zebrafish to identify the core region within SCE2 that drives enhancer activity in the brain. Sequencing SCE2-CE in an obese cohort identified four unique SNPs. Functional characterization of these variants in zebrafish showed that one variant, chr6:100658719 G>A, exhibited reduced enhancer activity, with statistically significant reduction in the hypothalamus during development.
In this study, we used comparative genomics to identify enhancers in the SIM1 locus. Although we identified enhancers that may regulate SIM1, there could well be additional enhancers in this locus that control the spatiotemporal expression of the gene. The use of techniques such as ChIP-seq on specific tissues (e.g. mouse hypothalamus) using a variety of enhancer marks, might help identify additional tissue-specific enhancers in this locus. Currently, no hypothalamus-specific ChIP-seq data sets have been published, especially in the context of energy homeostasis and the signaling cascade of which SIM1 is a member. Such future assays would help better characterize the regulatory landscape of this region.
The two enhancers we identified were active in the developing forebrain and midbrain during neurogenesis. SCE8 is active in a small region of the mammillary epithelium and the tegmentum (Supplementary Material, Fig. S2), both of which are known to express Sim1 at E12.5 (54). Spatiotemporal characterization of the SCE2 enhancer showed that its activity overlaps Sim1 expression in the earlier stages of neurogenesis (E9.5-E11.5) (Fig. 3). However, SCE2 did not show LacZ expression in the developing hypothalamus but was active in the adult hypothalamus (Fig. 3), suggesting that SCE2 may regulate Sim1 in the context of energy homeostasis during post-development. SCE2 was also found to be active in the hippocampus from E13.5 to adulthood (Fig. 3, Supplementary Material, Fig. S1) where Sim1 is not known to be expressed (56). This pattern of expression has been previously observed for the Pro-opiomelanocortin (Pomc)-GFP mouse model, a gene in the leptin–melanocortin pathway, but is primarily used to mark granule cells in the dentate gyrus/hippocampus (60). It is worth noting that this expression pattern could also be due to the site of integration of the transgene, being influenced by enhancers in that region (though it was observed in both founders) or background from the minimal promoter. Further studies, such as a mouse knockout of SCE2 or chromosome conformation capture, would be needed to determine whether SCE2 truly interacts and transcriptionally controls Sim1 and/or other genes in either the developmental or energy homeostasis context in the distinct neuronal subregions observed in our assays.
In comparison with SCE2, SCE8 is also active in the developing forebrain and midbrain and mammillary epithelium (data not shown), suggesting that there is redundancy in the potential regulation of Sim1 expression at E12.5 and perhaps during other time points. This could be attributed to phenomena termed ‘shadow enhancers’, where another enhancer could have a similar expression pattern so as to provide backup for the other enhancer (61).
Sequencing of SCE2-CE identified novel rare variants that were unique to the obese cohort and were not observed in either the lean cohort or in any of the populations analyzed from the 1000 Genomes Project (Supplementary Material, Table S7), despite not knowing any phenotypic data from the individuals who participated, some of whom could very well be obese. While the difference in prevalence of variants may not be significant between populations, the uniqueness of the SNPs identified in the obese cohort supports previous studies that report a higher prevalence of unique SIM1 variants in individuals with severe morbid obesity (23,27,28).
The observed changes in enhancer activity for SCE2-CE-100658719 G>A in zebrafish (Fig. 5) suggest that this variant could alter SIM1 expression. This variant was also the only variant appearing in an obese individual in a homozygous form. However, the zebrafish enhancer assay has an important limitation that needs to be taken into account. It is difficult to make quantitative conclusions due to the Tol2 transposase system since it allows for variable integration sites and copies of the transgene into the zebrafish genome (62). While we tried to correct for this by using an increased sample size (>100 eggs/per each injected variant), this caveat and poor survival rates of microinjected Casper embryos that can additionally skew numbers need to be kept in mind regarding our results.
