

Abstract
Duchenne muscular dystrophy (DMD) is caused by a lack of the dystrophin protein and has no effective treatment at present. Zebrafish provide a powerful in vivo tool for high-throughput therapeutic drug screening for the improvement of muscle phenotypes caused by dystrophin deficiency. Using the dystrophin-deficient zebrafish, sapje, we have screened a total of 2640 compounds with known modes of action from three drug libraries to identify modulators of the disease progression. Six compounds that target heme oxygenase signaling were found to rescue the abnormal muscle phenotype in sapje and sapje-like, while upregulating the inducible heme oxygenase 1 (Hmox1) at the protein level. Direct Hmox1 overexpression by injection of zebrafish Hmox1 mRNA into fertilized eggs was found to be sufficient for a dystrophin-independent restoration of normal muscle via an upregulation of cGMP levels. In addition, treatment of mdx5cv mice with the PDE5 inhibitor, sildenafil, which was one of the six drugs impacting the Hmox1 pathway in zebrafish, significantly increased the expression of Hmox1 protein, thus making Hmox1 a novel target for the improvement of dystrophic symptoms. These results demonstrate the translational relevance of our zebrafish model to mammalian models and support the use of zebrafish to screen for new drugs to treat human DMD. The discovery of a small molecule and a specific therapeutic pathway that might mitigate DMD disease progression could lead to significant clinical implications.
INTRODUCTION
Muscular dystrophy is a muscle-degenerative disease in which the muscle at first forms normally, but starts to degenerate faster than it can be repaired. The most common form of muscular dystrophy is Duchenne muscular dystrophy (DMD) representing over 90% of the diagnosed cases. In 1986, mutations in the dystrophin gene were found in our laboratory to be the cause of both Duchenne and Becker Muscular Dystrophy (1,2). The gene encodes a large 427-KDa protein that positions just inside of the sarcolemmal membrane and links the internal cytoskeleton with the muscle cell membrane (3,4). This linkage is vital to maintaining muscle membrane integrity during repeated cycles of cell contraction. Almost all known human dystrophin mutations that cause DMD typically result in the loss or degradation of the dystrophin protein at the sarcolemmal membrane.
Like mammals, zebrafish express dystrophin in skeletal muscle as part of a large complex whose stability depends on the presence of dystrophin (5). Dystrophin-deficient zebrafish, sapje and sapje-like fish (6,7), recapitulate the pathogenesis and severity of human DMD by displaying extensive muscle degeneration, inflammation and fibrosis (8). The zebrafish dystrophin-associated protein complex (DAPC) localizes to the muscle cell membrane in adult fish (9). Much evidence suggests that the DAPC proteins function similarly in zebrafish as in mammals and that mis-expression of these proteins gives rise to a muscle-specific phenotype that can be scored early in zebrafish development (5–7,10,11). The fish models for muscular dystrophies present decreased birefringence and motility defects in response to touch at 4 days post-fertilization (dpf). Birefringence measures the rotation of polarized light through the transparent zebrafish embryo at the highly ordered sarcomeric structure of the somitic muscle. These ‘dystrophic’ mutants all show similar phenotypes and exhibit muscle abnormalities and low birefringence at 4 dpf, which can be detected without harming the fish.
Zebrafish are a powerful tool for high-throughput drug screening in vivo, enabling the monitoring of drug effects on the entire organism (12,13). The birefringence property of intact muscle and its alterations in dystrophin-deficient muscle (14,15) offer a noninvasive means for detecting muscle improvement in treated zebrafish. The sapje and sapje-like fish are dystrophin-null models of a recessively inherited muscle degeneration phenotype (6,7). As such, ∼25% of the offspring from crosses between heterozygous mutant fish will have dystrophic skeletal muscle, which can be detected via muscle birefringence at 4 dpf. Therefore, small molecule treatments that lead to a significant reduction in the percentage of affected offspring, as determined by more fish with normal muscle birefringence and improved survival, could be highly indicative of promising DMD therapies (16).
Currently there are no effective long-term therapies for DMD, but cyclic guanosine monophosphate (cGMP) signaling is an emerging target for DMD treatment. Phosphodiesterase 5 (PDE5) inhibition has been shown to be effective in restoring normal muscular structure in a mouse model of the disease (17–21). Previously, we reported that aminophylline, a nonselective PDE inhibitor, had potential to improve dystrophic muscle in dystrophin-deficient fish (16). To identify additional compounds that improve the dystrophic phenotype in dystrophin-deficient fish and to unveil therapeutic pathways that might be targeted for DMD therapy, we used a drug-screening approach on two zebrafish models of dystrophin deficiency, sapje and sapje-like.
In this article, we have demonstrated that an additional four compounds all target heme oxygenase signaling, thereby rescuing the abnormal sapje and sapje-like muscle phenotype by the upregulation of the levels of heme oxygenase 1 protein (Hmox1). In addition, treatment of mdx5cv DMD model mice with one of these four drugs, the PDE5 inhibitor sildenafil, also significantly increased the expression of Hmox1 protein while alleviating the mouse dystrophic phenotype. These results point to Hmox1 as a novel target for the improvement of dystrophic symptoms. This also demonstrates the use of zebrafish in the discovery of small molecules and a specific therapeutic pathway that might mitigate DMD disease progression and could carry significant clinical implications.
RESULTS
Therapeutic drug screening using sapje and sapje-like fish
To identify additional candidate drugs that modulate the skeletal muscle phenotype in dystrophin-null fish, we screened two chemical libraries: the NINDS 2 Compound library (1040 chemicals) and the Institute of Chemistry and Cell Biology (ICCB) Bioactive molecule library (480 chemicals). Using a previously published screening strategy (16), eight additional compounds (Nos. 8–15 in Table 1) were identified that decreased the number of zebrafish with abnormal birefringence phenotype at 4 dpf following sapje heterozygous mating (Supplementary Material, Table S1). Similar to the previously published effective compounds (Nos. 1–7 in Table 1) (16), the eight newly identified chemicals resulted in improved muscle phenotype in both dystrophin-deficient sapje and sapje-like mutant zebrafish. These compounds were effective without restoring dystrophin expression, when compared with expression in non-treated sapje and sapje-like zebrafish (Supplementary Material, S1). Out of the total 2640 chemicals screened in this and previous publications (16), 15 chemicals showed potential to maintain normal muscle structure in the absence of dystrophin protein expression (Table 1).
