

Abstract
Collagen type IV alpha 1 and 2 (COL4A1 and COL4A2) are present in nearly all basement membranes. COL4A1 and COL4A2 mutations are pleiotropic, affecting multiple organ systems to differing degrees, and both genetic-context and environmental factors influence this variable expressivity. Here, we report important phenotypic and molecular differences in an allelic series of Col4a1 and Col4a2 mutant mice that are on a uniform genetic background. We evaluated three organs commonly affected by COL4A1 and COL4A2 mutations and discovered allelic heterogeneity in the penetrance and severity of ocular dysgenesis, myopathy and brain malformations. Similarly, we show allelic heterogeneity in COL4A1 and COL4A2 biosynthesis. While most mutations that we examined caused increased intracellular and decreased extracellular COL4A1 and COL4A2, we identified three mutations with distinct biosynthetic signatures. Reduced temperature or presence of 4-phenylbutyrate ameliorated biosynthetic defects in primary cell lines derived from mutant mice. Together, our data demonstrate the effects and clinical implications of allelic heterogeneity in Col4a1- and Col4a2-related diseases. Understanding allelic differences will be valuable for increasing prognostic accuracy and for the development of therapeutic interventions that consider the nature of the molecular cause in patients with COL4A1 and COL4A2 mutations.
INTRODUCTION
Collagen type IV alpha 1, COL4A1 (MIM 120130), and its binding partner COL4A2 (MIM 120090) are major constituents of nearly all basement membranes. COL4A1 and COL4A2 each contain a long, triple-helix-forming, collagenous domain flanked by a short 7S domain at the amino terminus and a globular, non-collagenous (NC1) domain at the carboxy terminus. Two COL4A1 peptides and one COL4A2 peptide associate via their NC1 domains (1,2) and assemble into heterotrimers within the endoplasmic reticulum (ER) before being secreted into the extracellular space. Heterotrimers polymerize to form a collagen IV network that is essential to the development, structure and function of normal tissues.
Consistent with the widespread distribution of these proteins, COL4A1 and COL4A2 mutations are pleiotropic and cause a broad spectrum of disorders affecting multiple organs, including the brain, eyes, kidneys and muscles, both in humans and in mice (3–6). Importantly, the severity of pathology is influenced by the genetic-context and environmental factors (7–9). Notably, we have shown previously that the presence and severity of Col4a1-induced ocular and muscular defects in mice is genetic-context dependent (8,9). In addition, accumulating evidence suggests that allelic heterogeneity also contributes to the variable expressivity of COL4A1 and COL4A2 mutations. Six families presenting with a syndrome referred to as HANAC (hereditary angiopathy, nephropathy aneurysms and cramps) have COL4A1 mutations clustered within 31 amino acids (10). This raises the possibility that COL4A1 mutations that affect specific functional domains may lead to distinct clinical outcomes (11). Furthermore, the specific nature of a mutation within a given protein domain may influence its biosynthetic consequences. Of note, there is little or no tolerance for amino acids other than glycine at every third position of the collagen triple helix; however, there is evidence that all glycine mutations are not functionally equivalent (12,13).
In this study, we investigate the contribution of allelic differences to phenotypic variability and to COL4A1 and COL4A2 biosynthesis using a series of Col4a1 and Col4a2 mutant mouse lines on a uniform C57BL/6J genetic background. We show that different Col4a1 and Col4a2 mutations have distinct molecular consequences that lead to ocular, cerebral and myopathic phenotypes of variable severity and penetrance and may reflect mechanistic heterogeneity. Understanding the role of allelic heterogeneity could provide valuable insight into COL4A1- and COL4A2-associated disorders. Moreover, allelic differences may reflect important functional subdomains in COL4A1 and COL4A2 and reveal distinct pathogenic mechanisms. Both of these could provide prognostic information to allow for better patient counseling and guide the development of targeted therapeutic interventions.
RESULTS
Col4a1 and Col4a2 mutations cause highly penetrant anterior and posterior ocular dysgenesis
To investigate if allelic heterogeneity contributes to phenotypic variability, we analyzed an allelic series of Col4a1 and Col4a2 mutant mice comprising a splice site mutation and seven glycine missense mutations within the triple-helix-forming domains (six in COL4A1, one in COL4A2), and one missense mutation in the globular NC1 domain of COL4A1 (Fig. 1A). We crossed each mutation onto a uniform, C57BL/6J genetic background and evaluated the effects of allelic differences on three organs that are commonly affected in patients with COL4A1 and COL4A2 mutations.
Figure 1.

Col4a1 and Col4a2 mutations cause highly penetrant ocular anterior segment dysgenesis. (A) Schematic representation of COL4A1 and COL4A2 domain structure and the positions of the mutations analyzed in this study; green boxes: putative integrin-binding sites. (B–G) Representative images from slit lamp examination of control (Col4a1+/+) and mutant (Col4a1+/mut) eyes showing ocular anterior segment dysgenesis in mutant mice, including open pupil (asterisk), enlarged and torturous iris vasculature, cataracts (arrowhead), iridocorneal adhesion (cross) and enlarged anterior chamber (arrows point to the anterior chamber). (H) The frequency of distinct ocular anterior segment dysgenesis phenotypes caused by different Col4a1 and Col4a2 mutations is shown in the table. The overall severity for each eye was ranked as mild (score = 1), moderate (score = 2) and severe (score = 3), and the average score of each strain is indicated. Note that in Col4a1+/Δex41 mice a severe open pupil phenotype prevented assessment of iris vasculature abnormalities in a subset of eyes. Between 14 and 32 eyes were analyzed per allele.
COL4A1 and COL4A2 mutations cause ocular defects including anterior segment dysgenesis (ASD) and optic nerve hypoplasia (ONH) in patients and mice. We have shown previously that Col4a1+/Δex41 mice have severe ASD and ONH when maintained on a C57BL/6J background (8). To determine if allelic differences influence ocular manifestations resulting from Col4a1 and Col4a2 mutations, we evaluated ASD and ONH phenotypes in our allelic series using slit lamp examination and histological analysis, respectively. All Col4a1 and Col4a2 mutations caused ASD involving the cornea (cloudiness, scarring, vascularization and iridocorneal adhesions), iris (large tortuous vessels, pigment dispersion), pupil (open, irregularly shaped, eccentric), lens (cataract, persistent pupillary membrane) and anterior chamber (enlargement) that were not observed in Col4a1+/+ mice (Fig. 1B–G). Abnormal iris vasculature was the most highly penetrant ocular phenotype observed amongst all Col4a1 and Col4a2 mutants. The expressivity of every mutation was variable and although the majority of mutant eyes displayed at least one abnormal feature, 5 out of 24 Col4a1+/S1582P eyes, 1 out of 22 Col4a1+/G658D eyes and 1 out of 22 Col4a1+/G912V eyes were without any obvious defects on exam. Since mice from each mutant strain are genetically identical and non-penetrance was usually unilateral, the phenotypic variability within each mutant strain likely results from stochastic molecular events. Importantly, both the penetrance and qualitative assessment of ASD severity were variable between mutant strains, suggesting that allelic heterogeneity influences the ocular phenotype resulting from Col4a1 and Col4a2 mutations (Fig. 1H). Of note, Col4a1+/Δex41 and Col4a1+/G1344D eyes generally had the most severe iris vasculature abnormalities and a subset of eyes from Col4a1+/G1344D mice had severely vascularized and scarred corneas. Across the board, Col4a1+/S1582P mice exhibited less penetrant and less severe ASD than other mice in the allelic series. Similarly, ONH was also observed in most, but not all, strains (Fig. 2). Optic nerves from Col4a1+/Δex41 mice were the most severely affected, whereas Col4a1+/S1582P and Col4a1+/G1180D mice did not have significantly decreased optic nerve cross-sectional areas compared with Col4a1+/+ controls. Collectively, these data show that Col4a1 and Col4a2 mutations cause highly penetrant anterior and posterior ocular dysgenesis. Our results suggest that allelic heterogeneity might influence penetrance and severity; however, the high variability of these phenotypes tempers the confidence of this conclusion.
Figure 2.

