

Abstract
Transposable elements (TEs) account for nearly one-half of the sequence content in the human genome, and de novo germline transposition into regulatory or coding sequences of protein-coding genes can cause heritable disorders. TEs are prevalent in and around protein-coding genes, providing an opportunity to impart regulation. Computational studies reveal that microRNA (miRNA) genes and miRNA target sites reside within TE sequences, but there is little experimental evidence supporting a role for TEs in the birth of miRNAs, or as platform for gene regulation by miRNAs. In this work, we validate miRNAs and target sites derived from TE families prevalent in the human genome, including the ancient long interspersed nuclear element 2 (LINE2/L2), mammalian-wide interspersed repeat (MIR) retrotransposons and the primate-specific Alu family. We show that genes with 3′ untranslated region (3′ UTR) MIR elements are enriched for let-7 targets and that these sites are conserved and responsive to let-7 expression. We also demonstrate that 3′ UTR-embedded Alus are a source of miR-24 and miR-122 target sites and that a subset of active genomic Alus provide for de novo target site creation. Finally, we report that although the creation of miRNA genes by Alu elements is relatively uncommon relative to their overall genomic abundance, Alu-derived miR-1285-1 is efficiently processed from its genomic locus and regulates genes with target sites contained within homologous elements. Taken together, our data provide additional evidence for TEs as a source for miRNAs and miRNA target sites, with instances of conservation through the course of mammalian evolution.
INTRODUCTION
Transposable elements (TEs or transposons) mobilize and reintegrate within a host organism's genome, and different TE families have diverse structural features, transposition mechanisms and evolutionary origins. Retrotransposons (Type I) replicate using a transcribed RNA intermediate as a template for reverse transcription and reintegration (1) and are further subcategorized according to the presence of long terminal repeats (LTRs). Most LTR-containing retrotransposons are ancient integrating retroviruses, which are no longer infectious. Non-LTR retrotransposons, including long and short interspersed nuclear elements (LINEs and SINEs, respectively), are the most abundant TE class in humans and account for >30% of the total DNA content (2). They are distributed throughout the genome, including in and near protein-coding loci. In some cases, transposition of TEs into genes induces monogenic disorders such as β-thalassemia, hemophilia and cystic fibrosis (3). In other instances, integration can induce genetic instability and cancer. Finally, the conservation of gene-proximal TEs has spawned research into whether or not retention reflects possible functional roles [reviewed in (3)].
Interestingly, a portion of the TE-driven impact on gene expression results from cellular pathways that defend against TE transposition. For example, CpG sequences in L1 promoters and Alu and SVA elements are sites for DNA methylation and heterochromatin formation, causing epigenetic silencing (4). Consequently, when these TEs integrate proximal into promoters, epigenetic silencing can spread into the promoters (5). If they are not epigenically silenced, promoter elements from Alus can drive expression (6). At the post-transcriptional level, the RNA interference (RNAi) pathway also plays a role in TE defense. Small non-coding PIWI-interacting RNAs (piRNAs) and endogenous siRNAs (endo-siRNAs) are loaded into Argonaute-family proteins (PIWI and AGO2, respectively) and guide silencing complexes to complementary TE sequences (7,8).
Similar to piRNAs and siRNAs, some microRNAs (miRNAs) are also processed from TE-derived genomic loci (6,9–11). Although miRNAs are not generally implicated in TE defense, the canonical targets for miRNA regulation, mRNA 3′ untranslated region (3′ UTRs), often contain TE sequences (12). Computational and some wet lab studies do support that TE-derived miRNAs can regulate target mRNAs through complementary target sites located within 3′ UTR-resident TE homologs (11,13). For example, analysis of degradome sequence tags in human cells shows that long interspersed nuclear element 2 (LINE2)-derived miR-28-5p and miR-151 regulate a subset of genes, including Ly6/Plaur domain-containing 3 (LYPD3) and ATP synthase mitochondrial F1 complex assembly factor 1 (ATPAF1), through non-canonical ‘centered-seed’ pairing to 3′ UTR-resident LINE2 elements (14). In this work, we show that miR-28-5p also regulates the expression of LYPD3 and E2F transcription factor 6 (E2F6) through conserved 3′ UTR-resident LINE2 sequences. We also found analogous interaction arising from the primate-specific Alu retrotransposon, using as a case study Alu-derived miR-1285 and its corresponding Alu-derived targets.
In additional work, we tested whether non-TE-derived miRNA families act on TE-derived miRNA target sites. Earlier work showed that adenosine-to-inosine (A-to-I) RNA editing, which most-commonly occurs in 3′ UTR-resident Alus, can create functional binding sites for primate-specific miR-513 and miR-769 in the 3′ UTR of DNA fragmentation factor alpha (15). Computational data have also demonstrated that edited and unedited 3′ UTR Alu loci harbor binding sites for well-conserved miRNAs (15,16). We tested whether expansion of some TE families created novel, lineage-specific binding sites for the highly conserved let-7, miR-24 and miR-122 families. We found that let-7 regulates several human genes through conserved sites found within mammalian-wide interspersed repeats (MIRs) and that Alu-derived binding sites for miR-24 and miR-122 are functional. Thus, TEs provide an important source of both miRNA genes and target sites, with evidence for some conserved through the course of mammalian evolution.
RESULTS
3′ UTR-resident TE sequences encode putative miRNA target sites
We first updated previously published information predicting miRNA-binding sites in human 3′ UTR-resident TEs using the most recent miRNA and 3′ UTR annotations (Supplementary Material, Table S1) (16,17). From this, we found that ∼60% of all TE-derived sites are located in Alu (∼35%), LINE1 (∼12%) and MIR (∼11%) elements, consistent with the prevalence of these TE families in 3′ UTRs (Supplementary Material, Fig. S1, Table S2). On average, 5–10% of a miRNA's target site repertoire is TE-derived; although it approaches 50% for some miRNA families (Supplementary Material, Fig. S2, Table S3). For >85% of miRNAs, the majority of TE-derived target sites overlap L1 and Alu elements (Supplementary Material, Fig. S2).
Target site prediction algorithms, which tend to over-predict functional sites, typically incorporate features beyond the minimal seed complement to improve accuracy. Such features include sequence conservation, binding thermodynamics and other local sequence features indicative of target site potency. Although conservation is a strong indicator of true target sites, Alu and L1 sequences in human 3′ UTRs are predominantly primate-specific, and therefore, target sites residing in these loci inherently lack conservation. Therefore, to find evidence for conserved and functional TE-derived target sites, we first searched for miRNA–target interactions derived from ancient TE families.