Table 1.
Candidate drugs from chemical screens using DMD model fish
| No. | Chemical name | Chemical library |
|---|---|---|
| 1 | Epirizole | Prestwick collection 1 |
| 2 | Homochlorcyclizine | Prestwick collection 1 |
| 3 | Conessine | Prestwick collection 1 |
| 4 | Aminophylline | Prestwick collection 1 |
| 5 | Equilin | Prestwick collection 1 |
| 6 | Pentetic acid | Prestwick collection 1 |
| 7 | Proscillaridin A | Prestwick collection 1 |
| 8 | Nitromide | NINDS 2 Compound library |
| 9 | Propantheline bromide | NINDS 2 Compound library |
| 10 | Androsterone acetate | NINDS 2 Compound library |
| 11 | Crassin acetate | NINDS 2 Compound library |
| 12 | Pomiferin | NINDS 2 Compound library |
| 13 | Sildenafil citrate | NINDS 2 Compound library |
| 14 | Cerulenin | ICCB Known Bioactives Library |
| 15 | 9a,11b-Prostaglandin F2 | ICCB Known Bioactives Library |
Candidate pathways impacted by six compounds from our screening
We screened 2640 FDA-approved drugs whose mechanisms of action are often well characterized, allowing us to identify converging molecular pathways. Of the fifteen total candidate drugs result from our drug screens, we identified six compounds that target heme oxygenase signaling (equilin, crassin acetate, prostaglandin, aminophylline, sildenafil citrate and cerulenin) (Fig. 1A). By targeting various components of the heme oxygenase pathway (Fig. 1A), these six drugs likely act to increase the levels of cGMP and cyclic adenosine monophosphate (cAMP). Equilin, crassin acetate and prostaglandin activate the inducible Hmox1; aminophylline is a nonselective PDE inhibitor; sildenafil citrate is a specific PDE5 inhibitor and cerulenin is involved in the regulation of PKA (16,17,22–25).
Figure 1.

Dystrophin-independent muscle improvement in sapje-like fish. Compounds targeting the heme oxygenase pathway found to be efficacious in sapje models of DMD were each administered to forty offspring of heterozygous sapje-like matings. Following 4 days of treatment, fish genotypes, muscle phenotypes and dystrophin expression were determined using Sanger sequencing, birefringence and immunostaining, respectively. (A) Efficacious drugs identified in high-throughput zebrafish screens converge on the heme oxygenase pathway. (B) Immunohistochemistry of the muscle of dystrophin−/− fish demonstrates that muscle improvement is independent of dystrophin expression. WT, wild type; NT, non-treatment and drug treatment listed. (C) 30–90% of dystrophin−/− fish were restored in at 4 dpf (P = 1.49e-02, chi2 test). Cerulenin was the most efficacious drug with 90% recovery rate (P < 1e-07, two proportion test).
To show that the improved muscle phenotype is independent of the specific genomic dystrophin mutation, these six efficacious drugs were first screened in sapje fish and then confirmed in the second allele of dystrophin deficiency, sapje-like. Immunohistochemistry and genotyping of treated fish demonstrated that mutant fish had become apparently unaffected through a mechanism that does not restore dystrophin expression; therefore, the treatment mechanism is dystrophin independent (Fig. 1B and C).
To determine whether the drugs were able to reverse abnormal muscle, dystrophin-null fish as determined by abnormal birefringence at 4 dpf were treated starting from 5 to 20 dpf with the selected drugs. Four of six candidate compounds (aminophylline, sildenafil citrate, cerulenin and crassin acetate) were effective in increasing the number of sapje fish, which survived long term (Fig. 2A). Interestingly, the treatment with a combination of aminophylline and sildenafil citrate had better survival results than either compound administered alone or in combination with any of the others (Fig. 2A). The surviving dystrophin-null fish were analyzed by immunostaining with anti-myosin heavy chain to confirm that chemically treated fish have improved muscle structure and also analyzed by immunoblotting to look at the expression of Hmox1. Myosin heavy chain staining indicated that aminophylline-, sildenafil citrate-, crassin acetate- and cerulenin-treated sapje fish showed improved muscle structure compared with those of untreated dystrophin-null fish (Fig. 2B). To analyze the effects on vasculogenesis in the dystrophic zebrafish using the candidate drugs, wild-type, untreated dystrophin-null fish and candidate chemical-treated dystrophin-null fish were immunostained with anti-VEGF, a marker of the developing vasculature in zebrafish (26). In dystrophin-null fish, the blood vessel structure was not clearly defined when compared with those of wild type. In chemically treated dystrophin-null fish, the structure was clearly defined and was similar to that of wild type (Supplementary Material, S2). These results indicated that the structure of blood vessels might be influenced indirectly by treatment with these drugs in the dystrophin-null fish when compared with the untreated dystrophin-null fish (Supplementary Material, S2). Moreover, immunoblotting of these chemically corrected fish clearly showed that improved fish also exhibited significantly increased expression of Hmox1 at 20 dpf (Fig. 2C). These results suggested that these four compounds impact the Hmox1 pathway by increasing Hmox1 expression and improving survival of dystrophin-null fish in the long term.
Figure 2.