Col4a1 and Col4a2 mutations cause variable optic nerve hypoplasia. (A) Representative images of optic nerve cross sections from Col4a1+/+ and Col4a1+/mut mice and (B) quantitative analysis of optic nerve cross-sectional area show variability in the size of mutant optic nerves, ranging from severely hypoplastic (Col4a1+/Δex41) to indistinguishable from control (Col4a1+/G1180D and Col4a1+/S1582P). Values are presented as mean ± SEM. Comparisons between Col4a1+/+ (WT) and each mutant strain were performed using Student's t-test (*P < 0.05). Comparison amongst different strains was performed using one-way ANOVA followed by Tukey's post hoc test. With the exception of Col4a1+/G1344D, Col4a1+/Δex41 was significantly different from all other Col4a1 mutations (†P < 0.05). Between 5 and 16 optic nerves were examined per allele.
Col4a1 and Col4a2 mutations cause variable brain malformations
Col4a1+/Δex41 mice have brain malformations including congenital cortical lamination defects (9). In addition, COL4A1 mutations were reported in three patients with cortical malformations, two of which were diagnosed with muscle-eye-brain disease/Walker-Warburg syndrome and one who presented with focal cortical dysplasia (6,9). To test if mice with different Col4a1 and Col4a2 mutations have distinct brain abnormalities, coronal brain sections collected at regular intervals were stained with cresyl violet and examined by light microscopy. All mutant brains examined exhibited cortical lamination defects (Fig. 3A–L). These included molecular layer heterotopia (MLH; clusters of neurons mislocalized to layer I of the cortex), midline fusion and disorganized lamination ranging from focal heterotopia (not restricted to layer I) to widespread heterotopia resembling extensive foliation. The number and size of cortical lamination defects were variable within and between mutant strains with the highest number of lesions observed in Col4a1+/Δex41, Col4a1+/G394V, Col4a1+/G912V and Col4a1+/G1038S brains (Fig. 3M). Col4a1+/S1582P brains had the lowest number of lesions and were not significantly different from Col4a1+/+ mice. Although, two out of five Col4a1+/+ brains had a single small MLH (not exceeding 140 μm along the rostrocaudal axis) (14), the MLH observed in mutant brains were always larger and, with the exception of Col4a1+/S1582P brains, were present in significantly higher numbers. The largest lesions, which spanned at least 1260 µm along the rostrocaudal axis, were more frequently observed in Col4a1+/G394V (five out of five), Col4a1+/G658D (four out five), Col4a1+/G912V (three out four), and Col4a1+/G1038S (three out of four) and never detected in Col4a1+/S1582P brains (data not shown). Most lesions, especially medium and large ones, occurred rostral to the hippocampus (plate 39, Paxinos and Franklin mouse brain atlas). Other brain malformations including enlarged ventricles, septal thinning and white matter defects, which were often associated with ventricular or cortical lamination defects, were also observed and the frequencies varied between strains (Fig. 3N) further supporting the contention of allelic heterogeneity.
Figure 3.

Col4a1 and Col4a2 mutations cause variable brain malformations. (A–L) Representative images of coronal brain sections from Col4a1+/+ and Col4a1+/mut mice stained with cresyl violet show diverse cortical defects in mutant mice. We observed molecular layer heterotopia (MLH) (arrows in C, D, H, K and L), midline fusion defects († in B and J), and focal (asterisks in D, F, G and L) or widespread heterotopias resembling extensive foliation (boxed area in B and higher magnification of the boxed area in J). (I–L) show higher magnification images of boxed area in A, B, C, and H, respectively. Other malformations include enlarged ventricles (F and G), septal thinning (arrowhead in F) and white matter defects (asterisk in D, F, and G) usually associated with disorganized lamination or with enlarged ventricles that manifest as thinning of white matter tracts in the external capsule (labeled as ‘ec’ in F and G). (M) Quantifying cortical neuronal lamination defects show that the number of lesions is influenced by allelic heterogeneity, with the greatest number of lesions observed in Col4a1+/G394V, Col4a1+/Δex41 and Col4a1+/G1038S brains, while Col4a1+/S1582P brains were not significantly different from Col4a1+/+ (WT). Comparisons between Col4a1+/+ (WT) and each mutant strain were performed using Student's t-test and, with the exception of the Col4a1+/S1582P brains, all Col4a1+/mut brains had a significantly higher number of lesions compared to Col4a1+/+ brain (P < 0.01, data not shown). Comparison amongst different strains was performed using one-way ANOVA followed by Tukey's post hoc test, Col4a1+/G394V, Col4a2+/G646D, Col4a1+/G912V, and Col4a1+/G1038S, Col4a1+/Δex41 brains had significantly more lesions than Col4a1+/+ brains (**P < 0.01), and Col4a1+/G394V, Col4a1+/G1038S and Col4a1+/Δex41 brains had significantly more lesions than Col4a1+/S1582P brains (†P < 0.05). (N) Penetrance of brain malformations other than lamination defects (EV: enlarged ventricles, ST: septal thinning, and WMD: white matter defect). Col4a1+/Δex41 and Col4a1+/S1582P brains showed the highest and lowest frequency of these malformations, respectively. Between 4 and 7 brains were analyzed per allele.
Col4a1 and Col4a2 allelic heterogeneity influences myopathy
Some patients with COL4A1 and COL4A2 mutations have myopathy ranging from elevated creatine kinase levels and muscle cramps to congenital muscular dystrophy (4,6,9,10,15–18), while other patients are within normal ranges or unreported. To determine the extent to which myopathy may be modulated by allelic difference, we evaluated the number of non-peripheral nuclei (a commonly used measure of myopathy indicative of degenerating and regenerating muscle fibers) in quadriceps sections from Col4a1 and Col4a2 mutant mice. By this measure, all mutations caused myopathy, with the exception of Col4a1+/S1582P (Fig. 4). Col4a1+/G394V mice exhibited the most severe myopathy, followed by Col4a1+/Δex41 mice. Comparison using one-way ANOVA revealed that the percentage of non-peripheral nuclei was significantly higher in Col4a1+/G394V muscles compared with all other mutant muscles.
Figure 4.
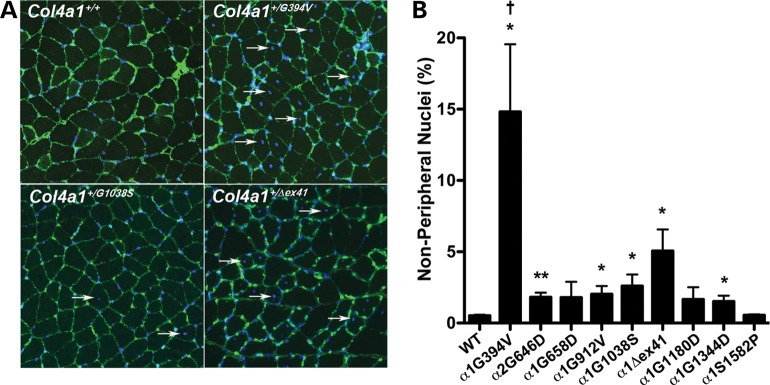
Col4a1 and Col4a2 allelic heterogeneity influences myopathy. (A) Representative Col4a1+/+ and Col4a1+/mut quadriceps muscle sections immunolabeled for COLIV and counterstained with DAPI showing variability in the number of non-peripheral nuclei (arrows) in mutant muscles. (B) Quantitative analysis of non-peripheral nuclei from all strains is shown in the bar graph. We counted a minimum of 4250 fibers in five muscles from five mice per allele and the values are presented as mean ± SEM. While Col4a1+/S1582P muscles are indistinguishable from Col4a1+/+ muscles, a considerable increase in the number of non-peripheral nuclei was observed in Col4a1+/G394V and Col4a1+/Δex41 muscles. All other mutations showed a modest increase in non-peripheral nuclei, not always reaching statistical significance compared to control muscles. The number of non-peripheral nuclei was significantly higher in Col4a1+/G394V muscles compared with all other Col4a1 mutant muscles. Comparisons between Col4a1+/+ (WT) and each mutant strain were performed using Student's t-test (*P < 0.05; **P < 0.01). Comparison amongst different strains was performed using one-way ANOVA followed by Tukey's post hoc test (†P < 0.01).
Collectively, the ocular, cerebral and muscular analyses of the Col4a1 and Col4a2 allelic series demonstrate that the different mutations evaluated in this study are not equivalent and that allelic heterogeneity in an otherwise uniform genetic context contributes to the penetrance and severity of various pathologies.