Ultimately, associating functional enhancer variants with obesity could further elucidate the genetic contributions to this phenotype. While coding exons have been the focus of obesity genetics, it is important to acknowledge that regulatory elements could also contribute to the genetic predisposition of human disease and phenotypes. With technological developments such as ChIP-seq (63), massively parallel reporter assays (64), whole-genome sequencing and other robust applications of advanced sequencing technologies, we will be able to further elucidate regulatory genetic elements that are associated with human variation and disease.
MATERIALS AND METHODS
Comparative genomics
Using ECR Browser (44), we selected intronic and intergenic ECRs between MCHR2 and ASSC3 that were ≥100 bp long with at least 70% sequence identity between human and mouse. This analysis generated 488 unique ECRs (Supplementary Material, Table S1). We manually filtered out repetitive sequences, expressed sequence tags (ESTs) and any other coding sequences using the UCSC Genome Browser (45). To prioritize ECRs for functional assays, the remaining 360 ECRs were then ranked by species conservation. A total of seventeen human and frog ECRs (labeled as SCEs) were chosen for enhancer assays (Supplementary Material, Table S2).
PCR amplification and cloning
Candidate sequences were PCR amplified from human genomic DNA (Roche) using TopTaq (Qiagen). Primers were designed to have an additional 100–200 bp flanking the ECR sequence (Supplementary Material, Table S2); previous experiments have shown this to be a reliable method for obtaining positive-enhancer activity (50). Inserts were first cloned into the pENTR-dTOPO vector (Life Technologies) following the manufacturer's protocol and then transferred using Gateway technology (Life Technologies) into the E1b-GFP-Tol2 (46) for the zebrafish-enhancer assays and into the Hsp68-LacZ vector (51) for mouse-enhancer assays. Orientation and sequence of the inserts were verified by restriction enzyme digest and sequencing. Plasmid DNA was generated for microinjections using the EndoFree Plasmid Midi Prep kit (Qiagen).
Enhancer assays
For zebrafish-enhancer assays, each construct was injected into Casper (47) embryos at the one-cell stage, following standard procedures along with Tol2 mRNA (62,65,66) to facilitate genomic integration. A minimum of 100 embryos per construct were annotated for GFP expression at 24–72 hpf and at least 12 fish were annotated at 1, 2 and 3 mpf. An enhancer was considered positive if 20% of the GFP-expressing fish showed a consistent expression pattern after subtracting tissue expression pattern percentages of the negative control (E1b-GFP empty vector) at all respective time points (Supplementary Material, Tables S3 and S4). To generate stable lines, embryos that exhibited GFP expression were selected to mature to adulthood. Once mature, zebrafish were backcrossed to Casper zebrafish; this allowed for the germline transmission of the enhancer-driven GFP pattern. At least two founders were used for the generation of stable lines in zebrafish.
For the mouse-enhancer assays, transgenic mice were generated by Cyagen Biosciences using standard procedures (52). For the SCE2 stable line, two founders were generated and analyzed in all time points. At embryonic time points, embryos were harvested and stained for LacZ using standard procedures (50). For the adult time point, P56, LacZ-enhancer assays were performed on mouse brains using previously described procedures (67). All animal work was approved by the UCSF Institutional Animal Care and Use Committee.
In situ hybridization
The mouse Sim1 vector (30) was used as a template to generate a digoxygenin-labeled probe. The Sim1 vector was digested with XhoI and T7 polymerase (Roche) was used to generate the probe; an illustra MicroSpin G-50 column (GE Healthcare) was utilized to purify the probe before quantifying. Embryonic mice were harvested and fixed in 4% PFA and Sim1 in situ hybridization assays were performed on whole embryos according to standard protocols (68,69).
SCE2-enhancer deletion series
Primers were designed around the SCE2-CE region and the flanking regions of the CE (Supplementary Material, Table S5). The three regions were cloned into the E1b-GFP vector and microinjected into Casper zebrafish embryos as previously described. Zebrafish embryos were annotated for GFP expression at three developmental time points (24–72 hpf).