Dystrophic muscle restoration via increased heme oxygenase signaling. (A) Treatment with multiple components of the Hmox1 signaling pathway results in increased survival of dystrophin-null fish, sapje fish (P = 1.16e-05, repeated-measures ANOVA. *For aminophylline, sildenafil: P ≤ 0.05, **for aminophylline + sildenafil: P ≤ 0.01, ***for cerulenin, crassin acetate: P ≤ 0.005, n = 10 in triplicate). (B) Normal skeletal muscle structure in treated dystrophin-null fish, sapje fish (20 dpf). Skeletal muscle in treated dystrophin-null fish was immunostained with anti-myosin heavy chain. Arrows point to common breaks in untreated dystrophic muscle. (C) Upregulation of Hmox1 expression in surviving chemically treated dystrophin-null fish. Hmox1 expression in surviving chemically treated dystrophin-null fish was analyzed with immunoblot (n = 3). Treatment with each candidate drug (aminophylline: P = 0.038, sildenafil: P = 0.012, crassin: P = 0.0095 and cerulenin: P = 0.045, compared with those of untreated dystrophin-null fish, t-test, n = 3) upregulated Hmox1 expression. (D) Birefringence phenotype rate in sapje-like fish and an anti-sense morpholino oligonucleotides-injected sapje-like fish after treatment with candidate drugs. Treatment with candidate drugs (aminophylline, sildenafil, cerulenin and crassin acetate) has significant effects on the improvement of reduced birefringence phenotypes in dystrophin-deficient skeletal muscle (40 fish in triplicate). After injection of an anti-sense morpholino oligonucleotides (MO) against the expression of zebrafish Hmox1 to inhibit zebrafish Hmox1 expression, the treatment with candidate drugs does not have significant effects on the improvement of reduced birefringence in skeletal muscle. (Uninjected fish versus Hmox1-MO-injected fish; aminophylline: P = 0.00079, sildenafil: P = 0.0043, crassin acetate: P = 0.027 and cerulenin: P = 0.028, t-test, n = 3). (E) Measurement of cGMP levels in sapje-like fish and Hmox1 MO-injected sapje-like fish after treatment with candidate drugs. Treatment with candidate drugs (aminophylline, sildenafil, crassin acetate and cerulenin) significantly has effects in increasing cGMP level (40 fish in triplicate). After injection of an MO against the expression of zebrafish Hmox1, the treatment with candidate drugs still shows strong effects in increasing cGMP levels to almost normalized levels of the uninjected fish. (Uninjected fish versus Hmox1-MO-injected fish; untreated fish: P = 0.040, aminophylline: P = 0.00028, sildenafil: P = 0.000018, crassin acetate: P = 0.044 and cerulenin: P = 0.010, t-test, n = 3).
To confirm that the candidate drugs influence on the cGMP levels and the muscle phenotypes, cGMP level and birefringence phenotypes in chemically treated dystrophin-null fish were analyzed and quantified. Treatment with our candidate drugs had significant effects in increased levels of cGMP and the improvement of abnormal birefringence in skeletal muscle (Fig. 2D and E). An anti-sense morpholino oligonucleotides (MO) against the expression of zebrafish Hmox1 was injected to 1 cell-stage of embryos from heterozygous sapje-like fish mating to inhibit the expression of Hmox1. With the zebrafish Hmox1 MO injection, Hmox1 expression and cGMP levels were reduced in MO-injected dystrophin-null fish (Supplementary Material, S3 and Fig. 2D). Treatment of Hmox1 morpholino-injected dystrophin-null fish with the candidate drugs did not improve the birefringence phenotype (Fig. 2E). The injection of a control morpholino did not change the results of chemical treatment with sildenafil (Supplementary Material, S4). These findings suggested that these candidate drugs acted to improve zebrafish muscle phenotype by influencing the levels of Hmox1 and cGMP.
Overexpression of zebrafish hmox1 mRNA in sapje and sapje-like fish
To determine whether increased Hmox1 expression alone is sufficient to induce the beneficial effects seen in dystrophic muscle, zebrafish hmox1 mRNA was injected into eggs from four matings of each heterozygous sapje fish and sapje-like fish. Overexpression of hmox1 in the injected sapje and sapje-like fish was confirmed by real-time PCR (Fig. 3A). Indeed, overexpression of hmox1 RNA normalized dystrophic skeletal muscle and reduced the fraction of affected fish as measured by birefringence (sapje, P = 3.48e-05; sapje-like, P = 1.0e-04; t-test, n = 3, Fig. 3B). The amount of cGMP and cAMP being expressed in the hmox1 RNA-injected sapje and sapje-like fish was also evaluated. The amount of total cGMP was significantly increased compared with uninjected sapje fish and sapje-like fish (sapje, P = 0.0078; sapje-like, P = 0.018; t-test, n = 3, (Fig. 3C). In contrast, the amount of cAMP in hmox1-injected sapje and sapje-like fish was not significantly different from normal controls (Supplementary Material, Fig. S5), suggesting that Hmox1 overexpression is likely to induce levels of cGMP and restore dystrophic muscle to normal. The improved dystrophin-null fish following hmox1 RNA injection were selected after genotyping and stained with anti-myosin heavy chain to examine their muscle structure. The muscle structure appeared similar to that of wild type, and they exhibited no abnormal muscle areas compared with untreated dystrophin-null fish (arrows in Fig. 3D).
Figure 3.

Hmox1 overexpression is sufficient for the observed muscle improvement. (A) RT-PCR confirmation of Hmox1 overexpression in sapje fish and sapje-like fish (*sapje, P = 9.09e-13; **sapje-like, P = 1.57e-04; t-test, n = 3). (B) Injecting 50 pg of zebrafish hmox1 mRNA into eggs from a heterozygous mating decreases the fraction of affected offspring (*sapje, P = 3.48e-05; **sapje-like, P = 1.0e-04; t-test, n = 3). (C) Measurement of the amount of cGMP in hmox1-overexpressed sapje fish and sapje-like fish (*sapje, P = 0.0078; **sapje-like, P = 0.018; t-test, n = 3). (D) Normal skeletal muscle structure in dystrophin-null sapje fish injected with 50 pg hmox1. Restored dystrophin-null fish have normal muscle structure similar to those of wild type. Dark midlines reflect pigmentation.
Hmox1 upregulation in sildenafil-treated mdx mice
To confirm that Hmox1 upregulation also occurs following sildenafil treatment of mdx5cv mice, sildenafil citrate was administered to mdx5cv mice at a previously reported dosage that resulted in beneficial effects in mouse muscle pathology (17,18). Sildenafil citrate was dissolved in sterile-filtered water (pH 3.0) to a concentration of 400 mg/l and administered ad libitum, with treatment beginning at 4 weeks of age, and lasting for 7–8 weeks. C57BL6/J and mdx5cv vehicle controls were given untreated sterile-filtered drinking water (pH 3.0). As previously published (21), PDE5 inhibitor treatment of mdx5cv significantly reduced the number of central nuclei in muscle fibers of the diaphragm and tibialis anterior compared with those of untreated mdx mice (Fig. 4A and B). Immunoblot results of muscle from the sildenafil-treated mdx5cv showed significantly increased expression of Hmox1 (Fig. 4C). Following 6 weeks of sildenafil treatment, mice were subjected to an exercise fatigue test. After a 15-min treadmill exercise regimen, the mice were monitored for 6 min for activity measures such as the time they spent resting (Fig. 5A), the distance they moved (Fig. 5B) and the number of rearing events (Fig. 5C). In each test, there were clear differences between vehicle-treated mdx5cvand wild-type mice. Sildenafil treatment enabled significant alleviation of exercise fatigue symptoms in the mdx5cvmice, demonstrating the drug's therapeutic effect and like seen in zebrafish increased expression of Hmox1.