Biosynthetic effects of Col4a1 and Col4a2 mutations
We hypothesized that the observed allelic heterogeneity may result from the effects of molecular differences between mutations on COL4A1 and COL4A2 biosynthesis. The pathogenicity of Col4a1 and Col4a2 mutations is commonly attributed to intracellular accumulation and impaired secretion of heterotrimers that incorporate mutant COL4A1 or COL4A2 proteins. Thus, we sought to test if the nine alleles from this series of Col4a1 and Col4a2 mutations lead to intracellular retention of COL4A1 and COL4A2. To this end, mouse embryonic fibroblasts (MEFs) isolated from each mutant strain were co-immunolabeled for COL4A1 or COL4A2 and HSP47, a collagen-binding protein in the ER. Most mutant MEFs showed greater co-localization of COL4A1 and HSP47 compared with Col4a1+/+ MEFs, suggesting increased COL4A1 in the ER of mutant cells (Fig. 5A). Similar results were observed with COL4A2 immunolabeling and with co-labeling of COL4A1 or COL4A2 with protein disulfide isomerase (PDI), another ER resident protein (data not shown). The degree of COL4A1 and HSP47 co-localization was mutation dependent. There was little, if any, increased co-localization of COL4A1 and HSP47 observed for the Col4a1+/G394V and Col4a1+/G658D MEFs, whereas there was striking co-localization in Col4a1+/Δex41 MEFs (Fig. 5A and data not shown). Other alleles exhibited intermediate levels for COL4A1 and COL4A2 (data not shown). The increased co-localization of COL4A1 and COL4A2 with HSP47 and PDI in Col4a1 and Col4a2 mutant MEFs suggests that mutations in Col4a1 and Col4a2 cause protein misfolding and subsequent retention within the ER. Despite this finding, we did not detect evidence of ER stress in mutant MEFs by western blot or qRT–PCR analyses (Supplementary Material, Fig. S1).
Figure 5.

Biosynthetic effects of Col4a1 and Col4a2 mutations. (A) Representative photomicrographs of Col4a1+/+ and Col4a1+/mut mouse embryonic fibrobalsts (MEFs) co-immunolabeled for COL4A1 and HSP47, a collagen-specific chaperone residing in the ER. Col4a1 mutations lead to variable increase in co-localization of COL4A1 and HSP47, suggesting that some mutations lead to more severe intracellular accumulation than others. (B–G) Western blot and densitometric analyses of intracellular and extracellular COL4A1 and COL4A2 levels in MEFs. (B) Soluble intracellular COL4A1 levels (or COL4A2 for Col4a2+/G646D) were normalized to an actin loading control and compared with samples from Col4a1+/+ littermates loaded on the same gel under reducing conditions. (C) Extracellular COL4A1 levels were determined by analyses of conditioned media from the same cell cultures used in (B). Loading volumes were normalized to the total protein concentration of the cellular fraction and the conditioned media from Col4a1+/mut cells were compared with that of cells from Col4a1+/+ littermates that were loaded on the same gel. In a separate experiment, laminin labeling was used to demonstrate that normalization to total protein concentration returned consistent results (Supplementary Material, Fig. S3). For (B) and (C), we used three to five independent MEF lines isolated from individual embryos for each mutant line. Values were compared using Student's t-test (*P < 0.05; **P < 0.01) and are presented as mean ± SEM. (D–G) To compare strains directly, reduced total intracellular or conditioned media from all strains were loaded on a single gel and probed for COL4A1 or COL4A2. We used five independent MEF lines derived from different embryos for each mutation and the values amongst strains run on the same gel were compared using a one-way ANOVA followed by a Tukey's post hoc test (†P < 0.01), arrow indicates a COL4A1 band of higher molecular weight. (H) Representative photomicrographs of western blot analyses of detergent-soluble and-insoluble intracellular COL4A1 in Col4a1+/+ (wt) and mutant (mut) MEFs showing variability in the proportions of intracellular COL4A1 monomers (M) and COL4A1-containing heterotrimers (H) in MEFs with different Col4a1 mutations. We used three to five independent MEF lines derived from different embryos per mutation.
To further evaluate the biosynthetic consequences of Col4a1 and Col4a2 mutations, we compared the intracellular and extracellular levels of COL4A1 and COL4A2 in MEFs using western blot analysis. First, we evaluated the effects of individual mutations by comparing COL4A1 or COL4A2 levels in MEFs isolated from Col4a1+/+ and Col4a1+/mut littermates within each strain. When we probed western blots for COL4A1 levels (or COL4A2 levels in the case of Col4a2+/G646D), we found that, compared with samples from Col4a1+/+ littermates that were run on the same gel, all mutant lines exhibited significantly increased levels of intracellular COL4A1 (or COL4A2 in the case of Col4a2+/G646D) with the exceptions of Col4a1+/G394V and Col4a1+/G658D (Fig. 5B). Conversely, all mutant MEF lines exhibited a statistically significant reduction in extracellular COL4A1 (or COL4A2) compared with Col4a1+/+ cells, with the exception of Col4a1+/G394V cells (Fig. 5C). Thus, from these experiments, there were two notable outliers: Col4a1+/G394V MEFs, that did not have significantly increased intracellular nor decreased extracellular levels of COL4A1, and Col4a1+/G658D MEFs, that did not have significantly increased intracellular but had dramatically decreased extracellular levels of COL4A1. To address the apparent discordance of the Col4a1G658D mutation, we performed qRT–PCR and detected reduced levels of Col4a1 and Col4a2 transcripts in Col4a1+/G658D compared with Col4a1+/+ MEFs (Supplementary Material, Fig. S2). Given that Col4a1 and Col4a2 are transcriptionally co-regulated (19), these data suggest that the reduced levels of COL4A1 protein observed in Col4a1+/G658D cells may result from transcriptional repression rather than degradation of the mutant Col4a1 transcript or protein. Furthermore, we detected decreased transcript levels for some (BiP, Atf4 and Atf6), but not all (CHOP), ER stress-related genes tested in Col4a1+/G658D MEFs (Supplementary Material, Fig. S1B). Together, these data suggest that there may be broad, but not universal, transcriptional repression in Col4a1+/G658D cells.
Next, we ran samples from all mutant lines on the same gels so that we could directly compare the relative effects of each mutation between strains. A striking pattern emerged of a position-dependent effect on the intracellular COL4A1 levels whereby mutations nearer the C-termini of the triple-helix-forming domains (Col4a1G1038S, Col4a1Δex41, Col4a1G1180D and Col4a1G1344D) tended to have greater intracellular levels of COL4A1 than did mutations nearer the N-termini of this domain (Col4a1G394V, Col4a2G646D, Col4a1G658D and Col4a1G912V) (Fig. 5D). For the mutations located near the N-termini with the lowest intracellular levels, we also detected a higher molecular-weight band (Fig. 5D, arrow) suggesting that these mutant proteins are also differentially posttranslationally modified compared with their counterparts nearer the C-terminus. When we tested the conditioned media from these samples, we did not observe such a trend. Instead, most mutations had greatly decreased extracellular levels of COL4A1 with the exception of Col4a1G394V, which was determined to be significantly different from all other mutations. When we repeated these experiments for intracellular and extracellular COL4A2 levels we observed similar results (Fig. 5E and G). The same pattern held true for all of the mutations within the triple-helix-forming domain, and the Col4a1G394V mutation was again significantly different from all others. However, this experiment revealed yet another notable exception. The NC1 domain mutation (Col4a1S1582P) had a disproportionately low level of intracellular COL4A2 compared with intracellular COL4A1.
The properties of aggregation and solubility of misfolded proteins are important for their pathogenic potential in other protein misfolding diseases (20). Therefore, we further characterized the solubility and nature (heterotrimers versus monomers) of the COL4A1 and COL4A2 proteins present in the intracellular fraction of mutant cells. To this end, we performed western blot analyses under non-reducing conditions to evaluate the relative proportions of monomers and heterotrimers in detergent-soluble and -insoluble fractions. In the soluble fraction, most mutations lead to a greater proportion of high-molecular-weight heterotrimers (Fig. 5H; Col4a1+/G658D is used as a representative example) with the exceptions of Col4a1+/G1344D and Col4a1+/S1582P, which had a greater proportion of monomers. In the insoluble fraction, again most mutations had a greater accumulation of heterotrimers (see Col4a1G658D in Fig. 5H) with the exception of Col4a1+/Δex41, which showed prominent presence of monomers. In general, mutations located toward the C-terminus of the triple-helix-forming domain tended to accumulate higher levels of monomers than those closer to the N-terminus. In addition, the relative levels of COL4A1 in the soluble and insoluble intracellular fractions varied according to the mutation (Fig. 6 and data not shown). Together, these data demonstrate by multiple measures that all mutations are not functionally equivalent and can have distinct effects on collagen expression and assembly.