SCE2-CE sequencing analysis
Sequencing of SCE2-CE was performed on previously reported obese and lean cohorts (58). Briefly, severely obese individuals (n = 510) had a mean BMI of 47.9 ± 8.3 kg/m2, age 48.3 + 12.1 years, 73% female and 85% Caucasian. Controls (n = 554) had an average BMI of 22.9 ± 1.4 kg/m2, and were age (51.3 +4.5 years), sex (68% female) and ethnically (82% Caucasian) matched. All work was conducted under approved protocols from the UCSF Committee on Human Research, and informed written consent was obtained from all patients.
Estimated allele frequencies for the SNPs in the SCE2-CE were calculated from the 1000 Genomes, March 2012 interim release (Supplementary Material, Table S7) (59). To reflect the ancestry of our population, we calculated them as a weighted average of 85% European ancestry (87 CEU: Utah residents with ancestry from Northern and Western Europe from Centre, 93 FIN: Finnish from Finland, 89 GBR: British individuals from England and Scotland, 14 IBS: Iberians in Spain, 98 TSI: Toscani in Italia), 10% African ancestry (97 LWK: Luhya in Webuye Kenya, 88 YRI: Yoruba in Ibadan) and 5% Latino race/ethnicity (60 CLM: Columbian in Medellin, Columbia, 66 MXL: Mexican in Los Angeles, 55 PUR: Puerto Rican in Puerto Rico).
Differential-enhancer activity of obesity-segregating SNPs
Unique SNPs identified in the obese cohort were generated by site-directed mutagenesis of the E1b-GFP plasmid containing the SCE2-CE insert using QuikChangeII (Agilent Technologies). SCE2-CE variants were purified and microinjected into one-cell Casper embryos as described above. The enhancer activity of each of the variants was annotated at three developmental time points (24 hpf, 48 hpf and 72 hpf); at least 100 surviving embryos were annotated at each time point. One-way ANOVA followed by Dunnett's multiple comparison test was performed on the variants' percent GFP compared with the reference sequence for statistical analysis using Prism 5.04 (GraphPad).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK090382 to N.A. and C.V.). N.A. is also supported by grants by the National Human Genome Research Institute (R01HG005058 and R01HG006768), National Institute of Child and Human Development (R01HD059862), National Institute of General Medical Sciences (GM61390) and National Institute of Neurological Disorders and Stroke (1R01NS079231). M.J.K. was also supported in part by the National Institutes of Health Training Grant (T32 GM007175), the Amgen Research Excellence in Bioengineering and Therapeutic Sciences Fellowship and the University of California San Francisco Quantitative Biosciences Consortium Fellowship for Interdiscplinary Research . N.O. was supported in part by the Dennis Weatherstone Pre-doctoral Fellowship from Autism Speaks. This publication was made possible by Grant Number UL1 RR024131 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NCRR or the NIH. This work was also supported by NIH/NIDDK-UCSF Diabetes and Endocrinology Research Center Grant Number P30 DK63720.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank John L.R. Rubenstein for his gift of the mSim1 in situ construct and assisting in murine brain annotations, Len A. Pennacchio and Axel Visel (LBL) for providing E11.5 SCE2 mouse transgenic enhancer embryos and the Ahituv lab for helpful comments on the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.National Center for Health Statistics. 2012. NCHS Data on Obesity.
- 2.World Health Organization. 2013. Overweight and obesity. Overweight Obes.
- 3.World Health Organization. 2013. Overweight: Situation and trends.