Figure 4.

Hmox1 expression and reduction of centrally located nuclei in mdx5cv mice following treatment with sildenafil. (A) Representative Hematoxylin and Eosin (H&E) staining of tibialis anterior (TA) and diaphragm (DIA) muscle sections in mdx5cv mouse treated with sildenafil citrate. Bars: 50 μm. (B) Mdx5cv mice showed significantly more centrally located nuclei in random non-overlapping fields of H&E-stained diaphragm muscle than in strain-matched wild type. Sildenafil showed significant reduction of central nuclei in TA and DIA muscle compared with the untreated mdx5cv TA and DIA. (n = 4, 11–12 weeks old). Mdx5cv mice were given water for 7–8 weeks at a 400 mg/l concentration of sildenafil Mdx5cv and strain-matched C57Bl6/J wild type received vehicle. *P = 0.0012, n = 4, t-test, **P = 8.98328E-07, t-test, n = 4. (C and D) Upregulation of Hmox1 expression in mdx5cv mice following treatment with sildenafil. Hmox1 expression in mice treated with sildenafil was analyzed via immunoblot (*P = 0.0015, versus wild type, t-test, n = 4).
Figure 5.
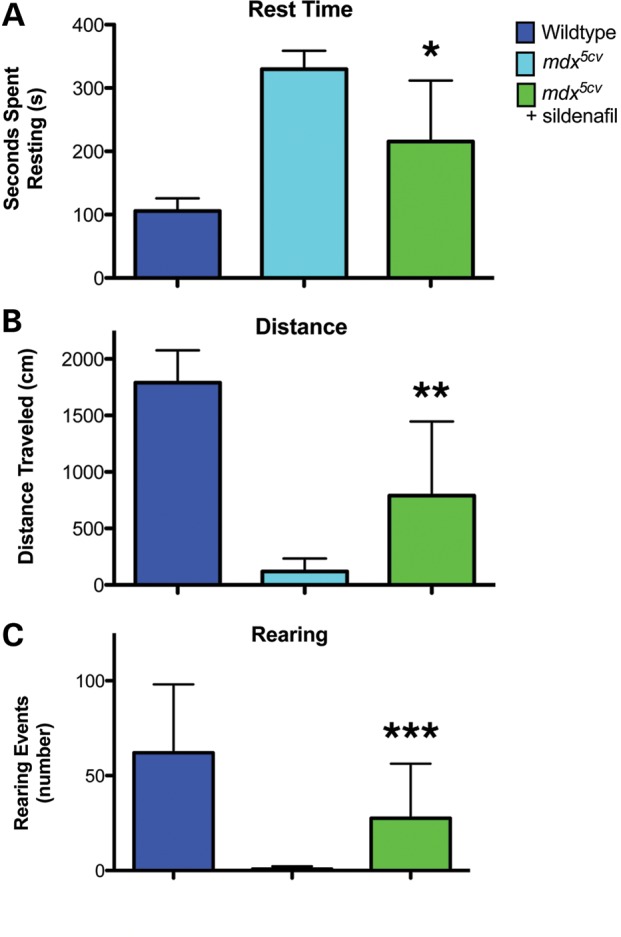
Total resting time (A), distance moved (B) and rearing events (C) improvement in exercise fatigue assay of sildenafil-treated mdx5cv mice. Activity was recorded in 10-week-old mice, following 6 weeks of drug treatment. Clear differentiation was seen in this assay after the 6-week treatment period (*P = 0.0016, **P = 0.0011, ***P = 0.03, repeated-measures ANOVA), with no large differences detected after 2 and 4 weeks of treatment. Mdx5cv mice were given water at a 400 mg/l concentration of sildenafil (green). Mdx5cv (light blue) and strain-matched C57Bl6/J wild type (dark blue) received vehicle (n = 4 mice with two females and two males.).
DISCUSSION
By using drug screens in dystrophin-deficient zebrafish, this study has identified a possible dystrophin-independent DMD therapy that converges on heme oxygenase signaling. A total of 15 drugs were identified that moderate the birefringence phenotype of two fish models of DMD, with six of these compounds are known to impact the Hmox1 pathway. Four of the Hmox1-related compounds significantly increased the number of surviving dystrophin-null fish compared with untreated dystrophin-null fish. Moreover, our findings indicate that these four compounds induce the expression of Hmox1 in surviving dystrophin-null fish. We also show that Hmox1 overexpression acts to improve zebrafish muscle phenotype and to increase the total levels of cGMP in dystrophin-null zebrafish. The compounds identified here functions to improve dystrophic muscle through the upregulation of Hmox1. The Hmox1 molecule functions by degrading the heme molecule into biliverdin-IX alpha, Fe (III) and monocarbon oxygen (27), which triggers multiple downstream effects, such as anti-inflammation, anti-oxidation and vasodilation (28–31). Oxidative stress has been shown to play an important role in the DMD disease process (4,17,20), and inflammation leads to reduced fibrosis and muscle weakness (28–32). It has been reported that the PDE5 inhibitor sildenafil induces an angiogenic response in human coronary arteriolar endothelial cells through Hmox1 and vascular endothelial growth factor, one of the important factors to induce regeneration of the vascular system and vasodilation (33). Hmox1 has also been reported to indirectly regulate the nuclear factor kappa-light-chain enhancer of activated B cells (NF-kappa B) activity (34,35), which is increased in expression in muscle biopsies of DMD patients (36). These finding combined with those presented here suggest that the modulation of the Hmox1 pathway could correct the skeletal muscle phenotype of dystrophin-deficient muscle. The modulation of the Hmox1 pathway in dystrophin-deficient muscle opens up the possibility that pathways related to anti-inflammatory, anti-oxidative and vasodilating can be additional mechanisms for treating DMD.