Figure 6.

Effects of reduced temperature and 4PBA on COL4A1 biosynthesis in Col4a1 mutant MEFs. Mutant MEFs were cultured under normal growth conditions at 37°C, at reduced temperature (26°C) or in the presence of 4PBA (10 mm) for 12 h, then serum deprived in the presence of 50 µg/ml ascorbic acid under the same conditions to promote assembly and secretion of COL4A1-containing heterotrimers. Protein lysate (soluble [SF] and insoluble [IF] intracellular fractions) and conditioned medium (E: extracellular) were subjected to western blot analysis. (A) Col4a1+/+ (WT) showed no difference in intracellular levels of COL4A1 with decreased temperature or 4PBA treatment, but demonstrated a decreased in extracellular levels of COL4A1 under decreased temperature conditions not seen with 4PBA treatment. While growing MEFs at reduced temperature decreased levels of intracellular COL4A1 in all selected Col4a1 mutant strains ((C) Col4a1 +/G658D, (E) Col4a1 +/Δex41, (G) Col4a1 +/G1344D and (I) Col4a1 +/S1582P), it only led to a small, non-significant increase in the levels of extracellular COL4A1 in Col4a1 +/G1344D and Col4a1 +/S1582P MEFs. In contrast, 4PBA decreased the levels of intracellular COL4A1 with a more pronounced reduction in the insoluble fraction and promoted secretion of COL4A1-containing heterotrimers. Values are presented as mean ± SEM. For statistical analysis, Student's t-tests were performed. *P < 0.05; **P < 0.01. N = 3–9 MEF lines per mutation. (B, D, F, H, J) Representative western blot images for the selected mutations and wild type. Extra: extracellular, Intra: intracellular, I: insoluble fraction, S: soluble fraction.
Protein folding-promoting conditions ameliorate COL4A1 accumulation in Col4a1 mutant MEFs
Mutations in Col4a1 and Col4a2 orthologs in C. elegans have also been shown to cause intracellular accumulation of COL4A1 and COL4A2 and lead to temperature sensitive lethality attributed to contraction-induced detachment of the body wall muscles. Rearing these mutant animals at reduced temperatures decreased intracellular accumulation, increased secretion and was sufficient to reduce lethality for some mutations (21). Of greater therapeutic relevance, treatment with 4-phenyl butyric acid (4PBA; an FDA-approved small molecule with chemical chaperone properties) is efficacious in promoting folding and preventing aggregation of misfolded protein associated with human diseases (22–24). Since most Col4a1 and Col4a2 mutations in our allelic series resulted in intracellular accumulation of mutant proteins at the expense of their secretion, we reasoned that reduced temperature or 4PBA treatment might decrease intracellular and increase extracellular COL4A1 and COL4A2 levels.
To improve our ability to observe an effect of reduced temperature or 4PBA, we first established the kinetics of COL4A1 secretion in Col4a1+/+ and Col4a1+/Δex41 MEFs. Although the levels of extracellular COL4A1 were consistently lower in Col4a1+/Δex41 compared with Col4a1+/+ MEFs, the time course for secretion was similar with a notable increase detected between 4 and 8 h after the induction of collagen secretion (by serum deprivation and ascorbic acid supplementation) with peak levels persisting up to 24 h (Supplementary Material, Fig. S4). Conversely, intracellular COL4A1 levels plateaued by 12 h post-induction in Col4a1+/Δex41 MEFs while intracellular levels remained low in Col4a1+/+ MEFs. Similar kinetics were also observed for other mutations tested (Col4a1+/G394V, Col4a1+/G658D and Col4a1+/G1038S; data not shown).
Next, we selected a subset of Col4a1 mutations with distinct characteristics and we evaluated the effects of reduced temperature or 4PBA treatment by measuring intracellular and extracellular COL4A1 levels 12 h after inducing collagen secretion. We first compared Col4a1+/+ cells grown under normal conditions to those grown in reduced temperature or with 4PBA (Fig. 6A and B). Col4a1+/+ MEFs grown at reduced temperature had a small but significant decrease of extracellular COL4A1. We observed a similar reduction in extracellular laminin suggesting that reduced secretion is a non-specific effect of growth at reduced temperature (Supplementary Material, Fig. S5). Reduced temperature had no effect on the intracellular levels of soluble COL4A1 in Col4a1+/+ cells and insoluble COL4A1 was not detected in these cells (indicated as n/a in Fig. 6A). 4PBA had no significant effect on intracellular or extracellular COL4A1 in Col4a1+/+ cells. In contrast, when we compared Col4a1+/mut MEFs grown under normal conditions to those grown at reduced temperature, we found significant reductions of soluble intracellular COL4A1 and all mutants had a trend towards reduced insoluble intracellular COL4A1 and increased extracellular COL4A1, although most did not reach statistical significance (Fig. 6C–J). Comparing Col4a1+/mut cells with or without 4PBA treatment yielded similar results with an interesting difference. While the decrease in intracellular COL4A1 levels in MEFs grown at reduced temperature was predominantly detected in the soluble fraction, 4PBA treatment significantly reduced the levels of insoluble COL4A1 for all mutations examined. There was also a trend toward reduced levels of soluble COL4A1 for all mutations but only Col4a1+/S1582P reached statistical significance. 4PBA treatment also increased extracellular COL4A1 for all mutations; two of which reached statistical significance. These data support the hypothesis that reduced temperature and 4PBA treatment have differential effects on soluble and insoluble COL4A1. The ability of 4PBA to decrease insoluble intracellular COL4A1 and increase extracellular COL4A1 is an encouraging observation for the potential therapeutic use of this treatment, or a similar approach, to reduce the penetrance or severity of pathology. It is important to note, however, that when directly comparing Col4a1+/+ and Col4a1+/mut cells on the same gels, 4PBA treatment did not completely restore intracellular and extracellular COL4A1 levels to those observed in Col4a1+/+ cells (Supplementary Material, Fig. S6). However, the magnitude of the change required for a beneficial effect on pathology is unknown and perhaps the levels of rescue achieved meet a therapeutic threshold.
DISCUSSION
COL4A1 and COL4A2 mutations are pleiotropic and cause a wide spectrum of disorders, including ocular dysgenesis, brain malformations and myopathy, of variable severity in both mice and humans. Here, we used an allelic series of Col4a1 and Col4a2 mutant mice to show that allelic heterogeneity contributes to phenotypic variability and identify at least one COL4A1 functional subdomain playing a role in disease expressivity. Furthermore, we demonstrate that different mutations have distinct effects on COL4A1 and COL4A2 biosynthesis and that these effects may be preventable under conditions that promote protein folding. Collectively, our findings provide the basis for targeted therapeutic strategies for patients with COL4A1 and COL4A2 mutations.