For this, we focused on genes with 3′ UTR-resident MIR elements, which represent transposition events that occurred early in mammalian evolution. The ∼1200 human genes with 3′ UTR-resident MIRs were used as input, and gene enrichment analysis done to identify functionally relevant miRNAs using the ToppFun algorithm (18). ToppFun incorporates seven data sets, two of which are validated miRNA–target interactions curated from the literature (miRTarbase and miRecords). The remaining (TargetScan, PITA, PicTar, MSigDB and miRSVR) are data generated from in silico target prediction algorithms. Although no significant interactions were found in the validated targets databases, target genes for >80 miRNAs were significantly enriched (P ≤ 0.05) in the 3′ UTR-MIR gene set according to at least one target data set. For all but four miRNAs, evidence for enrichment came from only one of the data sets (Fig. 1; bottom). Two data sets yielded significant enrichment for miR-610, miR-214 and miR-146b-3p target genes. Strikingly, let-7 targets were significantly enriched according to all five algorithms. Also, let-7 was the only miRNA with targets enriched in the miRSVR conserved, high efficacy (miRSVR C/HE) category, which represents the highest-confidence target sites for this program.
Figure 1.

Genes containing 3′ UTR-embedded MIR elements are significantly enriched for conserved and high-confidence let-7a target sites. (Top Left) A pie graph summarizes the prevalence of let-7 target sites predicted in 3′ UTR-TEs. Each section displays the number of let-7 target sites tabulated after grouping TEs into the indicated families. (Top Right) The subset of 3′ UTR-resident MIRs is shown, grouped according to whether or not a let-7 target sit is predicted within the TE. The red sections of the two pie charts indicate common information, as they each depict the prevalence of MIR-derived let-7 target sites. Gene functional enrichment analysis was performed on the genes with 3′ UTR-MIRs using the ToppFun algorithm. (Bottom) Functional enrichment significance (-log10 P-value) is plotted, representing miRNAs whose targets are significantly enriched (P ≤ 0.05; Bonferroni correction) in genes with 3′ UTR-resident MIRs. Enrichment of targets predicted by Target-scan, PITA, MSigDB, PicTar and mirSVR are plotted and P-values corresponding to let-7 targets highlighted in red. For mirSVR (C = conserved, NC = non-conserved, HE = high efficacy (predicted), LE = low efficacy).
While the ToppFun analysis revealed that genes with 3′ UTR-MIRs were enriched for let-7 target sites, it did not show whether these sites resided within the MIR element. Intersecting let-7 and 3′ UTR-MIR coordinates showed that ∼8% of the MIRs contain a let-7 site. Conversely, analyzing the composition of TE families overlapping let-7 target sites revealed that MIR-derived target sites account for nearly 40% of let-7′s TE-derived target sites (Fig. 1; top left). Interestingly, we found that some of the target loci, such as those found in Myosin 1F (MYO1F) and E2F transcription factor 6 (E2F6), are highly conserved (Fig. 2A). These computational data suggest that active MIR element transposition early in the course of mammalian evolution provided a platform for let-7 regulation that is conserved in extant species.
Figure 2.

Assessment of conserved, MIR-derived target sites for let-7. (A) Let-7 target sites (yellow box) overlapping a MIR element (red boxes) annotated by RepeatMasker. MYO1F and E2F6 are two candidates where: (i) no let-7 site is present in the 3′ UTR aside from the MIR-derived site shown and (ii) PhyloP conservation scores (Mammal Cons track) showed sequence conservation coincident with the target site. (B) 3′ UTRs of MYCBP, MFSD4, E2F6 and MYO1F, each containing a single MIR-derived let-7 site, were cloned into dual-luciferase reporters and co-transfected into HEK293 cells with a synthetic let-7 mimic (Pre-miR™ at the doses indicated). Reactions were balanced to 1.0 nM with a non-targeting Pre-miR™ (ctrl). Luciferase activity is plotted as a percent of Renilla:Firefly ratio measured from the 0 nM let-7 (1.0 nM ctrl) treatment group. (C) HeLa cells, which express high levels of endogenous let-7a, were co-transfected with the luciferase reporters and a let-7a Anti-miR inhibitor (25, 50 nM), balanced to 50 nM with a negative control Anti-miR oligo (ctrl). Luciferase activity is presented as the Renilla:Firefly ratios measured 48 h post-transfection and normalized to the 50 nM ctrl dose of the corresponding reporter. N = 3 biological replicates with three technical replicates per assay; error bars = SD. *P ≤ 0.05 (Student's t-test; two-tailed). (D) Cumulative distribution functions are shown summarizing gene expression changes (log2 fold change) in response to let-7 over-expression (si-let-7), relative to the control siRNA (si-GFP). Transcripts were grouped according to annotations of 3′ UTR-MIRs and let-7 target sites, as indicated in the figure legends. (E) As in D, except gene expression changes were measured in response to let-7 inhibition (let-7 2′OMe) relative to a control oligo (Luc. 2′-OMe).
We next cloned the 3′ UTRs of human MYO1F, E2F6, MYC-binding protein (MYCBP) and major facilitator superfamily domain-containing protein 4 (MFSD4) into luciferase reporter plasmids to validate the functionality of conserved MIR-derived let-7 target sites. Each 3′ UTR harbored a single conserved MIR-derived let-7 target site. When co-transfected with a synthetic let-7 mimic, the 3′ UTR reporters, but not the empty PsiCHECK-2 negative control (CTRL), responded with a dose-dependent reduction in luciferase activity (Fig. 2B). While these data demonstrate that let-7a-mediated knockdown depends on the MIR-containing 3′ UTRs, transfection of the let-7a mimics can exceed physiologically relevant expression levels. However, it should also be noted that in this case, significant knockdown was observed at let-7 mimic concentrations as low as 0.1 nM, which is 500-fold less than the manufacturer-recommended dose. To test whether endogenous levels of let-7a regulate expression of the 3′ UTRs, HeLa cells were transfected with the luciferase constructs and let-7a or non-targeting (ctrl) oligonucleotide inhibitors (Anti-miRs). Inhibition of endogenous let-7a resulted in a significant increase in luciferase activity at the intermediate (25 nM) concentration for the E2F6 reporter, and at the highest (50 nM) dose for E2F6, MYCBP and MYO1F (Fig. 2C). No significant change was observed in the negative control (CTRL) or MFSD4 constructs. Together, these data show that 3′ UTRs containing conserved, MIR-derived target sites can respond to let-7 regulation.