Previously, phosphodiesterase inhibition has been shown to improve disease course in the mdx mouse model of DMD (17–19,37). Our sildenafil-treated mdx mice revealed that this PDE5 inhibitor upregulates Hmox1 expression whereas it improves the phenotype of dystrophic skeletal muscle. Our results of the sildenafil treatment for mdx5cv mice replicate those previously reported, which also showed improvement of the diaphragm muscle pathology and exercise activities in mdx mouse (17,37). Furthermore, the restoration of the dystrophin-mediated neuronal nitric oxide synthase (nNOS) anchoring to the sarcolemma of the mdx mouse was found to enhance exercise performance by restoring activity-induced vasomodulation and subsequently minimizing muscle damage (20,38). Here we identify Hmox1 as a likely link that can trigger multiple beneficial effects. Hmox1 is an nitric oxide (NO)-inducible heme oxygenase known for its cytoprotective role, which acts as a potent anti-inflammatory and anti-oxidative stress agent, in part by elevating cGMP production (30,39). This study demonstrates that Hmox1 is a viable and sufficient target for the improvement of dystrophic muscle in zebrafish and mouse models. We have also identified several FDA-approved compounds that target multiple components of the Hmox1-mediated cGMP signaling pathway as a potential novel therapy for the treatment of dystrophin-deficient muscles.
Altogether, this study highlights the use of zebrafish to screen for disease-relevant drug compounds and it additionally defines Hmox1 upregulation as an effective pathway for both fish and mouse models for DMD. The results in these studies point to Hmox1 as a potential effective therapeutic pathway for DMD. The Hmox1 pathway and its relevant candidate drugs will likely be informative for the development of therapeutics to treat human patients of DMD.
MATERIALS AND METHODS
Zebrafish strains and fish cultures
Two zebrafish models of DMD were used for chemical screening: the sapje (stop codon in DMD exon 4) and sapje-like (splice site mutation in exon 62) fish (6,15). The muscle degeneration phenotype of these mutant fish is transmitted in a recessive manner such that 25% of the offspring show degenerative muscle symptoms after 3 dpf. Twenty pairs of heterozygous sapje or sapje-like fish were mated, and the fertilized eggs were cultured at 28.5°C. Zebrafish embryos were collected and raised at 28.5°C according to standard procedures and standard criteria under the guidelines of the Boston Children's Hospital Institutional Animal Care and Use Committee, Boston Children's Hospital animal ARP protocol (12-09-2286R).
Small molecule screening
In all, 2640 FDA-approved drugs were screened, including those in the ICCB at Harvard Medical School's NINDS 2 Compound library (1040 chemicals; Discover Systems, Inc., Gaylordsville, CT, USA), BIOMOL ICCB-Longwood Known Bioactives 3 (480 chemicals; Harvard ICCB, Boston, MA, USA), and the previously reported Prestwick 1 Collection (16). These libraries contain diverse molecules of known action, bioavailability and safety in humans and cover ∼47% of all FDA-approved drugs. Libraries were first screened in pools of 8 compounds for 4 days using 20 fish, in duplicate, as previously described (16). Muscle phenotypes were detected by a birefringence assay, which measures the rotation of polarized light through the transparent zebrafish embryos at the highly ordered sarcomeric structure of the somitic muscle (14). Following the identification of pools that had reduced fractions of fish with abnormal birefringence, each compound from these pools was tested individually on both sapje and sapje-like fish (each 20 fish) for 4 days in duplicate. Eight chemicals that significantly reduced the number of dystrophic fish were identified in the individual screens (Table 1). Aminophylline and equilin were identified in a previous screen (16) and further analyzed here as efficacious compounds targeting heme oxygenase signaling.
Long-term treatment of sapje fish with candidate chemicals
Pairs of heterozygous sapje fish were mated, and fertilized eggs were cultured at 28.5°C. Zebrafish embryos were collected and raised at 28.5°C according to standard procedures and criteria. Mutant fish showing abnormal birefringence were identified under the dissection scope at 4 dpf and placed in a new plate to be treated with individual candidate chemicals from 4 to 20 dpf. Affected and unaffected embryos were arrayed in 24-well plates and cultured in 1 ml of fish water containing individual chemicals at 28.5°C starting at 4 dpf, ten embryos each in triplicate. From 5 to 20 dpf, the fish were cultured in 100 ml of fish water containing the individual chemicals (aminophylline, crassin acetate and sildenafil citrate of 2.5 μg/ml, cerulenin of 5 μg/ml) and combination of aminophylline (2.5 μg/ml) and sildenafil citrate (2.5 μg/ml) at room temperature. The number of surviving fish was counted and recorded every other day.
Genotyping sapje and sapje-like fish
Genomic DNA was extracted from chemically treated fish using 100 mm Tris–HCl, pH 8.5; 5 mm EDTA, pH 8.0; 0.2% SDS; 200 mm NaCl. Capillary sequencing was used to detect the specific dystrophin mutation in sapje (forward primer 5′ CTGGTTACATTCTGAGAGACTTTC 3′, reverse primer 5′ AGCCAGCTGAACCAATTAACTCAC 3′) and sapje-like fish (forward primer 5′ TCTGAGTCAGCTGACCACAGCC 3′, reverse primer 5′ ATGTGCCTGACATCAACATGTGG 3′). PCR was performed with the primers noted above at 52°C for 35 cycles, purified using QIAGEN's QIAquick PCR Purification Kit, and bidirectionally sequenced on Applied Biosystems' 3730 DNA Analyzers (Life Technologies, Carlsbad, CA, USA) at The Boston Children's Hospital Molecular Genetics Core Facility.
Cloning of zebrafish heme oxygenase 1 (hmox1) cDNA
Total RNA was extracted from 4 dpf wild-type zebrafish embryos using QIAGEN's RNeasy micro kit and converted to cDNA with SuperScript III First-Strand System for RT-PCR (Life Technologies) according to the manufacturer’s protocols. The following primers were used to amplify the fish hmox1 cDNA coding sequence and added a myc tag and an EcoRI site: 5′ GAATTCATGGACTCCACCAAAAGC 3′ (forward) and 5′ GAATTCTTACAGATCCTCTTCTGAGATGAGTTTTTGTTCAAAAGCGTAAACTCCCATGCC 3′ (reverse, with myc tag and EcoRI site). The PCR product of the full-length hmox1 cDNA was cloned into a pCRII plasmid (Life Technologies). All PCR products and cloned fragments were subject to confirmatory capillary sequencing on Applied Biosystems' 3730 DNA Analyzers at The Boston Children's Hospital Molecular Genetics Core Facility.