We compared the phenotypes and molecular consequences of a series of nine distinct Col4a1 and Col4a2 mutations. As is the case for COL4A1 and COL4A2 mutations reported to cause disease in humans, the majority of mutations analyzed in our study were glycine missense mutations occurring in the triple-helix-forming domain and therefore serve as valuable and relevant models to study the pathogenic mechanisms of disease (Fig. 1A) (3). The allelic series also included one splice site mutation that results in a 17 amino acid deletion in the triple-helix-forming domain (Col4a1Δex41) and one non-glycine missense mutation occurring in the NC1 domain (Col4a1S1582P). We found that all Col4a1 and Col4a2 mutations caused highly penetrant ocular, cerebral and muscular phenotypes (summarized in Table 1). Assessment of ocular dysgenesis is primarily qualitative and the phenotypes are variable, which make putative differences between alleles difficult to resolve. Quantitative measurements of optic nerve cross-sectional area revealed that nerves from the Col4a1+/Δex41 strain were significantly smaller compared with all other mutations. The sizes of Col4a1+/G1180D and Col4a1+/S1582P optic nerves were not significantly different from those of Col4a1+/+ optic nerves, suggesting that this phenotype demonstrates allelic heterogeneity. We have shown previously that Col4a1+/Δex41 mice have significantly fewer retinal ganglion cells which is explained, at least in part, by excess cell death during development (8,9). However, we have not excluded the possibility that Col4a1 mutant mice have defective myelination or other factors that might also contribute. When we quantified the number of structural brain malformations, we found that Col4a1+/G394V, Col4a1+/G1038S and Col4a1+/Δex41 were the most severe strains, whereas Col4a1+/S1582P mice were notable for being much milder than the other strains. Finally, in quantitative analysis of non-peripheral nuclei in sections of quadriceps, Col4a1+/G394V and Col4a1+/Δex41 mutations again stood out as the most severe, while Col4a1+/S1582P quadriceps were essentially normal. Overall, subject to our ability to quantify these phenotypes, Col4a1S1582P consistently led to the mildest phenotypes for all phenotypes examined and Col4a1Δex41, Col4a1G394V and Col4a1G1038S mutations tended to cause the most severe pathology. Notably, the ranking of the severity of pathology for each mutation across different tissues was not consistent suggesting that there may also be tissue-specific differences in expressivity.
Table 1.
Summary of results comparing severity of alleles
| ASD | ONH | Neuronal lamination defects | Myopathy | Increased intracellular COL4A1 | Decreased extracellular COL4A1 | |
|---|---|---|---|---|---|---|
| a1G394V | +++ | + | ++++ | ++++ | + | − |
| a2G646D | ++ | + | +++ | ++ | ++ | ++ |
| a1G658D | + | + | ++ | + | + | + |
| a1G912V | ++ | + | ++++ | ++ | ++ | ++ |
| a1G1038S | +++ | ++ | ++++ | ++ | +++ | +++ |
| a1Dex41 | ++++ | +++ | ++++ | +++ | +++ | +++ |
| a1G1180D | ++ | − | ++ | + | +++ | ++ |
| a1G1344D | +++ | ++ | ++ | ++ | +++ | ++++ |
| a1S1582P | + | − | + | − | ++++ | + |
Careful characterization of the phenotypic and molecular effects of different mutations in an otherwise uniform genetic context allowed us to make a number of important observations. Indeed, our data suggest that the consequences of a mutation might involve the domain, position within a domain, existence of functional subdomains, the type of mutation, and other, yet unidentified properties. First, the Col4a1S1582P mutation lies within the NC1 domain and all phenotypes examined were consistently less severe for this strain compared with mice with Col4a1 mutations located in the triple-helix-forming domain. A difference was also reflected in the biosynthetic consequences of the mutation. Whereas intracellular levels of COL4A1 and COL4A2 were concordant for each of the glycine missense mutations within the triple-helix-forming domain (Fig. 5D and E), the levels were discordant for the Col4a1S1582P mutation, with disproportionate levels of COL4A1 relative to COL4A2. The NC1 domain is a globular domain that initiates heterotrimer assembly by driving the association of the NC1 domain from COL4A2 with NC1 domains from two COL4A1 proteins. Therefore, one possible distinguishing feature of this mutation is that the mutant NC1 domains do not fold or function properly and thus, the mutant proteins do not participate in heterotrimer formation. In contrast, proteins with normal NC1 domains and with missense mutations in the triple-helix-forming domain are expected to be incorporated into heterotrimers during their assembly. This explanation is consistent with the observation that intracellular COL4A1 for Col4a1+/S1582P MEFs was predominantly soluble monomers whereas for most other mutations, intracellular COL4A1 had significant contributions of soluble and insoluble heterotrimers (Fig. 5H). Although we cannot make definitive conclusions based on a single allele and other explanations are possible, these data are consistent with a domain-specific effect whereby mutations within the NC1 domain cause less severe pathology than do mutations within the triple-helix-forming domain.
Second, we observed a position-dependent effect on heterotrimer trafficking for mutations within the triple-helix-forming domain. Analyses of intracellular COL4A1 and COL4A2 demonstrated a trend of increased intracellular COL4A1 and COL4A2 for mutations closer to the carboxy end of this domain. It is not clear why this trend was not as apparent for the extracellular levels of each protein and if this discordance is a true reflection of the biology or due to technical issues in detection or degradation of the extracellular proteins. Immunofluorescence of Col4a1+/G394V MEFs showed less co-localization of COL4A1 with the ER resident protein HSP47 compared with Col4a1+/Δex41 MEFs (Fig. 5A). This indicates more successful trafficking of COL4A1 from the ER to the Golgi in Col4a1+/G394V compared with Col4a1+/Δex41. Moreover, mutations nearer the amino end of the triple-helix-forming domain that had the lowest levels of intracellular COL4A1 (Col4a1G394V, Col4a2G646D, Col4a1G658D and Col4a1G912V) also had a higher molecular-weight band detected on western blots which is consistent with further posttranslational processing of these proteins. Notably, the position effect on intracellular levels of COL4A1 and COL4A2 also appears to apply to the Col4a2 mutation. One may have predicted that mutations in Col4a2 would not increase intracellular protein levels as much as a similarly positioned mutation in Col4a1 because of the stoichiometric relationship of COL4A1 and COL4A2 in heterotrimers (2:1 ratio of COL4A1:COL4A2). Assuming random assortment of the normal and mutant peptides in a Col4a2+/mut cell, 50% of the heterotrimers should be normal and 50% should incorporate the mutant COL4A2 protein. Conversely, in Col4a1+/mut cells only 25% of heterotrimers should be normal, 25% should include two mutant COL4A1 proteins and 50% should include one mutant COL4A1 protein. Based upon our data, the Col4a2 mutation behaves similarly to a position-matched mutation in Col4a1.
Third, and most strikingly, despite the correlation between the position of a mutation within the triple-helix-forming domain and the extent to which mutant heterotrimers advance through the secretory pathway, reduced trafficking does not translate into increased disease severity, at least for myopathy and cerebral cortical malformations. Therefore, properties other than position influence the severity of pathology. This effect is clearly illustrated by comparing the Col4a1Δex41 and Col4a1G1180D strains. Col4a1Δex41 is a splice acceptor site mutation that causes the 51-nucleotide exon 41 to be removed during mRNA processing and results in an internal, in-frame deletion of amino acids 1169–1185. Thus, to the extent to which a point mutation and a deletion can be compared, Col4a1Δex41 and Col4a1G1180D are position matched. Intracellular COL4A1 and COL4A2 levels between these two mutations are similar and are consistent with what might be expected based upon position. However, by all measures of pathology, the Col4a1Δex41 mutation is among the most severe alleles. Also consistent with a position-independent effect on disease severity is the observation that, despite having very low levels of intracellular COL4A1 and virtually normal levels of extracellular COL4A1, the Col4a1G394V mutation was associated with severe myopathy and cerebral cortical malformations. These data suggest that the position of a mutation within the triple-helix-forming domain exerts an effect on heterotrimer trafficking and pathology, but that there are other, allele-specific effects on pathogenesis.
What, then, does the position-dependent effect on heterotrimer trafficking, but not disease severity, reveal about the molecular mechanisms of COL4A1 and COL4A2 pathogenesis? We and others have previously proposed a role for ER stress in pathogenesis. We have detected ER stress in some experimental paradigms but not in others, which fosters uncertainty about the role of this pathway as a primary pathogenic insult (8,25–27). Here, we clearly show that the severity of myopathy and cortical malformations do not correlate with levels of intracellular COL4A1 or COL4A2 and we failed to detect ER stress even in the primary cells with the greatest levels of intracellular proteins. It is possible that this finding is a result of the in vitro setting used in this study (8,26) or that ER stress plays a more important role in the etiology of some phenotypes more than others. It may also be that the anomalous behavior of the Col4a1G658D mutation (very low levels both intracellular and extracellular COL4A1 and a broad, but not universal, transcriptional attenuation) indeed reflects previous activation of an ER stress response. For example, ER stress and activation of the unfolded protein response can trigger an IRE1-mediated degradation of particular classes of mRNA (28–30); however, further study is required to understand the vagaries of this allele. Regardless, our data suggest that even if an ER stress response is activated, it is unlikely to be a primary pathogenic mechanism for myopathy and cortical malformations.