We next analyzed microarray data representing gene expression changes following let-7 over-expression (si-let-7) or inhibition (let-7 2′OMe) to determine whether endogenous transcripts harboring MIR-resident target sites also respond to let-7 activity (Supplementary Material, Table S4; GSE2918, S. Bhattacharya, unpublished). Microarray data are used extensively for studying miRNA function, because miRNAs primarily work by triggering the destabilization of target mRNAs (19). Fold changes in expression (log2-scaled) were calculated for si-let-7 and let-7 2′OMe relative to each respective negative control. Transcripts were grouped according to the presence of a 3′ UTR-MIR or let-7 target site and whether the target and MIR sites overlapped (Supplementary Material, Table S4). Compared with genes containing no let-7 target site, over-expression of let-7a mimics (si-let-7) significantly reduced expression of genes harboring only a MIR-derived let-7a site [(Kolmogorov–Smirnov test (K-S test)]; d = 0.1281, P = 0.020) or only a non-MIR site (K-S test; d = 0.0844, P = 2.48e-117), as demonstrated in the cumulative distribution function (CDF) plots for the target-containing transcripts (Fig. 2D). Genes with MIR-derived target sites were silenced to a slightly lesser extent than their non-MIR-derived counterparts, although this difference was insignificant (K-S test; d = 0.1145, P = 0.053) (Fig. 2D). Conversely, when let-7a was inhibited, target site-containing transcripts were up-regulated (CDFs shifted to the right) significantly relative to those with no site, both for MIR-derived sites (K-S test; d = 0.1671, P = 0.001) and non-MIR-derived sites (K-S test; d = 0.0697, P = 5.92e-08) (Fig. 2E). Surprisingly, in the over-expression data set, we also observed that genes harboring a MIR element, yet lacking a let-7 target site, had lower expression levels in general, than genes with no MIR and no target site, although we have no explanation for this observation (K-S test; d = 0.0885, P = 1.66e-10) (Fig. 2D). No significant difference was seen with the non-targeted, MIR-containing genes in the Anti-miR treatment (K-S test; d = 0.027, P = 0.122) (Fig. 2E). Taken together, these data support the reporter experiments demonstrating that 3′ UTR-MIR elements with let-7 target sites respond to let-7 regulation. Furthermore, finding conserved and functional sites enriched in an abundant TE family serves as precedent for similar events resulting from the primate-specific expansion of Alu elements.
MiRNAs with high Alu-target site frequency targetspecific regions in the Alu
While this work was in preparation, Hoffman et al. used publically available microarray data to assess the activity of Alu-derived miRNA-binding sites in human cells to test for evidence of Alu-derived miRNA-binding sites in the human genome (20). They found that seed matches to miRNA families were highly prevalent in 3′ UTRs harboring Alus but that they were largely nonfunctional. However, because Hoffman et al. focused on seed complements to positions 2–8 alone, target sites with a 7mer-A1 architecture were not predicted. 7mer-A1 sites are known to provide functional targets (21). By including this site type, we found ∼1400 Alu-derived targets for miR-122, whereas only 62 were reported by Hoffman et al. Several miRNAs, including miR-122 and miR-24, had a high frequency of Alu-derived targets (Supplementary Material, Table S2); for miR-24 and miR-122, >80% of TE-derived target sites were found within 3′ UTR-resident Alus (1948 and 1402 Alu targets, respectively), most of which are 7mer-1A sites (Fig. 3A and Supplementary Material, Table S2).
Figure 3.

Assessment of primate-specific, Alu-derived target sites for miR-122 and miR-24, present in the Alu consensus sequence. (A). Pie charts summarizing the prevalence of the most prominent TE families are presented, with values representing the number of unique (left) miR-24 or (right) miR-122 sites tabulated. (B) RepeatMasker annotations were used to calculate miR-24, miR-122 and miR-125-3p target site positions relative to the Alu consensus sequence, tabulating target prevalence at each position. Target site frequencies are plotted, representing the fraction of all 3′ UTR Alus found with the target sequence at that position. (C) The Primate Conservation track indicates an increase in conservation score coincident with the miR-24 target overlapping the AluSp family sequence. (D) Microarray data representing gene expression changes in response to miR-122 over-expression in human cells were analyzed, and log2 expression changes relative to the miRNA control set calculated. Transcripts were grouped according the presence of 3′ UTR Alus or miR-122 sites and the cumulative distributions plotted for each group as indicated. (E) Luciferase reporters expressing EIF2S3 and MAP3K9 3′ UTRs were co-transfected into HEK293 cells with Pre-miR™ miR-24 mimics (0, 1, 10 nM Pre-miR™ doses) and luciferase activity measured 24 h later. Renilla:firefly ratios are plotted, normalizing to the ratio calculated for the corresponding 0 nM treatment set. N = 4 biological replicates with three technical replicates per transfection; error bars = SD. *P ≤ 0.05 (Student's t-test; two-tailed).
We reasoned that if high target site frequencies (as compared with other TEs) were due to miRNAs targeting regions with little sequence divergence from the parent Alu, target sites for these miRNAs would be enriched at specific positions in the Alu element. Using genomic coordinates and RepeatMasker annotations from the overlapping Alu and miRNA target site features, we calculated the position of each site in relation to the parental Alu. This analysis showed that target site locations for miR-24, miR-122 and others were highly uniform (Fig. 3B). This also suggests that these target sites existed in the parental Alu and, therefore, were present at the point of integration and were not formed through subsequent mutations.
MiR-24 directly regulates transcripts through Alu-derived target sites
We next tested whether any of the Alu-derived target sites create functional platforms for miRNA regulation. Although Alu sequences inherently lack conservation, we prioritized candidates where the target site had greater conservation than the host Alu (e.g. MAP3K9, Fig. 3C). We selected five candidate genes that fit this criterion [platelet f11 receptor (F11R), carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 6 (CHST6), procadherin beta 11 (PCDHB1) and eukaryotic translation initiation factor 2, subunit 3 gamma (EIF2S3), mitogen-activated protein 3-kinase 9 (MAP3K9)], and cloned the human 3′ UTR of each into a luciferase reporter. The 3′ UTR of MFSD4, which has an Alu-derived miR-24 site in addition to the MIR-derived let-7a target site, was also tested. Dose-dependent luciferase reduction resulted in response to a miR-24 mimic for EIF2S3 and MAP3K9, as well as the artificial miR-24 perfect target control miR-24_2xT (Fig. 3D, Supplementary Material, Fig. S3). At the doses used (1 and 10 nM), no significant knockdown was observed in the other constructs tested or in the psiCHECK™-2 no-target control (data not shown and Fig. 3D). Blocking endogenous miR-24 with an anti-miR resulted in a significant dose-dependent increase in luciferase activity only in the artificial target positive control, consistent with the low validation rate seen in the over-expression experiments and the general low degree of functionality shown in a study by Hoffman et al. (data not shown). Nonetheless, the over-expression data reveal that given sufficient miR-24 expression levels, EIF2S3 and MAP3K9 can be regulated by miR-24.