Generation of expression constructs and in vitro transcription
Fish hmox1 full-length mRNAs with a myc tag were cloned into the pCS2+ expression vector after digestion with EcoRI. RNAs were synthesized from Asp718-digested pCS2+ plasmids using the sp6 mMessage mMachine kit (Life Technologies). Transcripts were quantified on the Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Phenol red (0.1%) was added to the RNA solution as a tracer, and 50 pg hmox1 mRNA was injected into 1-cell-stage fertilized embryos from heterozygous sapje matings. Embryos were then cultured in the Children's Hospital aquatic system at 28.5°C.
Immunohistochemistry
Immunohistochemical staining of whole mutant fish bodies was performed as previously described (16). Embryos were incubated separately with anti-dystrophin (1: 25, Sigma–Aldrich, St. Louis, MO, USA) or anti-myosin heavy chain (1: 25, F59; Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA) or anti-VEGF (1: 50, R&D system, Inc., Minneapolis, MN, USA) at 4°C overnight. After washing three times, samples were incubated with secondary antibodies and examined as previously described (16).
Western blotting
Fish or fish embryos and mouse muscle sections were homogenized in Tris-buffered saline containing RIPA buffer (Sigma–Aldrich), protease inhibitors and phosphatase inhibitors (Roche, Indianapolis, IN, USA). Standard western blotting was performed with anti-Hmox1 (1: 200, Aviva System Biology, San Diego, CA, USA), anti-beta-actin (1: 500, Sigma–Aldrich) and anti-alpha beta tubulin antibodies (1: 1000, Cell Signaling Technology, Inc., Beverly, MA, USA). Relative band intensities were quantified by densitometry using ImageJ, and fold differences determined between the Hmox1/beta-actin ratio of each treatment and that of the untreated and wild-type fish.
Morpholino oligonucleotide injection
A anti-sense morpholino oligonucleotides (MO) (GENE TOOLS LLC., Philomath, OR, USA) targeted to interfere with zebrafish Hmox1 translation was designed using the 5′ sequence around the putative translation start site of the cloned zebrafish Hmox1 mRNA. The morpholino sequences were Hmox1 MO: 5′ TTGTGCTGTAGATGTCCTGTCTTTC 3′. For control MO, we used a control morpholino (standard control oligo, GENE TOOLS): 5′ CCTCTTACCTCAGTTACAATTTAT 3′.
Morpholino was re-suspended in 1× Danieau solution [58 mm NaCl, 0.7 mm KCl, 0.4 mm MgSO4, 0.6 mm Ca(NO3)2, 5 mm HEPES; pH 7.6] with 0.1% phenol red used as an injection indicator. Morpholinos (1.5 ng) were injected into the yolk of 1-cell-stage fertilized embryos from heterozygous sapje-like fish matings. Embryos were then cultured in the Children's Hospital aquatic system at 28.5°C.
Determination of cellular cAMP and cGMP levels
cAMP and cGMP levels in hmox1 mRNA-injected embryos were determined with a competitive protein-binding technique by using Cyclic GMP XPAssay Kit and Cyclic AMP XPAssay Kit from Cell Signaling Technology following the manufacturer's instructions.
RNA expression analysis with real-time PCR
For analysis of expression in different tissues, 10-fold serial dilutions of the cDNA were analyzed on 96-well optical plates using the Power SYBR Green Mix system (Life Technologies) and analyzed on the ABI 7900HT real-time PCR machine (Life Technologies) following the manufacturer's instructions using with primer sets of zebrafish Hmox1; forward primer for zebrafish Hmox1 cDNA (NM_001127516): 5′ ATGGACTCCACCAAAAGC 3′, reverse primer for zebrafish Hmox1 cDNA: 5′ CCTTTCTGGTAGCTGAGCATC 3′; primer sets of zebrafish elongation factor 1 (EF1) alpha; forward: 5′ CTGGAGGCCAGCTCAAACAT 3′, reverse: 5′ ATCAAGAAGAGTAGTACCGCTAGCATTAC 3′. Cycle times were normalized to EF1 alpha as a loading control. Relative fold expression and changes were calculated using the 2−ΔΔCt method.
Sildenafil treatment of DMD mouse models
C57BL6/J wild-type control mice and mdx5cvmice were originally purchased from Jackson Laboratories (Bar Harbor, ME, USA) (39). All mouse husbandry and treatment was approved by the Boston Children's Hospital Animal Facilities/Institutional Animal Care and Use Committee and maintained in pathogen-free cages following federal standard of care practices. All mice were fed a 5P00 Prolab RMH 3000 standard chow diet (5% fat; LabDiet, PMI Nutrition International, St. Paul, MN, USA) and had access to food ad libitum. All mouse lines and subsequent breeding were maintained on a C57BL6/J background under the Boston Children's Hospital ARCH animal protocol (12-10-2287R). Sildenafil was administered to mdx5cv mice at a dosage and with a protocol that have been previously studied and found therapeutically effective in the mdx mice (17,18). Sildenafil citrate (powder; Pfizer, Inc.) was dissolved in acidified, sterile-filtered water (pH 3.0) to a concentration of 400 mg/l and administered ad libitum. Previous publications report that this administration technique and dosage result in the ingestion of ∼80 mg/kg/day, with an IC50 of 10 nM for sildenafil inhibition of 50% of PDE5A (18). C57BL6/J and mdx5cv vehicle controls were given untreated acidified, sterile-filtered drinking water (pH 3.0) via the same administration protocol. Each cohort consisted of two males and two females, with treatment beginning at 4 weeks of age, and lasting for 7–8 weeks. At the end of each treatment period, the mice were euthanized and the diaphragm and the tibialis anterior muscles were harvested and snap-frozen in 100% acetone chilled with dry ice. Muscle sections were used for protein extraction and were stained with Hematoxylin and Eosin to visualize areas of necrosis and inflammation, as well as the myofibers, their cross sectional area and the number and location of their nuclei. The muscle samples were also used for western blotting and determination of hmox1 levels.