Moreover, the observation that Col4a1G394V causes severe myopathy and cortical malformations despite having relatively little intracellular and normal levels of extracellular COL4A1 implies that the pathology is caused by an extracellular insult. Using immunofluorescence, we previously showed that homozygous Col4a1Δex41/Δex41 mice did not have detectable levels of COL4A1 in Reichert's membrane (25). We interpret this data as evidence that heterotrimers comprised of two mutant COL4A1 peptides (the only type that can form in homozygous mutant animals) are secreted at extremely low levels, if at all. However, the fate of heterotrimers with one normal and one mutant COL4A1 peptide remains an open question. If it is true that normal heterotrimers are secreted and heterotrimers with two mutant COL4A1 proteins are not, then it follows that the differences in intracellular and extracellular COL4A1 levels between mutant alleles must be the result of differential trafficking efficiency of the 50% of heterotrimers with one normal and one mutant COL4A1 peptide and suggests that this class of heterotrimers might indeed be secreted into the extracellular space. The severe myopathy and cortical malformations in Col4a1+/G394V mice that have essentially normal levels of extracellular COL4A1 and COL4A2 strongly supports that these heterotrimers are being secreted. These data suggest that pathogenesis involves the extracellular mutant proteins that interfere with the normal structure or function of basement membranes.
Mutations in genes coding for laminins, another major class of basement membrane proteins, can cause similar ocular, cortical and muscular phenotypes in humans and model organisms (31–38) that may result from altered interactions with dystroglycan or integrin receptors (39). Integrin- and dystroglycan-mediated adhesion and signaling are particularly important for muscle development and function and for cerebral cortex formation (40–42). This raises the possibility that Col4a1 pathology is secondary to laminin-mediated molecular events. It is important to note, however, that the Col4a1G394V mutation occurs within a putative integrin-binding site and may directly perturb integrin-mediated adhesion or signaling (Fig. 1A) (11), suggesting that the effect of the Col4a1G394V mutation may be direct. Furthermore, these data are consistent with the reports of six families with HANAC syndrome that have COL4A1 mutations that cluster within 31 amino acids that may also disrupt integrin-binding domains. Together, these data reflect the identification of a functional subdomain within the triple-helix-forming domain and support the hypothesis that impaired integrin adhesion or signaling may therefore be the primary molecular insult for myopathy and cerebral cortical malformations caused by Col4a1 and Col4a2 mutations.
Despite our suggestion that heterotrimers that include mutant peptides are secreted and contribute to pathogenesis, there is evidence that conditions that promote trafficking and secretion might be efficacious in preventing, delaying or diminishing disease. Col4a1+/mut C. elegans reared under reduced temperatures had decreased intracellular and increased extracellular COL4A1 and COL4A2 (21). This presumed shift in heterotrimer fate prevented body wall muscles from detaching during contractions and led to survival of animals that would have otherwise died. To model this result in a more therapeutically relevant system, we treated Col4a1+/mut cells with 4PBA. We found that 4PBA decreased intracellular COL4A1, particularly in the detergent-insoluble fraction, and increased extracellular COL4A1. It is unclear if the increase in extracellular COL4A1 includes mutant protein, and if so, what the effects of mutant protein are in the extracellular space. Based upon the promising proof-of-principle experiment in C. elegans, it is possible that we are not simply shifting the fate of mutant heterotrimers but also qualitatively altering them to functionally rescue the extracellular effect. Chemical chaperones may not be efficacious for the most severe alleles (for example, those like Col4a1G394V that may directly impact an integrin binding site); nevertheless, they hold therapeutic potential for at least a subset of Col4a1 and Col4a2 mutations. In vivo studies using these newly developed Col4a1 and Col4a2 mutant mouse models will help to clarify these issues and personalized medicine may predict in advance, which patients are most likely to benefit from the development of such therapies.
Collectively, our study underscores distinct roles of allelic heterogeneity on heterotrimer trafficking and disease severity in a series of Col4a1 and Col4a2 mutant mice and gives novel and important insight into the molecular mechanisms underlying myopathy and malformations of the cerebral cortex. Col4a1 and Col4a2 mutant mice constitute a valuable tool for further understanding the biological importance of allelic differences on the variable expressivity of disease caused by mutations in genes encoding type IV collagens. Determining the distinct biological consequences of different classes of mutations may improve accuracy of prognoses and guide genetic and reproductive counseling decisions based on genotype/phenotype information. These same insights may allow the development of personalized therapeutic options for preventing, delaying or diminishing COL4A1- and COL4A2-related disorders.
MATERIALS AND METHODS
Animals
Ethics statement: All experiments were conducted in compliance with protocols approved by the Institutional Animal Care and Use Committee at University of California San Francisco (protocol AN082706).
Eight mouse strains with distinct mutations in Col4a1 and one mouse strain with a mutation in Col4a2 (25,43) were backcrossed to C57BL/6J mice for a minimum of five generations to generate an allelic series of Col4a1 and Col4a2 mutant mice on a uniform genetic background (Supplementary Material, Table S1). All mutant mice used in this study were heterozygous for a given Col4a1 or Col4a2 mutation, and include both males and females ranging in age from 7.5 to 9.5 months old unless otherwise specified. Genotyping was performed by sequencing genomic DNA from tail biopsies digested with Proteinase K (Invitrogen) with the primers indicated in Supplementary Material, Table S1.
Phenotypic analyses
Slit lamp examination
Ocular anterior segment examinations were performed on 1.0- to 3.6-month-old heterozygous mutant mice and wild-type littermates using a slit lamp biomicroscope (Topcon SL-D7) attached to a digital SLR camera (Nikon D200). Observers were masked to mouse genotypes while evaluating clinical phenotypes.
Optic nerve analysis
Mice were trans-cardially perfused with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) at pH 7.4 and their optic nerves were harvested and stored in the same fixative at 4°C until use. Optic nerves were rinsed with phosphate buffer (0.01 M NaH2PO4, 0.04 M Na2HPO4, 5% sucrose), post-fixed with 2% osmium tetroxide, rinsed in distilled water and dehydrated with a graded series of ethanol concentrations. After embedding in Eponate-12 resin, 1-µm sections were collected, stained with 1% paraphenylenediamine (PPD) for 20 min and imaged by light microscopy. The cross-sectional area of optic nerves was measured using ImageJ software (National Institutes of Health).
Cerebral analysis
As above, brains were collected after trans-cardiac perfusion of 4% PFA and were post-fixed in the same fixative at 4°C overnight or were immersion fixed for 24 h at 4°C. Brains were cryoprotected in 30% sucrose in PBS overnight at 4°C, embedded in OCT compound (Sakura Finetek) and flash frozen using dry ice/ethanol. For Nissl staining, coronal cryosections (35 µm, collected at regular intervals of 140 µm) were rehydrated through 100 and 95% alcohol to distilled water and stained for 10 min in 0.1% cresyl violet solution. Sections were then rinsed in distilled water and incubated in 95% ethyl alcohol for 10 min before being dehydrated, cleared in xylene and mounted with Permount (Fisher). A minimum of 45 sections were examined per brain.
Muscle analysis
Quadriceps muscles were harvested by flash freezing in isopentane chilled in liquid nitrogen. The numbers of non-peripheral nuclei were evaluated in cryosections (10 µm) that were collected from the central portion of the muscle at regular intervals (200 µm) and labeled with Collagen IV antibody (Southern Biotech, raised in goat, 1:200) followed by an anti-goat AlexaFluor-488 secondary antibody. The nuclei were labeled with DAPI (2 µg/ml). Observers masked to genotype counted between 564 and 1998 fibers per muscle.
Microscopy
Images were captured using AxioVision software and an AxioImager M1 microscope equipped with an AxioCam MRm digital camera for fluorescence or AxioCam ICc3 for bright field (Zeiss).