To test whether Alu-dependent miRNA regulation is apparent on a global level, we analyzed microarray data measuring mRNA transcriptional changes in response to miR-122 over-expression [the miR-24 over-expression microarray data in human cells are not useful for this work as no seed-mediated changes in gene expression can be documented (data not shown)]. Genes were annotated according to whether they contained (i) a 3′ UTR Alu, (ii) an Alu-derived target site (including 7mer-1A sites) or (iii) a canonical (non-TE-derived) target site for miR-122 (Supplementary Material, Table S5). Analysis of CDFs for all groups revealed significant repression in genes containing an Alu-derived target site in its 3′ UTR, relative to genes whose 3′ UTRs lacked both Alus and target sites (K-S test; d = 0.1451, P = 6.17e-10), but not to the degree seen in genes with non-TE-derived miRNA target sites relative to no site (K-S test; d = 0.1744, P < 2.2e-16) (Fig. 3D). This difference observed comparing Alu and non-Alu-derived targets was significant (K-S test; d = 0.0795, P = 0.0081). We repeated this analysis using microarray data for additional miRNAs and found that Alu-derived sites were on average less down-regulated than genes with canonical sites (data not shown). This supports the work by the Hoffman et al., and the luciferase data presented for miR-24, in that most Alu-derived sites lack functionality or are weakly responsive to miRNA regulation. However, we also note that in spite of the generally weaker knockdown of Alu target sites, given a sufficiently large set, the small fraction of responsive Alu-derived sites still represent 20–30% of the down-regulated target list.
Proliferation of Alu and B1 SINEs caused convergent acquisition of miRNA targets in their respective primate and murine lineages
To test the impact of recently evolved lineage-specific TEs, we repeated our target prediction analysis using mouse 3′ UTR-resident TE sequences. As suggested in the study by Hoffman et al., B1 elements in rodents are homologous to Alus in primates and often harbor analogous miRNA-binding sites. To select candidates for functional validation, we searched for the convergent acquisition of TE-derived target sites to determine whether murine and primate orthologs would independently gain regulatory sites for the same miRNA (Fig. 4A). For this, we gathered coordinates for mouse 3′ UTR-resident TE sequences and used the ‘lift-over’ utility on the Galaxy web server to convert to the corresponding human coordinates. Mouse 3′ UTR sequences that overlapped TEs with no mappable human counterpart were then selected, as were human 3′ UTR sequences overlapping TEs with no mappable mouse counterpart. Target sites were predicted using miRNA families and target transcripts present in both species. We tested human and mouse solute carrier family 12, member 8 (SLC12A8), sideroflexin2 (SFXN2), UBX domain-containing protein 2B (UBXN2B) and CDGSH iron sulfur domain 2 (CISD2), using 3′ UTR reporters. The 3′ UTR of chimpanzee SFXN2 (ptrSFXN2) was also cloned because the miR-24 seed match contained a single base mutation in the target site. Both mouse and human SFXN2 and SLC12A8 showed significant repression when co-expressed with 15 or 30 nM of miR-24 mimic (Fig. 4B). Chimpanzee SFXN2 showed no significant response. No significant response was seen with UBXN2B, CISD2 in either species (Fig. 4B and data not shown). Again, the low validation rate of Alu-derived sites corroborates the findings of Hoffman et al. However, finding that human and mouse but not chimpanzee SFXN2 responded to miR-24 activity provides evidence for another Alu-derived site in humans and shows that analogous mechanisms lead to target site creation in other species.
Figure 4.
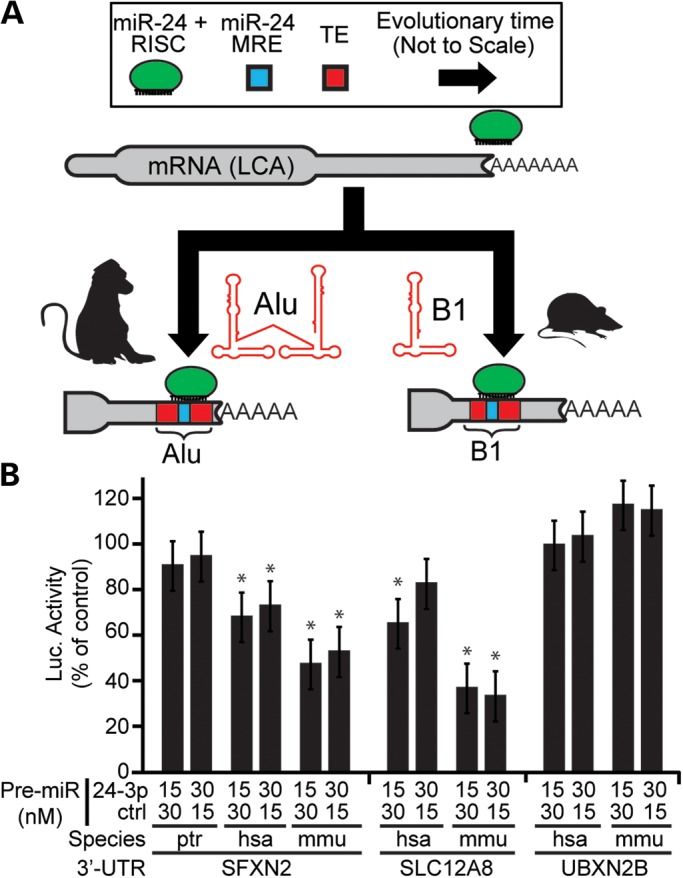
Convergent evolution of miR-24-mediated regulation in rodents and primates through lineage-specific TE homologs. (A) A cartoon schematic demonstrating a hypothetical example of miR-24 target sites gained independently in primate and rodent evolutionary lineages, via transposition of Alu and B1 elements, respectively. Arrows representing evolutionary time are not drawn to scale. The hypothetical mRNA of the last common ancestor (LCA) of these two lineages is drawn lacking miR-24 binding sites. (B) Luciferase reporters with miR-24 target 3′ UTRs from human (hsa), mouse (mmu) or chimpanzee (ptr) were co-transfected with miR-24 Pre-miRs at the doses indicated. Luciferase activity was calculated as the Renilla:Firefly ratios measured 24 h after transfection, normalized to the corresponding 0 nM treatment group. N = 3 biological replicates with three technical replicates per assay; error bars = SD. *P ≤ 0.05 (Student's t-test; two-tailed).