Exercise fatigue assay
This assay (40) was modeled after the 6-min walk test, a human patient clinical endpoint measure used to assess mobility and efficacy of therapeutics. After exercise, mdx5cv mice are found to have decreased rearing, ambulation and distance traveled, as well as increased rest time (41). Mice activity was recorded in individual Actitrack Locomotor arena chambers for a 6-min interval immediately after mild exercise, tracking movement via infrared beam interruptions. Mild exercise consisted of running on a 15° downhill track at 10 mpm for 10 min, with acclimatization at 3 mpm for 5 min (20,41).
SUPPLEMENTARY MATERIAL
FUNDING
This study is supported by the NINDS grant 5P50NS040828-10, the Muscular Dystrophy Association, Pfizer, and the Bernard F. and Alva B. Gimbel Foundation. M.S.A. is supported by a Muscular Dystrophy Association (MDA) Development Grant MDA255059. Sequencing was performed by the Intellectual and Developmental Disabilities Research Center Molecular Core Laboratory, which was supported by the National Institute of Child Health and Human Development grant 2P30HD018655-30. Behavioral analysis was performed in the BCH Neurodevelopmental Behavioral Core (supported by BCH IDDRC grant, NIH-P30-HD-18655).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. Beggs and Gussoni, members of the Kunkel laboratory, Boston Children's Hospital Fish Facility and Harvard Medical School's Institute of Chemistry and Cell Biology.
Conflict of Interest statement. This work was partly supported by Pfizer, Inc. We declare no other potential conflict of interest. L.M.K. is a consultant for Pfizer, Inc., Summit Corporation PLC, Shire Human Genetic Therapies and Tarix Pharmaceuticals for muscle disease drug therapies.
REFERENCES
- 1.Monaco A.P., Neve R.L., Colletti-Feener C., Bertelson C.J., Kurnit D.M., Kunkel L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. doi:10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 2.Burghes A.H., Logan C., Hu X., Belfall B., Worton R.G., Ray P.N. A cDNA clone from the Duchenne/Becker muscular dystrophy gene. Nature. 1987;328:434–437. doi: 10.1038/328434a0. doi:10.1038/328434a0. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. doi:10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida M., Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 5.Guyon J.R., Mosley A.N., Zhou Y., O'Brien K.F., Sheng X., Chiang K., Davidson A.J., Volinski J.M., Zon L.I., Kunkel L.M. The dystrophin associated protein complex in zebrafish. Hum. Mol. Genet. 2003;12:601–615. doi:10.1093/hmg/ddg071. [PubMed] [Google Scholar]
- 6.Guyon J.R., Goswami J., Jun S.J., Thorne M., Howell M., Pusack T., Kawahara G., Steffen L.S., Galdzicki M., Kunkel L.M. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum. Mol. Genet. 2009;18:202–211. doi: 10.1093/hmg/ddn337. doi:10.1093/hmg/ddn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassett D.I., Bryson-Richardson R.J., Daggett D.F., Gautier P., Keenan D.G., Currie P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. 2003;130:5851–5860. doi: 10.1242/dev.00799. doi:10.1242/dev.00799. [DOI] [PubMed] [Google Scholar]
- 8.Berger J., Berger S., Hall T.E., Lieschke G.J., Currie P.D. Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul. Disord. 2010;20:826–832. doi: 10.1016/j.nmd.2010.08.004. doi:10.1016/j.nmd.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Chambers S.P., Dodd A., Overall R., Sirey T., Lam L.T., Morris G.E., Love D.R. Dystrophin in adult zebrafish muscle. Biochem. Biophys. Res. Commun. 2001;286:478–483. doi: 10.1006/bbrc.2001.5424. doi:10.1006/bbrc.2001.5424. [DOI] [PubMed] [Google Scholar]
- 10.Gupta V., Kawahara G., Gundry S.R., Chen A.T., Lencer W.I., Zhou Y., Zon L.I., Kunkel L.M., Beggs A.H. The zebrafish dag1 mutant: a novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011;20:1712–1725. doi: 10.1093/hmg/ddr047. doi:10.1093/hmg/ddr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall T.E., Bryson-Richardson R.J., Berger S., Jacoby A.S., Cole N.J., Hollway G.E., Berger J., Currie P.D. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. doi:10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White R.M., Cech J., Ratanasirintrawoot S., Lin C.Y., Rahl P.B., Burke C.J., Langdon E., Tomlinson M.L., Mosher J., Kaufman C., et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. doi: 10.1038/nature09882. doi:10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphey R.D., Stern H.M., Straub C.T., Zon L.I. A chemical genetic screen for cell cycle inhibitors in zebrafish embryos. Chem. Biol. Drug. Des. 2006;68:213–219. doi: 10.1111/j.1747-0285.2006.00439.x. doi:10.1111/j.1747-0285.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- 14.Berger J., Sztal T., Currie P.D. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res Commun. 2012;423:785–788. doi: 10.1016/j.bbrc.2012.06.040. doi:10.1016/j.bbrc.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 15.Bassett D.I., Currie P.D. The zebrafish as a model for muscular dystrophy and congenital myopathy. Hum. Mol. Genet. 2003;12:265–270. doi: 10.1093/hmg/ddg279. doi:10.1093/hmg/ddg279. [DOI] [PubMed] [Google Scholar]