- 4.Haslam D.W., James W.P.T. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 5.McTigue K.M., Hess R., Ziouras J. Obesity in older adults: a systematic review of the evidence for diagnosis and treatment. Obesity. 2006;14:1485–1497. doi: 10.1038/oby.2006.171. [DOI] [PubMed] [Google Scholar]
- 6.Guh D.P., Zhang W., Bansback N., Amarsi Z., Birmingham C.L., Anis A.H. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88. doi: 10.1186/1471-2458-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comuzzie A., Higgins P., Voruganti S., Cole S. Cutting the fat: the genetic dissection of body weight. Prog. Mol. Biol. Transl. Sci. 2010;94:194–212. doi: 10.1016/s1877-1173(10)94007-6. [DOI] [PubMed] [Google Scholar]
- 8.Maes H., Neale M., Eaves L. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 9.Stunkard A., Harris J. The body-mass index of twins who have been reared apart. New Engl. J. 1990;322:1483–1487. doi: 10.1056/NEJM199005243222102. [DOI] [PubMed] [Google Scholar]
- 10.Barsh G.S., Farooqi I.S., O'Rahilly S. Genetics of body-weight regulation. Nature. 2000;404:644–651. doi: 10.1038/35007519. [DOI] [PubMed] [Google Scholar]
- 11.Farooqi S., O'Rahilly S. Genetics of obesity in humans. Endocr. Rev. 2006;27:710–718. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 12.Nambu J.F., Franks R.G., Hu S., Crews S.T. The single-minded gene of Drosophila for the expression of genes important development of CNS midline cells is required for the. Cell. 1990;63:63–75. doi: 10.1016/0092-8674(90)90288-p. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L., Xiao H., Nambu J.R. CNS midline to mesoderm signaling in Drosophila. Mech. Dev. 1997;67:59–68. doi: 10.1016/s0925-4773(97)00107-x. [DOI] [PubMed] [Google Scholar]
- 14.Holder J., Butte N., Zinn A. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum. Mol. Genet. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 15.Turleau C., Demay G., Cabanis M.O., Lenoir G., de Grouchy J. 6Q1 Monosomy: a distinctive syndrome. Clin. Genet. 1988;34:38–42. doi: 10.1111/j.1399-0004.1988.tb02613.x. [DOI] [PubMed] [Google Scholar]
- 16.Villa A., Urioste M., Bofarull J.M., Martinez-Frias M.-L. De Novo interstitial deletion q16.2q21 on Chromsome 6. Am. J. Hum. Genet. Med. Genet. 1995;383:379–383. doi: 10.1002/ajmg.1320550326. [DOI] [PubMed] [Google Scholar]
- 17.Gilhuis H.J., Ravenswaaij C.M.A.V.A.N., Hamel B.E.N.J.C., Ls È. Interstitial 6q deletion with a Prader ± Willi-like phenotype: a new case and review of the literature cytogenetic studies. Eur. J. Paediatr. Neurol. 2000;4:39–43. doi: 10.1053/ejpn.1999.0259. [DOI] [PubMed] [Google Scholar]
- 18.Faivre L., Cormier-Daire V., Lapierre J., Colleaux L., Jacquemont S., Genevieve D., Saunier P., Munnich A., Turleau C., Romana S., et al. Deletion of the SIM1 gene (6q16.2) in a patient with Prader-Willi-like phenotype. J. Med. Genet. 2002;39:594–596. doi: 10.1136/jmg.39.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varela M.C., Simões-Sato A.Y., Kim C.A., Bertola D.R., De Castro C.I.E., Koiffmann C.P. A new case of interstitial 6q16.2 deletion in a patient with Prader-Willi-like phenotype and investigation of SIM1 gene deletion in 87 patients with syndromic obesity. Eur. J. Med. Genet. 2006;49:298–305. doi: 10.1016/j.ejmg.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Bonaglia M.C., Ciccone R., Gimelli G., Gimelli S., Marelli S., Verheij J., Giorda R., Grasso R., Borgatti R., Pagone F., et al. Detailed phenotype-genotype study in five patients with chromosome 6q16 deletion: narrowing the critical region for Prader-Willi-like phenotype. Eur. J. Hum. Genet. 2008;16:1443–1449. doi: 10.1038/ejhg.2008.119. [DOI] [PubMed] [Google Scholar]