Potentially active Alu loci contain miRNA binding motifs
Although the majority of 3′ UTR Alus do not respond to miRNA activity, the few validated cases presented here and those suggested in the Hoffman et al. show that given the proper context, miRNAs can regulate genes through 3′ UTR-resident Alus. Furthermore, because Alus are transpositionally active in humans, they remain potential sources of novel miRNA-binding sites. Given that target sites for some miRNAs correspond to parts of the Alu consensus sequence, we hypothesized that a subset of active Alus also harbor target sites. A recent study assessed the features of active Alu elements and found 124 key positions that were 100% conserved in active elements (22). To test whether these potentially active Alus harbor miRNA-binding sites, we searched for seed complements in the ∼12 000 human Alus that retained all 124 sequence features (Supplementary Material, Table S6). We found that miRNAs with a high frequency of Alu-derived 3′ UTR sites, including miR-24 and miR-122, also had a high frequency of sites in the potentially active Alu sequences (Supplementary Material, Table S6). In addition, binding sites for several other miRNAs were present in well over 90% of the potentially active sequences. These data show that de novo integration of Alu elements will likely contain seed complements for a subset of miRNAs. If an Alu integrates into a 3′ UTR in an appropriate context, one or more functional miRNA target sites could be created.
miRNAs processed from TE sequences can regulate target genes containing homologous elements
In the examples presented thus far, the miRNA's origin precedes that of the corresponding target sites. In other words, target sites created during a period of active TE expansion would represent novel, lineage-specific targets for a miRNA family with previously established regulatory functions. In addition to miRNA target sites, data also suggest that a subset of miRNA genes are themselves TE-derived (6,10,11,23). Because miRNAs act through complementary base-pair interactions, target sites for these miRNAs could arise through the transposition of homologous TEs into 3′ UTRs. To search for such interactions, we focused specifically on miRNAs where the sequence alignment to the TE overlaps the seed of the miRNA guide strand, updating the list of TE-derived miRNAs previously described (6,10,11,23). In line with previous observations, ∼20% of human miRNA genes overlapped RepeatMasker track annotations (Supplementary Material, Fig. S4).
While most miRNAs with TE homology are of relatively recent origin, one notable example with broad conservation is miR-28 (11). Inspection of this locus indicates that tandem inverted copies of the 3′ end of a LINE2c retrotransposon formed the 5′ and 3′ arms of the miRNA precursor (Supplementary Material, Fig. S5A). A recently published study suggested that miR-28-5p binds to and guides endonucleolytic cleavage of the LYPD3 transcript, interacting with the transcript through a ‘centered-seed’ binding site residing in a homologous LINE2 element (14). LINE2 retrotransposons also accounted for the greatest fraction of miR-28-5p TE-derived sites (Supplementary Material, Fig. S5B). In support of the previous observations, a luciferase reporter expressing the LYPD3 3′ UTR was repressed when co-transfected with a miR-28-5p mimic (Supplementary Material, Figs. S5C and S5D). Similar responses were observed using 3′ UTR reporters for E2F6, within which miR-28-5p is also predicted to bind through an L2 sequence (Supplementary Material, Figs. S5C and S5D). These data demonstrate that conserved miRNA-mediated regulation can arise through concomitant, TE-dependent miRNA and target site creation.
Validation of Alu-derived miR-1285
Although miR-28 is a functional, TE-derived miRNA, most TE-derived miRNAs are not well-conserved (11). To test the activity of primate-specific TE-derived miRNAs, we used Alu-derived miR-1285 as a case study. There are two proposed hsa-miR-1285 loci (miR-1285-1 and miR-1285-2; Fig. 5A). The two miRNAs share a common mature sequence and homology with Alu elements but differ in the primary sequence and secondary structure of their stem-loop precursors (Fig. 5B). Small RNA sequencing data available from MiRBase included 533 reads mapping to the mature miR-1285 sequence, common to both miR-1285 paralogs (Fig. 5B). Because one cannot discriminate between miR-1285-1 and miR-1285-2 based on the mature miRNA reads, we used reads that mapped uniquely to precursors as evidence for expression. A total of 312 reads mapped specifically to miR-1285-1, whereas only 57 mapped to miR-1285-2, suggesting that expression was primarily from the miR-1285-1 locus (Fig. 5B).
Figure 5.

Alu-derived hsa-miR-1285-1 regulates homologous Alu-derived target sites. (A) Genome browser views of miR-1285-1 and miR-1285-2 loci are shown, including any overlapping transcript or RepeatMasker annotations. The miR-1285 precursor locus (light blue rectangle with arrows) is drawn to include the additional ∼200 flanking bases (light blue, no arrows). The miR-1285-1 isoform is located within an intron of the KRIT1 mRNA. (B) The most stable predicted secondary structure for human miR-1285-1 and miR-1285-2 precursors is reproduced from those provided in the miRBase repository. High-throughput sequencing reads mapping to the two loci were also taken from miRBase, and the reads mapping to the common mature miRNA sequence are shown, along with the total read counts. The mature miRNA sequence, as reported in miRBase, is highlighted in green. The Venn diagram summarizes the number of reads uniquely mapping to each locus. The area of the circles and region of overlap is drawn in proportion to the number of reads contained in each group. (C) A cartoon representation of the miR-1285 expression constructs is drawn. The coloring scheme is reproduced from Figure 5A. (D) Luciferase reporters expressing artificial 3′ UTR target sites for miR-1285 (miR1285_2xT), or a seed-mutant control (MUT-miR1285-2xT), were transfected into HEK293 cells along with the indicated doses of the miR-1285-1 (1285) and control (ctrl) expression plasmids (shown in A), and luciferase activity was measured 48 h later. Luciferase activity is represented as the measured ratio between Renilla and firefly luciferase. Predicted hybridizations between the miR-1285 mature sequence and the artificial target and mutant sites are shown below the corresponding luciferase data. (E) Luciferase reporters expressing the 3′ UTR of putative miR-1285 targets, ADAMTS17, CHST6 and EIF2S3, were co-transfected with the indicated concentrations of an artificial miR-1285 (1285) or control (ctrl) Pre-miR oligo. (C–E) N = 3 biological replicates with three technical replicates per transfection; error bars = SD. *P ≤ 0.05 (ANOVA; Tukey's post hoc).