Molecular analyses
Isolation and culture of primary MEFs
MEFs were isolated from embryonic day (E) 14.5 mouse embryos. Embryos were minced and trypsinized to produce single-cell suspensions (44). MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with glutamine (2 mm), penicillin/streptomycin (0.2 mm), and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in 5% CO2 humidified atmosphere. When cells reached 90–100% confluence, they were serum-deprived for 2–72 h in the presence of ascorbic acid (50 µg/ml) before subsequent analysis. For analysis of intracellular and extracellular levels of COL4A1 and COL4A2, cells were harvested after 24 h. In treatment experiments, cells were cultured at decreased temperatures (26°C) or in the presence of 4-phenylbutyric acid (4PBA, 10 mm) for 12 h followed by serum deprivation and ascorbic acid supplementation for an additional 12 h before being harvested.
Immunofluorescence
MEFs were fixed in 4% PFA in PBS and incubated overnight at 4°C with anti-COL4A1 (1:100 dilution, rat monoclonal clone H11, Shigei Medical Research Institute) and anti-HSP47 (1:500 dilution, mouse monoclonal clone M16.10A1, Stressgen Biotechnologies) antibodies in PBS containing 0.1% Triton-X (PBS-T). Immunolabeling was visualized using AlexaFluor 594 or 488 conjugated secondary antibodies raised in donkey (1:500, Invitrogen-Molecular Probes) in PBS-T. Slides were mounted using Mowiol containing DAPI (2 µg/ml).
Western blot analysis
MEFs were harvested and collected by centrifugation and the conditioned medium was collected and supplemented with protease inhibitors (Pierce). Cell pellets were resuspended directly in Laemmli buffer for semi-quantitative whole-cell lysate analysis, or proteins were isolated using a protein extraction buffer containing 0.05 m Tris–HCl, pH 8.0, 0.15 m NaCl, 5.0 mm EDTA, 1% NP-40 and protease inhibitors at 4°C for subsequent qualitative and semi-quantitative analyses of detergent-soluble and-insoluble intracellular fractions. Protein concentration in the soluble fraction was determined using a detergent-compatible colorimetric protein assay (Bio-Rad). Total protein (60 µg) was separated on a 4–15% gradient SDS–PAGE gel under non-reducing conditions for whole-cell lysate, insoluble fraction and conditioned medium samples, and under reducing conditions for soluble fraction samples and then transferred to polyvinylidene fluoride membranes (Bio-Rad). For the conditioned media, the volume loaded on the gel was adjusted based on the protein concentration of the soluble fraction of the corresponding sample. Similarly, insoluble fractions were resuspended in a volume of Laemmli buffer proportional to the protein concentration of the soluble fraction and equal volumes were loaded on the gel.
For COL4A1 or COL4A2 immunoblotting, membranes were blocked for 2 h at room temperature in 5% non-fat milk diluted in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBS-T), and overnight at 4°C in 3% bovine serum albumin (BSA) diluted in TBS. Membranes were then incubated with rat anti-COL4A1 (H11) or rat anti-COL4A2 (H22) monoclonal antibodies (1:100, Shigei Medical Research Institute, Japan) in 1% BSA diluted in TBS for 3 h at room temperature and washed in TBS-T. For ER stress analysis, membranes were blocked with 5% non-fat milk in TBS-T for 1 h at room temperature, incubated with anti-IRE1 (1:2000, ab37073, Abcam), anti-ATF6 (1:1000, ab371149, Abcam), anti-CHOP (1:250, ab11419, Abcam), anti-BiP (1:2000, 3177S, Cell Signalling) in blocking buffer overnight at 4°C, and washed in TBS-T. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (donkey anti-mouse or -rabbit IgG, 1:10 000, Jackson ImmunoResearch) diluted in 5% non-fat milk in TBS-T for 1 h at room temperature. Anti-laminin 1 + 2 (1:2000, ab7463, Abcam), anti-actin (1:2000, A3853, Sigma) and anti-tubulin (1:5000, T6557, Sigma) antibodies were used as extracellular and intracellular loading controls, respectively. Immunoreactivity was visualized by chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific). Densitometric analysis was performed on low exposure images using ImageJ (National Institutes of Health).
Quantitative real-time–polymerase chain reaction
RNA was extracted from MEFs using RNeasy Mini Kit (Qiagen), DNase treated (Promega) and reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad). qRT–PCR was performed on a Bio-Rad C1000 Thermal Cycler/CF96 Real-Time System using SsoFast EvaGreen Supermix (Bio-Rad) and the primers indicated in Supplementary Material, Table S2. Samples were run in duplicate with 5 ng of RNA per reaction and Col4a1, Col4a2, BiP, ATF6, ATF4, CHOP mRNA levels were normalized to Gapdh or Hprt1.
Statistical analysis
Control and mutant samples from each strain in independent experiments were compared using a Student's t-test. Comparison between mutant strains was performed using a one-way ANOVA followed by a Tukey's post hoc test. P-values of <0.05 were considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by National Institutes of Health (EY019887 to D.B.G.); Muscular Dystrophy Association (D.B.G.); Research to Prevent Blindness (D.B.G.); Howard Hughes Medical Institute (D.S.K.) and Knights Templar Eye Foundation (M.M.). Additional support was provided by a core grant from National Institute of Health (EY02162) and Research to Prevent Blindness unrestricted grant both to UCSF Department of Ophthalmology. Funding to pay the Open Access publication charges for this article was provided by That Man May See.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jeff Jorgensen for technical assistance.
Conflict of Interest statement. None.
REFERENCES
- 1.Boutaud A., Borza D.B., Bondar O., Gunwar S., Netzer K.O., Singh N., Ninomiya Y., Sado Y., Noelken M.E., Hudson B.G. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 2000;275:30716–30724. doi: 10.1074/jbc.M004569200. [DOI] [PubMed] [Google Scholar]
- 2.Khoshnoodi J., Pedchenko V., Hudson B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo D.S., Labelle-Dumais C., Gould D.B. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum. Mol. Genet. 2012;21:R97–110. doi: 10.1093/hmg/dds346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonduti D., Pichiecchio A., La Piana R., Livingston J.H., Doherty D.A., Majumdar A., Tomkins S., Mine M., Ceroni M., Ricca I., et al. COL4A1-related disease: raised creatine kinase and cerebral calcification as useful pointers. Neuropediatrics. 2012;43:283–288. doi: 10.1055/s-0032-1325116. [DOI] [PubMed] [Google Scholar]
- 5.Lemmens R., Maugeri A., Niessen H.W., Goris A., Tousseyn T., Demaerel P., Corveleyn A., Robberecht W., van der Knaap M.S., Thijs V.N., et al. Novel COL4A1 mutations cause cerebral small vessel disease by haploinsufficiency. Hum. Mol. Genet. 2013;22:391–397. doi: 10.1093/hmg/dds436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoneda Y., Haginoya K., Kato M., Osaka H., Yokochi K., Arai H., Kakita A., Yamamoto T., Otsuki Y., Shimizu S., et al. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann. Neurol. 2013;73:48–57. doi: 10.1002/ana.23736. [DOI] [PubMed] [Google Scholar]
- 7.Gould D.B., Phalan F.C., van Mil S.E., Sundberg J.P., Vahedi K., Massin P., Bousser M.G., Heutink P., Miner J.H., Tournier-Lasserve E., et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 8.Gould D.B., Marchant J.K., Savinova O.V., Smith R.S., John S.W. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol. Genet. 2007;16:798–807. doi: 10.1093/hmg/ddm024. [DOI] [PubMed] [Google Scholar]