- 21.Rosenfeld J.A, Amrom D., Andermann E., Andermann F., Veilleux M., Curry C., Fisher J., Deputy S., Aylsworth A.S., Powell C.M., et al. Genotype-phenotype correlation in interstitial 6q deletions: a report of 12 new cases. Neurogenetics. 2012;13:31–47. doi: 10.1007/s10048-011-0306-5. [DOI] [PubMed] [Google Scholar]
- 22.Izumi K., Housam R., Kapadia C., Stallings V.A., Medne L., Shaikh T.H., Kublaoui B.M., Zackai E.H., Grimberg A. Endocrine phenotype of 6q16.1-q21 deletion involving SIM1 and Prader-Willi syndrome-like features. Am. J. Med. Genet. Part A. 2013;9999:1–7. doi: 10.1002/ajmg.a.36149. [DOI] [PubMed] [Google Scholar]
- 23.Ahituv N., Kavaslar N., Schackwitz W., Ustaszewska A., Martin J., Hebert S., Doelle H., Ersoy B., Kryukov G., Schmidt S., et al. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung C.-C.C., Luan J., Sims M., Keogh J.M., Hall C., Wareham N.J., O'Rahilly S., Farooqi I.S. Studies of the SIM1 gene in relation to human obesity and obesity-related traits. Int. J. Obes. 2007;31:429–434. doi: 10.1038/sj.ijo.0803443. [DOI] [PubMed] [Google Scholar]
- 25.Ghoussaini M., Stutzmann F., Couturier C. Analysis of the SIM1 contribution to polygenic obesity in the French population. Obesity. 2010;18:1670–1675. doi: 10.1038/oby.2009.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traurig M., Mack J., Hanson R.L., Ghoussaini M., Meyre D., Knowler W.C., Kobes S., Froguel P., Bogardus C., Baier L.J. Common variation in SIM1 is reproducibly associated with BMI in Pima Indians. Diabetes. 2009;58:1682–1689. doi: 10.2337/db09-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnefond A., Raimondo A., Stutzmann F., Ghoussaini M., Ramachandrappa S., Bersten D.C., Durand E., Vatin V., Balkau B., Lantieri O., et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi–like features. J. Clin. Invest. 2013;123:1–5. doi: 10.1172/JCI68035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramachandrappa S., Raimondo A., Cali A.M.G., Keogh J.M., Henning E., Saeed S., Thompson A., Garg S., Bochukova E.G., Brage S., et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J. Clin. Invest. 2013;1:1–9. doi: 10.1172/JCI68016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zegers D., Beckers S., Hendrickx R., Van Camp J.K., de Craemer V., Verrijken A., Van Hoorenbeeck K., Verhulst S.L., Rooman R.P., Desager K.N., et al. Mutation screen of the SIM1 gene in pediatric patients with early-onset obesity. Int. J. Obes. 2013 doi: 10.1038/ijo.2013.188. http://www.nature.com/ijo/journal/vaop/ncurrent/full/ijo2013188a.html . [DOI] [PubMed] [Google Scholar]
- 30.Fan C., Kuwana E., Bulfone A., Fletcher C.F., Copeland N.G., Jenkins N.A., Crews S., Martinez S., Puelles Ø.L., Rubenstein J.L.R., et al. Expression patterns of two murine homologs of. Mol. Cell. Neurosci. 1996;16:1–16. doi: 10.1006/mcne.1996.0037. [DOI] [PubMed] [Google Scholar]
- 31.Moffett P., Dayo M., Reece M., McCormick M., Pelletier J. Characterization of msim, a murine homologue of the Drosophila sim transcription factor. Genomics. 1996;35:144–155. doi: 10.1006/geno.1996.0333. [DOI] [PubMed] [Google Scholar]
- 32.Yamaki A., Noda S., Kudoh J., Shindoh N., Maeda H., Minoshima S., Kawasaki K., Shimizu Y., Shimizu N. The mammalian single-minded (SIM) gene: mouse cDNA structure and diencephalic expression indicate a candidate gene for Down syndrome. Genomics. 1996;35:136–143. doi: 10.1006/geno.1996.0332. [DOI] [PubMed] [Google Scholar]