We next tested whether a functional miRNA is generated for either putative miRNA. Recent work validating mouse miRNA annotations suggested that a miRNA must be processed from the context of its genomic locus and loaded into silencing complexes to be considered a bona fide miRNA (24). We employed a similar strategy to assess miR-1285 functionality and cloned the precursor hairpins from miR-1285-1 and miR-1285-2, including ∼200 nt of flanking sequence into expression plasmids (Fig. 5C). HEK-293 cells were transfected with 0, 100 or 200 ng of either miR-1285-1 or miR-1285-2, balanced with a control plasmid lacking a miRNA. All plasmids were co-transfected with their corresponding artificial target site or seed-mutant reporters (Supplementary Material, Fig. S3). In agreement with the small RNA sequencing data, we found that miR-1285-1 but not miR-1285-2 reduced expression of the artificial reporter (Fig. 5D). Neither miR-1285-1 nor miR-1285-2 significantly altered luciferase activity from the seed-mutant reporter, demonstrating that miR-1285-1 construct functions as a miRNA, silencing in a seed-dependent manner (Fig. 5D). Together, these data suggest that miR-1285-1 and not miR-1285-2 is a functional miRNA.
We next tested whether putative target genes respond to miR-1285-1 over-expression. The majority of miR-1285-1 target sites were located in Alus (Supplementary Material, Table S3), and so luciferase reporters with Alu-derived target sites, including EIF2S3, CHST6 and CBFA2T2, were assessed for knockdown. As predicted, over-expression of a miR-1285-1 mimic significantly reduced expression of the three reporter plasmids tested (Fig. 5E). Together, these data demonstrate that a miRNA gene and corresponding target sites can arise from the transposition of homologous Alu elements.
DISCUSSION
In this work, we demonstrate that the most prevalent TE families in the human genome, namely Alu, MIR and LINE2 elements, provide a platform for miRNA-mediated regulation when resident in mRNA 3′ UTRs. We also found that while the majority of TE-derived target sites in human 3′ UTRs reside in primate-specific L1 and Alu elements, sequence conservation and potent activity were also evident in the MIR-derived let-7 target sites.
Recent efforts have moved from in silico miRNA target prediction algorithms to directly profiling miRNA–target interactions using crosslinking and immunoprecipitation (e.g. HITS-CLIP) technologies coupled with high-throughput sequencing (25). In these studies, after crosslinking ribonucleoprotein complexes, AGO proteins are immunoprecipitated along with any associated RNA molecules. The associated RNAs, which are subjected to RNA library preparation and high-throughput sequencing, provide a profile of miRNA-binding sites across the transcriptome. With relevance to this work, one complication in managing HITS-CLIP (or data from similar protocols) data is with reads mapped to multiple positions, something that will arise when studying repetitive sequences. One common solution is to ignore reads that map to multiple loci. However, given that we identify functional target sites residing in repetitive loci, it is possible that the filtering step would cause some TE-derived sites to be missed.
Further inquiry into the extent of Alu-derived target site efficacy will undoubtedly benefit from high-throughput approaches of measuring gene expression changes after modulating miRNA levels, such as that seen in the microarray experiments queried in this work. The low degree of sequence divergence among the 3′ UTR-resident Alus leads to a preponderance of predicted sites for some miRNAs. As a consequence of their limited divergence from parental Alu sequences, distinct miRNAbinding sites cluster in specific Alu primary sequence regions (Fig. 3B). Although on average, Alu-derived sites had lower potency than canonical (non-TE-derived) sites, evidence from the array data and our luciferase results show that some are functional.
The Hoffman et al. study reported relatively low potency of Alu-derived target sites and explored, computationally, possible explanations for their observations. They reported that Alus tend to reside towards the center of 3′ UTRs, whereas earlier studies of miRNA function showed that potent target site loci most often lie in the 5′ or 3′ ends of the 3′ UTRs. They also show that the secondary structure of the Alu sequences makes some target sites relatively inaccessible to the miRNA machinery. Aside from secondary structure, inactivity of Alu target sites could result from the fact that Alus associate with Signal Recognition Particle (SRP9/14) proteins (26–28). If SRP binding occurs in the putative target sites, miRNA access may be shielded. In a previous study, in vitro transcription of chloramphenicol acetyltransferase mRNAs with artificial 5′ or 3′ UTR Alus transcribed in the sense orientation was bound by the SRP complex (26). If SRP binding occurs in the setting of the endogenous transcript and blocks miRNA association, we would predict that miRNAs predominantly targeting the antisense Alu would be less affected. Future work to characterize SRP9/14 binding activity in human mRNAs would have intriguing implications for gene regulation and would allow for direct testing of whether miRNA associations are affected.
Considering miRNA genes, of the ∼1.2 million Alu copies present in the human genome, <20 are expected to produce mature miRNAs, and the functionality of most Alu-derived miRNAs remains untested. In this work, we found that of the two miR-1285 loci, only miR-1285-1 produces an active mature sequence. Further support for TE-derived miRNA function was found with LINE2-derived miR-28, which is well-conserved and silences LINE2-resident target sites. Thus, Alu-derived miRNAs or other miRNAs with low apparent sequence conservation deserve closer scrutiny. One difficulty in correctly annotating Alu-derived miRNAs arises from a recent observation that DICER1 degrades Alu RNAs (29). Thus, some Alu-derived small RNAs are DICER-dependent degradation products rather than miRNAs. These results emphasize the importance of wet lab experiments for validating Alu-derived miRNAs, such as those presented here.
In summary, we provide evidence that several TE-derived miRNAs and miRNA-binding sites are conserved and capable of mediating silencing, with evidence from reporters and global transcript expression profiles. Taken together, our data support that TEs have been important in the evolution of human miRNA interactions and suggest that novel miRNA functions may continue to arise as active transposition persists.
MATERIALS AND METHODS
Annotation of 3′ UTR-TEs and TE-derived target sites
Gene coordinates, sequences and annotations for human (GRCh37/hg19) and mouse (NCBI37/mm9) were obtained from the UCSC Genome Browser tracks. Information regarding TE coordinates and related information was taken from the RepeatMasker track. Simple repeats, low complexity regions and other non-TE repeats were not included in downstream analyses. Tools at the Galaxy web server were used to intersect TE and 3′ UTR genomic coordinates (www.usegalaxy.org).