- 9.Labelle-Dumais C., Dilworth D.J., Harrington E.P., de Leau M., Lyons D., Kabaeva Z., Manzini M.C., Dobyns W.B., Walsh C.A., Michele D.E., et al. COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 2011;7:e1002062. doi: 10.1371/journal.pgen.1002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plaisier E., Gribouval O., Alamowitch S., Mougenot B., Prost C., Verpont M.C., Marro B., Desmettre T., Cohen S.Y., Roullet E., et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 11.Parkin J.D., San Antonio J.D., Pedchenko V., Hudson B., Jensen S.T., Savige J. Mapping structural landmarks, ligand binding sites, and missense mutations to the collagen IV heterotrimers predicts major functional domains, novel interactions, and variation in phenotypes in inherited diseases affecting basement membranes. Hum. Mutat. 2011;32:127–143. doi: 10.1002/humu.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibley M.H., Graham P.L., von Mende N., Kramer J.M. Mutations in the alpha 2(IV) basement membrane collagen gene of Caenorhabditis elegans produce phenotypes of differing severities. EMBO J. 1994;13:3278–3285. doi: 10.1002/j.1460-2075.1994.tb06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Agtmael T., Schlötzer-Schrehardt U., McKie L., Brownstein D.G., Lee A.W., Cross S.H., Sado Y., Mullins J.J., Pöschl E., Jackson I.J. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum. Mol. Genet. 2005;14:3161–3168. doi: 10.1093/hmg/ddi348. [DOI] [PubMed] [Google Scholar]
- 14.Lipoff D.M., Bhambri A., Fokas G.J., Sharma S., Gabel L.A., Brumberg J.C., Richfield E.K., Ramos R.L. Neocortical molecular layer heterotopia in substrains of C57BL/6 and C57BL/10 mice. Brain Res. 2011;1391:36–43. doi: 10.1016/j.brainres.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 15.Plaisier E., Chen Z., Gekeler F., Benhassine S., Dahan K., Marro B., Alamowitch S., Paques M., Ronco P. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am. J. Med. Genet. A. 2010;152A:2550–2555. doi: 10.1002/ajmg.a.33659. [DOI] [PubMed] [Google Scholar]
- 16.Livingston J., Doherty D., Orcesi S., Tonduti D., Piechiecchio A., La Piana R., Tournier-Lasserve E., Majumdar A., Tomkins S., Rice G., et al. COL4A1 mutations associated with a characteristic pattern of intracranial calcification. Neuropediatrics. 2011;42:227–233. doi: 10.1055/s-0031-1295493. [DOI] [PubMed] [Google Scholar]
- 17.Shah S., Ellard S., Kneen R., Lim M., Osborne N., Rankin J., Stoodley N., van der Knaap M., Whitney A., Jardine P. Childhood presentation of COL4A1 mutations. Dev. Med. Child Neurol. 2012;54:569–574. doi: 10.1111/j.1469-8749.2011.04198.x. [DOI] [PubMed] [Google Scholar]
- 18.Yoneda Y., Haginoya K., Arai H., Yamaoka S., Tsurusaki Y., Doi H., Miyake N., Yokochi K., Osaka H., Kato M., et al. De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly. Am. J. Hum. Genet. 2012;90:86–90. doi: 10.1016/j.ajhg.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollner R., Schmidt C., Fischer G., Kühn K., Pöschl E. Cooperative and competitive interactions of regulatory elements are involved in the control of divergent transcription of human Col4A1 and Col4A2 genes. FEBS Lett. 1997;405:31–36. doi: 10.1016/s0014-5793(97)00152-x. [DOI] [PubMed] [Google Scholar]
- 20.Walsh D.M., Selkoe D.J. A beta oligomers - a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 21.Gupta M.C., Graham P.L., Kramer J.M. Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J. Cell Biol. 1997;137:1185–1196. doi: 10.1083/jcb.137.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Almeida S.F., Picarote G., Fleming J.V., Carmo-Fonseca M., Azevedo J.E., de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J. Biol. Chem. 2007;282:27905–27912. doi: 10.1074/jbc.M702672200. [DOI] [PubMed] [Google Scholar]
- 23.Bonapace G., Waheed A., Shah G.N., Sly W.S. Chemical chaperones protect from effects of apoptosis-inducing mutation in carbonic anhydrase IV identified in retinitis pigmentosa 17. Proc. Natl. Acad. Sci. USA. 2004;101:12300–12305. doi: 10.1073/pnas.0404764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yam G.H., Gaplovska-Kysela K., Zuber C., Roth J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest. Ophthalmol. Vis. Sci. 2007;48:1683–1690. doi: 10.1167/iovs.06-0943. [DOI] [PubMed] [Google Scholar]
- 25.Gould D.B., Phalan F.C., Breedveld G.J., van Mil S.E., Smith R.S., Schimenti J.C., Aguglia U., van der Knaap M.S., Heutink P., John S.W.M. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 26.Firtina Z., Danysh B.P., Bai X., Gould D.B., Kobayashi T., Duncan M.K. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J. Biol. Chem. 2009;284:35872–35884. doi: 10.1074/jbc.M109.060384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeanne M., Labelle-Dumais C., Jorgensen J., Kauffman W.B., Mancini G.M., Favor J., Valant V., Greenberg S.M., Rosand J., Gould D.B. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am. J. Hum. Genet. 2012;90:91–101. doi: 10.1016/j.ajhg.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 29.Gaddam D., Stevens N., Hollien J. Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol. Biol. Cell. 2013;24:14–20. doi: 10.1091/mbc.E12-06-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollien J., Lin J.H., Li H., Stevens N., Walter P., Weissman J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zenker M., Aigner T., Wendler O., Tralau T., Müntefering H., Fenski R., Pitz S., Schumacher V., Royer-Pokora B., Wühl E., et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 32.Semina E.V., Bosenko D.V., Zinkevich N.C., Soules K.A., Hyde D.R., Vihtelic T.S., Willer G.B., Gregg R.G., Link B.A. Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev. Biol. 2006;299:63–77. doi: 10.1016/j.ydbio.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Wühl E., Kogan J., Zurowska A., Matejas V., Vandevoorde R.G., Aigner T., Wendler O., Lesniewska I., Bouvier R., Reis A., et al. Neurodevelopmental deficits in Pierson (microcoria-congenital nephrosis) syndrome. Am. J. Med. Genet. A. 2007;143:311–319. doi: 10.1002/ajmg.a.31564. [DOI] [PubMed] [Google Scholar]
- 34.Bredrup C., Matejas V., Barrow M., Bláhová K., Bockenhauer D., Fowler D.J., Gregson R.M., Maruniak-Chudek I., Medeira A., Mendonça E.L., et al. Ophthalmological aspects of Pierson syndrome. Am. J. Ophthalmol. 2008;146:602–611. doi: 10.1016/j.ajo.2008.05.039. [DOI] [PubMed] [Google Scholar]
- 35.Matejas V., Hinkes B., Alkandari F., Al-Gazali L., Annexstad E., Aytac M.B., Barrow M., Bláhová K., Bockenhauer D., Cheong H.I., et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 2010;31:992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barak T., Kwan K.Y., Louvi A., Demirbilek V., Saygı S., Tüysüz B., Choi M., Boyacı H., Doerschner K., Zhu Y., et al. Recessive LAMC3 mutations cause malformations of occipital cortical development. Nat. Genet. 2011;43:590–594. doi: 10.1038/ng.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta V.A., Kawahara G., Myers J.A., Chen A.T., Hall T.E., Manzini M.C., Currie P.D., Zhou Y., Zon L.I., Kunkel L.M., et al. A splice site mutation in laminin-α2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PloS ONE. 2012;7:e43794. doi: 10.1371/journal.pone.0043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radner S., Banos C., Bachay G., Li Y.N., Hunter D.D., Brunken W.J., Yee K.T. β2 and γ3 laminins are critical cortical basement membrane components: ablation of Lamb2 and Lamc3 genes disrupts cortical lamination and produces dysplasia. Dev. Neurobiol. 2013;73:209–229. doi: 10.1002/dneu.22057. [DOI] [PubMed] [Google Scholar]
- 39.Gawlik K.I., Mayer U., Blomberg K., Sonnenberg A., Ekblom P., Durbeej M. Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 2006;580:1759–1765. doi: 10.1016/j.febslet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 40.Carmignac V., Durbeej M. Cell-matrix interactions in muscle disease. J. Pathol. 2012;226:200–218. doi: 10.1002/path.3020. [DOI] [PubMed] [Google Scholar]
- 41.Schmid R.S., Anton E.S. Role of integrins in the development of the cerebral cortex. Cereb. Cortex. 2003;13:219–224. doi: 10.1093/cercor/13.3.219. [DOI] [PubMed] [Google Scholar]
- 42.Moore S.A., Saito F., Chen J., Michele D.E., Henry M.D., Messing A., Cohn R.D., Ross-Barta S.E., Westra S., Williamson R.A., et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- 43.Favor J., Gloeckner C.J., Janik D., Klempt M., Neuhäuser-Klaus A., Pretsch W., Schmahl W., Quintanilla-Fend L. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–736. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson E.J. Embryo-derived stem cell lines. In: Robertson E.J., editor. Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. Oxford, UK: IRL Press; 1987. pp. 71–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.