- 16.Kawahara G., Karpf J.A., Myers J.A., Alexander M.S., Guyon J.R., Kunkel L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2011;108:5331–5336. doi: 10.1073/pnas.1102116108. doi:10.1073/pnas.1102116108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Percival J.M., Whitehead N.P., Adams M.E., Adamo C.M., Beavo J.A., Froehner S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012;228:77–87. doi: 10.1002/path.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adamo C.M., Dai D.F., Percival J.M., Minami E., Willis M.S., Patrucco E., Froehner S.C., Beavo J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2010;107:19079–19083. doi: 10.1073/pnas.1013077107. doi:10.1073/pnas.1013077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khairallah M., Khairallah R.J., Young M.E., Allen B.G., Gillis M.A., Danialou G., Deschepper C.F., Petrof B.J., Des Rosiers C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. USA. 2008;105:7028–7033. doi: 10.1073/pnas.0710595105. doi:10.1073/pnas.0710595105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi Y.M., Rader E.P., Crawford R.W., Iyengar N.K., Thedens D.R., Faulkner J.A., Parikh S.V., Weiss R.M., Chamberlain J.S., Moore S.A., et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456:511–515. doi: 10.1038/nature07414. doi:10.1038/nature07414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asai A., Sahani N., Kaneki M., Ouchi Y., Martyn J.A., Yasuhara S.E. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One. 2007;2:e806. doi: 10.1371/journal.pone.0000806. doi:10.1371/journal.pone.0000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsushima H., Tanaka H., Mizumoto N., Takashima A. Identification of crassin acetate as a new immunosuppressant triggering heme oxygenase-1 expression in dendritic cells. Blood. 2009;114:64–73. doi: 10.1182/blood-2009-02-204297. doi:10.1182/blood-2009-02-204297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mies F., Spriet C., Heliot L., Sariban-Sohraby S. Epithelial Na+ channel stimulation by n-3 fatty acids requires proximity to a membrane-bound A-kinase-anchoring protein complexed with protein kinase A and phosphodiesterase. J. Biol. Chem. 2007;282:18339–18347. doi: 10.1074/jbc.M611160200. doi:10.1074/jbc.M611160200. [DOI] [PubMed] [Google Scholar]
- 24.O'Brien J.J., Baglole C.J., Garcia-Bates T.M., Blumberg N., Francis C.W., Phipps R.P. 15-deoxy-Delta12,14 prostaglandin J2-induced heme oxygenase-1 in megakaryocytes regulates thrombopoiesis. J. Thromb. Haemost. 2009;7:182–189. doi: 10.1111/j.1538-7836.2008.03191.x. doi:10.1111/j.1538-7836.2008.03191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcantoni E., Di Francesco L., Dovizio M., Bruno A., Patrignani P. Novel insights into the vasoprotective role of heme oxygenase-1. Int. J. Hypertens. 2012:127910. doi: 10.1155/2012/127910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bahary N., Goishi K., Stuckenholz C., Weber G., Leblanc J., Schafer C.A., Berman S.S., Klaqsbrun M., Zon L.I. Duplicate VegfA genes and orthologues of the KDR receptor tyrosine kinase family mediate vascular development in the zebrafish. Blood. 2007;110:3627–3636. doi: 10.1182/blood-2006-04-016378. doi:10.1182/blood-2006-04-016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Platt J.L., Nath K.A. Heme oxygenase: protective gene or Trojan horse. Nat. Med. 1998;4:1364–1365. doi: 10.1038/3947. doi:10.1038/3947. [DOI] [PubMed] [Google Scholar]
- 28.Chora A.A., Fontoura P., Cunha A., Pais T.F., Cardoso S., Ho P.P., Lee L.Y., Sobel R.A., Steinman L., Soares M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Invest. 2007;117:438–447. doi: 10.1172/JCI28844. doi:10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L., Lu H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010;69:771–776. doi: 10.1097/NEN.0b013e3181e9a34b. doi:10.1097/NEN.0b013e3181e9a34b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryter S.W., Alam J., Choi A.M. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. doi:10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 31.Vercherat C., Chung T.K., Yalcin S., Gulbagci N., Gopinadhan S., Ghaffari S., Taneja R. Stra13 regulates oxidative stress mediated skeletal muscle degeneration. Hum. Mol. Genet. 2009;18:4304–4316. doi: 10.1093/hmg/ddp383. doi:10.1093/hmg/ddp383. [DOI] [PubMed] [Google Scholar]
- 32.Heydemann A., McNally E. NO more muscle fatigue. J. Clin. Invest. 2009;119:448–450. doi: 10.1172/JCI38618. doi:10.1172/JCI38618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidavalur R., Penumathsa S.V., Zhan L., Thirunavukkarasu M., Maulik N. Sildenafil induces angiogenic response in human coronary arteriolar endothelial cells through the expression of thioredoxin, hemeoxygenase and vascular endothelial growth factor. Vascul. Pharmacol. 2006;45:91–95. doi: 10.1016/j.vph.2006.03.010. doi:10.1016/j.vph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Seldon M.P., Silva G., Pejanovic N., Larsen R., Gregoire I.P., Filipe J., Anrather J., Soares M.P. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J. Immunol. 2007;179:7840–7851. doi: 10.4049/jimmunol.179.11.7840. [DOI] [PubMed] [Google Scholar]
- 35.Brunt K.R., Tsuji M.R., Lai J.H., Kinobe R.T., Durante W., Claycomb W.C., Ward C.A., Melo L.G. Heme oxygenase-1 inhibits pro-oxidant induced hypertrophy in HL-1 cardiomyocytes. Exp. Biol. Med. (Maywood) 2009;234:582–594. doi: 10.3181/0810-RM-312. doi:10.3181/0810-RM-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monici M.C., Aguennouz M., Mazzeo A., Messina C., Vita G. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. doi:10.1212/01.WNL.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- 37.Burdi R., Rolland J.F., Fraysse B., Litvinova K., Cozzoli A., Giannuzzi V., Liantonio A., Camerino G.M., Sblendorio V., Capogrosso R.F., et al. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J. Appl. Physiol. 2009;106:1311–1324. doi: 10.1152/japplphysiol.90985.2008. doi:10.1152/japplphysiol.90985.2008. [DOI] [PubMed] [Google Scholar]
- 38.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009;119:624–635. doi: 10.1172/JCI36612. doi:10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gozzelino R., Jeney V., Soares M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. doi:10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi Y.M., Rader E., Crawford R.W., Campbell K. Endpoint measures in the mdx mouse relevant for muscular dystrophy pre-clinical studies. Neuromuscul. Disord. 2012;22:34–42. doi: 10.1016/j.nmd.2011.08.001. doi:10.1016/j.nmd.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beastrom N., Lu H., Macke A., Canan B., Johnson E., Penton C., Kaspar B.K., Rodino-Klapac L.R., Zhou L., Janssen P.M.L., et al. mdx5cv mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am. J. Pathol. 2011;179:2464–2474. doi: 10.1016/j.ajpath.2011.07.009. doi:10.1016/j.ajpath.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.