Target site prediction and feature annotation
MiRNA target site predictions in human 3′ UTRs were generated for all human miRNA seed families using the standalone implementation of TargetScan 5.1 (30). From the TargetScan output, genomic coordinates were calculated and duplicate sites removed. Target coordinates were intersected with RepeatMasker intervals using the Galaxy server, requiring at least one base-pair of overlap (31). Intervals corresponding to Alu-derived target sites were selected using the TE, target and RepeatMasker alignment information to calculate target site positions relative to the Alu consensus alignment. Briefly, target positions within the host Alu feature were calculated using the genomic coordinates for each, in addition to the orientation and alignment information provided in the RepeatMasker track annotations.
Alu-derived target site positional enrichment relative to Alu consensus
After TargetScan binding sites were intersected with RepeatMasker annotations, target site positions were calculated in relation to the consensus TE alignment. RepeatMasker track data contain consensus alignment positional information. The target site position relative to this consensus alignment was calculated by finding the target position in relation to the TE feature and then adjusting to account for the TE alignment to the consensus.
Generating unique miRNA target site coordinates
When using unique miRNA seed families and sequences in the initial target prediction, target site coordinate redundancy (i.e. same chromosome, start, end and strand) results from overlapping mRNA isoforms with distinct accession numbers. With TE-derived miRNA predictions, a second source is the partial overlap of RepeatMasker annotations. Here, redundant target sites were collapsed, with the exception of those resulting from TE overlaps.
TE annotations of miRNA genes
Genomic coordinate and sequence data for human miRNA hairpins was obtained from the miRBase FTP repository (Version 15). These sequences represent the pre-miRNA plus additional flanking sequence 3′ and 5′ of the DROSHA cleavage site, but are not intended to represent the full pri-miRNA. To determine TE overlap with the mature miRNA, TE and pre-miRNA coordinates were intersected using genomic interval functions on the Galaxy web server. Finally, the local positions of the mature miRNA within the precursor were used to find miRNAs for which the TE completely overlapped the miRNA seed sequence (positions 2–8 of the mature miRNA).
Detailed positional analysis of Alu-derived miRNAs
Genomic coordinates for Alu-derived target sites were extracted, along with RepeatMasker track annotations for the associated TE. These annotations provide a summary of a sequence alignment between the Alu and the corresponding Alu family consensus. The target site position relative to the genomic Alu start position was first calculated and then adjusted according to the alignment start/stop positions provided in the track data.
Target prediction in active Alus
Genomic coordinates and sequences of potentially active Alu elements were taken from Supplementary Material, Table S4 published by Bennet et al. (22). Target sites were predicted in the Alu sequences, as well as their reverse complements, using the methods described earlier.
Microarray analysis
With the exception of the let-7 experiment, preprocessed microarray fold change values were obtained from the Supplementary Data 4 table in Garcia et al. (32). The original data are available from NCBI GEO using the accession numbers given in Supplementary Material, Table S7. Data series GSE8501 contains the experimental data for miR-122 (GSM210901), miR-128 (GSM210903) and miR-132 (GSM210904). GSE2075 contains data for miR-1 (GSM37599). Experimental data for the let-7 experiment (GSE2918) were obtained from the NCBI Gene Expression Omnibus and analyzed using the GEO2R tool. Gene expression changes (log2 scale) were calculated for let-7 over-expression (GSM63477, GSM63479 and GSM63480) or inhibition (GSM63471, GSM63472 and GSM63473) relative to the corresponding control conditions (GSM63481, GSM63482, GSM63483 and GSM63474, GSM63475, GSM63476, respectively). Probe sets were annotated and grouped according to TE and target site presence or absence in the associated RefSeq 3′ UTRs. K-S tests for significant differences between groups were performed using R.
Cloning 3′ UTR reporters
All 3′ UTR reporter constructs were based on the psiCHECK™-2 (Invitrogen) dual-luciferase system, with the 3′ UTR of interest cloned into the XhoI/NotI cloning site 3′ of the Renilla luciferase stop codon and 5′ of the SV40 poly-A signal. 3′ UTR sequences were cloned from genomic DNA isolated from HEK293 (human) or BEND3 (mouse) cell lines, using Qiagen DNA extraction kits. PCR primers with appropriate restriction sites were designed to flank the longest RefSeq-annotated isoform containing the TE/target of interest (Supplementary Material, Table S8). Phusion® Hi-Fidelity DNA Polymerase (New England Biolabs) was used to perform the PCR amplification. Standard cloning protocols were subsequently followed to restriction-digest and then ligate the vector and inserts. Proper insert sequence and orientation were confirmed both by analytical restriction digests and direct Sanger sequencing.
Artificial miRNA target sites were all based on two tandem copies of the reverse-complemented mature miRNA sequence of interest, separated by a short linker sequence containing an AgeI restriction site to facilitate downstream screening (Supplementary material, Fig. S5). The sequence was modified to introduce mismatches near the center of each site and in any locations where other miRNAs had potential seed pairing. The resulting sequences were ordered as pairs of synthetic DNA oligonucleotides (integrated DNA technologies) that when subsequently annealed formed the artificial sites with 5′XhoI/3′NotI half-sites. T4-polynucleotide kinase (T4-PNK) was used to phosphorylate the 5′ ends of the annealed pairs, which then served as the insert for the downstream cloning protocol in the same manner as mentioned earlier.
Cloning endogenous microRNAs
Endogenous miRNAs were PCR-amplified from human genomic DNA (HEK293 cells) using primers designed to flank the 5′ and 3′ ends of the annotated hairpin by at least 200bp on each end. PCR products were subcloned into PCR Blunt II TOPO plasmids, using standard protocols. After sequence verification, the TOPO plasmids served as a template for a second PCR reaction using primers nested within the original insert, containing the XhoI and SalI restriction sites and producing a product containing the miRNA hairpin ± 200 bp. Standard cloning protocols were then followed to clone the insert into the CMV promoter-driven expression plasmid (pFB-AAV-miRNA-pA).
Cell culture and transfections
HEK293 and HeLa cells were cultured in DMEM (10% FBS) without antibiotics. Approximately 24 h prior to transfection, cells were seeded onto 24-well plates. Transfections were performed in triplicate, using 5 ng luciferase reporter per reaction. Artificial miRNA mimics (Pre-miRs™) or Anti-miRs™ (Ambion®) were transfected using Lipofectamine 2000 in Optimem, at final concentrations ranging between 0 and 50 nM. All reactions were balanced with a negative control (NC#1) such that the final concentration of the combined oligonucleotides equaled that of the highest dose of the test miRNA. Media was completely removed from cells prior to adding the transfection complexes, which were combined with an equal volume of DMEM (10% FBS) just before plating. Cells co-transfected with miRNA mimics and luciferase reporters were harvested 24 h later. For all other conditions, cells were harvested 36–48 h post-transfection.