- 33.Michaud J.L., Rosenquist T., May N.R., Fan C.-M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998;12:3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holder J.L., Zhang L., Kublaoui B.M., DiLeone R.J., Oz O.K., Bair C.H., Lee Y.-H., Zinn A.R. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am. J. Physiol. Endocrinol. Metab. 2004;287:E105–E113. doi: 10.1152/ajpendo.00446.2003. [DOI] [PubMed] [Google Scholar]
- 35.Michaud J.L., Boucher F., Melnyk A., Gauthier F., Goshu E., Lévy E., Mitchell G. A., Himms-Hagen J., Fan C.M. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 2001;10:1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 36.Huszar D., Lynch C. A., Fairchild-Huntress V., Dunmore J.H., Fang Q., Berkemeier L.R., Gu W., Kesterson R.A., Boston B. A., Cone R.D., et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 37.Butler A.A., Marks D.L., Fan W., Kuhn C.M., Bartolome M., Cone R.D. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat. Neurosci. 2001;4:605–611. doi: 10.1038/88423. [DOI] [PubMed] [Google Scholar]
- 38.Tolson K.P., Gemelli T., Gautron L., Elmquist J.K., Zinn A.R., Kublaoui B.M. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J. Neurosci. 2010;30:3803–3812. doi: 10.1523/JNEUROSCI.5444-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang C., Gagnon D., Vachon P., Tremblay A., Levy E., Massie B., Michaud J.L. Adenoviral-mediated modulation of Sim1 expression in the paraventricular nucleus affects food intake. J. Neurosci. 2006;26:7116–7120. doi: 10.1523/JNEUROSCI.0672-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xi D., Gandhi N., Lai M., Kublaoui B.M. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS One. 2012;7:e36453. doi: 10.1371/journal.pone.0036453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher S., Grice E., Vinton R., Bessling S., Urasaki A., Kawakami K., AS M. Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat. Protoc. 2006;1:1297–1305. doi: 10.1038/nprot.2006.230. [DOI] [PubMed] [Google Scholar]
- 42.Ahituv N., Prabhakar S., Poulin F., Rubin E.M., Couronne O. Mapping cis-regulatory domains in the human genome using multi-species conservation of synteny. Hum. Mol. Genet. 2005;14:3057–3063. doi: 10.1093/hmg/ddi338. [DOI] [PubMed] [Google Scholar]
- 43.Ghoussaini M., Vatin V., Lecoeur C., Abkevich V., Younus A., Samson C., Wachter C., Heude B., Tauber M., Tounian P., et al. Genetic study of the melanin-concentrating hormone receptor 2 in childhood and adulthood severe obesity. J. Clin. Endocrinol. Metab. 2007;92:4403–4409. doi: 10.1210/jc.2006-2316. [DOI] [PubMed] [Google Scholar]
- 44.Ovcharenko I., Nobrega M.A., Loots G.G., Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent W., Sugnet C., Furey T., Roskin K., Pringle T., Zahler A., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q., Ritter D., Yang N., Dong Z., Li H., Chuang J., Guo S. A systematic approach to identify functional motifs within vertebrate developmental enhancers. Dev. Biol. 2009;337:484–495. doi: 10.1016/j.ydbio.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.White R.M., Sessa A., Burke C., Bowman T., LeBlanc J., Ceol C., Bourque C., Dovey M., Goessling W., Burns C.E., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–189. doi: 10.1016/j.stem.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eaton J.L., Glasgow E. The zebrafish bHLH PAS transcriptional regulator, single-minded 1 (sim1), is required for isotocin cell development. Dev. Dyn. 2006;235:2071–2082. doi: 10.1002/dvdy.20848. [DOI] [PubMed] [Google Scholar]
- 49.Eaton J.L., Glasgow E. Zebrafish orthopedia (otp) is required for isotocin cell development. Dev. Genes Evol. 2007;217:149–158. doi: 10.1007/s00427-006-0123-2. [DOI] [PubMed] [Google Scholar]