Luciferase assays
Luciferase assays were carried out using the Dual-Luciferase Assay Kit (Promega) using the standard protocol. Briefly, transfected cells on a 24-well plate were lysed by removing the media and adding 100 µl of 1× Passive Lysis Buffer to each well. Cells were rocked gently for 15 min. And then 10 µl of lysate was taken from each well and added to the bottom of opaque, flat-bottom 96-well plates. Luciferase substrates for firefly (1× Luciferase Assay Reagent II) and Renilla (1× Stop & Glo) were prepared as indicated in the manual. A Glomax 96-well Plate injector/reader (Promega) was used to inject 50 µl of substrate sequentially, reading for 2 s after each injection.
CONTRIBUTIONS
R.M.S. and B.L.D. conceived of the project and wrote the manuscript, and with C.O. designed and interpreted the experiments. R.M.S. performed the computational analyses and with C.O. performed the laboratory experiments.
SUPPLEMENTARY MATERIAL
FUNDING
Support for this was from the National Institute of Health and the Roy J. Carver Trust.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of the Davidson, McCray and Xing labs for their suggestions and helpful insights, particularly Sarah Fineberg, Ji Wan and Glen Borchert helped in the early stages of this project. We also thank Evelyn Anderson, Zachary Bursetin, Allison Carrol, Nate Davidson and Brian Wall for their technical contributions.
Conflict of Intrest statement: None declared.
REFERENCES
- 1.Xing J., Witherspoon D.J., Ray D.A., Batzer M.A., Jorde L.B. Mobile DNA elements in primate and human evolution. Am J Phys Anthropol. 2007 doi: 10.1002/ajpa.20722. Suppl 45, 2–19. [DOI] [PubMed] [Google Scholar]
- 2.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Cordaux R., Batzer M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubin C.M., VandeVoort C.A., Teplitz R.L., Schmid C.W. Alu repeated DNAs are differentially methylated in primate germ cells. Nucleic Acids Res. 1994;22:5121–5127. doi: 10.1093/nar/22.23.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estécio M.R., Gallegos J., Dekmezian M., Lu Y., Liang S., Issa J.P. SINE retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol. Cancer Res. 2012;10:1332–1342. doi: 10.1158/1541-7786.MCR-12-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borchert G.M., Lanier W., Davidson B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 7.Zamore P.D. RNA silencing: genomic defence with a slice of pi. Nature. 2007;446:864–865. doi: 10.1038/446864a. [DOI] [PubMed] [Google Scholar]
- 8.Okamura K., Lai E.C. Endogenous small interfering RNAs in animals. Nat. Rev. Mol. Cell Biol. 2008;9:673–678. doi: 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smalheiser N.R., Torvik V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–326. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Piriyapongsa J., Jordan I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One. 2007;2:e203. doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piriyapongsa J., Marino-Ramirez L., Jordan I.K. Origin and evolution of human microRNAs from transposable elements. Genetics. 2007;176:1323–1337. doi: 10.1534/genetics.107.072553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krichevsky A.M., Metzer E., Rosen H. Translational control of specific genes during differentiation of HL-60 cells. J. Biol. Chem. 1999;274:14295–14305. doi: 10.1074/jbc.274.20.14295. [DOI] [PubMed] [Google Scholar]
- 13.Filshtein T.J., Mackenzie C.O., Dale M.D., Dela-Cruz P.S., Ernst D.M., Frankenberger E.A., He C., Heath K.L., Jones A.S., Jones D.K., et al. OrbId: origin-based identification of microRNA targets. Mob. Genet. Elements. 2012;2:184–192. doi: 10.4161/mge.21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin C., Nam J.W., Farh K.K., Chiang H.R., Shkumatava A., Bartel D.P. Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell. 2010;38:789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borchert G., Gilmore B., Spengler R., Xing Y., Lanier W., Bhattacharya D., Davidson B. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet. 2009;18:4801–4807. doi: 10.1093/hmg/ddp443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smalheiser N.R., Torvik V.I. Alu elements within human mRNAs are probable microRNA targets. Trends Genet. 2006;22:532–536. doi: 10.1016/j.tig.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Smalheiser N.R., Torvik V.I. Complications in mammalian microRNA target prediction. Methods Mol. Biol. 2006;342:115–127. doi: 10.1385/1-59745-123-1:115. [DOI] [PubMed] [Google Scholar]
- 18.Chen J., Bardes E.E., Aronow B.J., Jegga A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nuc. Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman Y., Dahary D., Bublik D.R., Oren M., Pilpel Y. The majority of endogenous microRNA targets within Alu elements avoid the microRNA machinery. Bioinformatics. 2013;29:894–902. doi: 10.1093/bioinformatics/btt044. [DOI] [PubMed] [Google Scholar]
- 21.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 22.Bennett E.A., Keller H., Mills R.E., Schmidt S., Moran J.V., Weichenrieder O., Devine S.E. Active Alu retrotransposons in the human genome. Genome Res. 2008;18:1875–1883. doi: 10.1101/gr.081737.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borchert G.M., Holton N.W., Williams J.D., Hernan W.L., Bishop I.P., Dembosky J.A., Elste J.E., Gregoire N.S., Kim J.A., Koehler W.W., et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elements. 2011;1:8–17. doi: 10.4161/mge.1.1.15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chi S.W., Zang J.B., Mele A., Darnell R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu K., Chang D.Y., Maraia R.J. Human signal recognition particle (SRP) Alu-associated protein also binds Alu interspersed repeat sequence RNAs. Characterization of human SRP9. J. Biol. Chem. 1995;270:10179–10186. doi: 10.1074/jbc.270.17.10179. [DOI] [PubMed] [Google Scholar]
- 27.Chang D.Y., Hsu K., Maraia R.J. Monomeric scAlu and nascent dimeric Alu RNAs induced by adenovirus are assembled into SRP9/14-containing RNPs in HeLa cells. Nucl. Acids Res. 1996;24:4165–4170. doi: 10.1093/nar/24.21.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bovia F., Wolff N., Ryser S., Strub K. The SRP9/14 subunit of the human signal recognition particle binds to a variety of Alu-like RNAs and with higher affinity than its mouse homolog. Nucl. Acids Res. 1997;25:318–326. doi: 10.1093/nar/25.2.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaneko H., Dridi S., Tarallo V., Gelfand B.D., Fowler B.J., Cho W.G., Kleinman M.E., Ponicsan S.L., Hauswirth W.W., Chiodo V.A., et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010 doi: 10.1002/0471142727.mb1910s89. Chapter 19, Unit 19 10 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.