- 50.Pennacchio L. A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D., et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 51.Kothary R., Clapoff S., Brown A., Campbell R. A transgene containing lacZ inserted into the dystonia locus is expressed in neural tube. Nature. 1988;335:435–437. doi: 10.1038/335435a0. [DOI] [PubMed] [Google Scholar]
- 52.Nagy A., Gertsenstein M., Vintersten K., Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual 3rd edn. NY: Cold Spring Harbor Press, Cold Spring Harbor; 2003. [Google Scholar]
- 53.Marion J.-F., Yang C., Caqueret A., Boucher F., Michaud J.L. Sim1 and Sim2 are required for the correct targeting of mammillary body axons. Development. 2005;132:5527–5537. doi: 10.1242/dev.02142. [DOI] [PubMed] [Google Scholar]
- 54.Shimogori T., Lee D.A., Miranda-Angulo A., Yang Y., Wang H., Jiang L., Yoshida A.C., Kataoka A., Mashiko H., Avetisyan M., et al. A genomic atlas of mouse hypothalamic development. Nat. Neurosci. 2010;13:767–775. doi: 10.1038/nn.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Visel A., Minovitsky S., Dubchak I., Pennacchio L.A. VISTA Enhancer Browser: a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.New York, NY: GENSAT Gene Expr. Nerv. Syst. Atlas Proj. NINDS Contract. N01NS02331 HHSN271200723701C to Rockefeller Univ. [Google Scholar]
- 57.Visel A., Blow M., Li Z., Zhang T., Akiyama J. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Calton M.A., Ersoy B.A., Zhang S., Kane J.P., Malloy M.J., Pullinger C.R., Bromberg Y., Pennacchio L.A., Dent R., McPherson R., et al. Association of functionally significant melanocortin-4 but not melanocortin-3 receptor mutations with severe adult obesity in a large North American case-control study. Hum. Mol. Genet. 2009;18:1140–1147. doi: 10.1093/hmg/ddn431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G. A. An integrated map of genetic variation from 1, 092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Overstreet L.S., Hentges S.T., Bumaschny V.F., de Souza F.S.J., Smart J.L., Santangelo A.M., Low M.J., Westbrook G.L., Rubinstein M. A transgenic marker for newly born granule cells in dentate gyrus. J. Neurosci. 2004;24:3251–3259. doi: 10.1523/JNEUROSCI.5173-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong J.-W., Hendrix D.A., Levine M.S. Shadow enhancers as a source of evolutionary novelty. Science (80-.). 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawakami K. Transposon tools and methods in zebrafish. Dev. Dyn. 2005;234:244–254. doi: 10.1002/dvdy.20516. [DOI] [PubMed] [Google Scholar]
- 63.Furey T.S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haberle V., Lenhard B. Dissecting genomic regulatory elements in vivo. Nat. Biotechnol. 2012;30:504–606. doi: 10.1038/nbt.2266. [DOI] [PubMed] [Google Scholar]
- 65.Westerfield M. The Zebrafish Book, A Guide for the Laboratory use of Zebrafish (Danio Rerio) 5th ed. Eugene: University of Oregon, Press; 2007. [Google Scholar]
- 66.Nüsslein-Volhard C., Dahm R. Zebrafish: A Practical Approach. Oxford University Press, New York; 2002. [Google Scholar]
- 67.Schmidt A., Tief K., Foletti A., Hunziker A., Penna D., Hummler E., Beermann F. lacZ transgenic mice to monitor gene expression in embryo and adult. Brain Res. Protoc. 1998;3:54–60. doi: 10.1016/s1385-299x(98)00021-x. [DOI] [PubMed] [Google Scholar]
- 68.Flandin P., Zhao Y., Vogt D., Jeong J., Long J. Lhx6 and Lhx8 coordinately induce neuronal expression of Shh that controls the generation of interneuron progenitors. Neuron. 2011;70:939–950. doi: 10.1016/j.neuron.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Conlon R., Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.