

Abstract
Abuse of methamphetamine (METH) is a major and significant societal problem in the US, as a number of studies have suggested that METH is associated with increased cerebrovascular events, hemorrhage or vasospasm. Although cellular and molecular mechanisms involved in METH-induced toxicity are not completely understood, changes in brain O2 may play an important role and contribute to METH-induced neurotoxicity including dopaminergic receptor degradation. Given that O2 is the terminal electron acceptor for many enzymes that are important in brain function, the impact of METH on brain tissue pO2 in vivo remains largely uncharacterized. This study investigated striatal tissue pO2 changes in male C57BL/6 mice (16–20g) following METH administration using EPR oximetry, a highly sensitive modality to measure pO2 in vivo, in situ and in real time. We demonstrate that 20 min after a single injection of METH (8 mg/kg i.v.), the striatal pO2 was reduced to 81% of the pretreatment level and exposure to METH for 3 consecutive days further attenuated striatal pO2 to 64%. More importantly, pO2 did not recover fully to control levels even 24 hrs after administration of a single dose of METH. and continual exposure to METH exacerbates the condition. We also show a reduction in cerebral blood flow associated with a decreased brain pO2 indicating an ischemic condition. Our findings suggests that administration of METH can attenuate brain tissue pO2, which may lead to hypoxic insult, thus a risk factor for METH-induced brain injury and the development of stroke in young adults.
Keywords: Methamphetamine, EPR oximetry, Neurotoxicity, Hypoxia, Cerebral Blood Flow, Brain Oxygen
Introduction
Methamphetamine (METH) abuse continues to be a significant, but not adequately addressed, societal problem in the US. While METH-induced neurotoxicity has been studied for decades, the cellular and molecular mechanisms underlying this toxicity are poorly understood. Acute and chronic METH abuse damages multiple organs; however, neurotoxicity in the brain is the more prominent. In vitro, ex vivo and clinical studies suggest METH abuse and neurotoxicity is associated with hypoxia as a result of hypoperfusion and vasoconstriction (Kousik et al., 2011), cerebrovascular incidents, such as hemorrhagic and ischemic stroke (Perez et al., 1999; Wang et al., 2001; Ho et al., 2009) and oxidative stress (Cadet et al., 1998; Riddle et al., 2006). Studies have suggested attenuation of oxygen (O2) in the brain to be associated with uncoupling of mitochondria after METH administration (Shiba et al., 2001). Additionally, it has been shown that METH displaces vesicular dopamine and serotonin, which are oxidized to dopamine-type quinones in the striatum. Thus, METH-induced dopaminergic receptor degeneration in addition to pathways that elicit mitochondrial toxicity and increased glutamine release may play an important role its neurotoxicity (Yamamoto and Zhu, 1998; Pubill et al., 2005).
Molecular O2 plays a central role in the control of brain physiology, e.g. O2 is the terminal acceptor for enzymes that are crucial to the biosynthesis of the neurotransmitters, dopamine, serotonin and norepinephrine. Thus, changes in tissue O2 directly impact homeostasis and brain tissue is highly sensitive to changes in availability of O2, in which pathological changes can occur if there are significant changes in the delivery or utilization of O2 in the brain (Love, 1999; Zauner et al., 2002; Liu and Rosenberg, 2005). While it is well established that decreased levels of O2 in the brain occur following stroke and traumatic brain injury, only in recent years have preliminary ex vivo data surfaced to suggest that METH-neurotoxicity may be linked to changes in O2 levels (Shiba et al., 2001; Wang et al., 2001; Kousik et al., 2011) and as a risk factor for the development of stroke in young adults (Perez et al., 1999; Ho et al., 2009). Despite these extensive results, there is no direct in vivo in situ and in real time evidence regarding METH-induced alterations in brain tissue partial pressure of oxygen (pO2). It remains unclear whether METH is a predisposing factor to ischemic brain injury and, if so, whether changes in pO2 are a significant contributing factor to METH-induced cerebral damage in vivo.
Surprisingly, given the significance of O2 in brain physiology, the accurate measurement of O2 in the brain using different modalities is not a trivial task. For instance, O2 in biological systems has been determined by more invasive methods such as Clark-type electrodes and fluorescence quenching of a ruthenium dye and Blood oxygen level-dependent (BOLD) MRI imaging provides only quantitative estimate of O2 (Baudelet and Gallez, 2002; Elas et al., 2003). Experimental efforts are also hampered by the physical difficulty of measuring O2–delivery to the tissue at the microvascular level. With development of in vivo electron paramagnetic resonance (EPR) oximetry (Dunn and Swartz, 2003; Ahmad and Kuppusamy, 2010), we have measured cerebral interstitial pO2 in several ischemic rodent models using the minimally invasive O2 sensitive paramagnetic probe, lithium phthalocyanine (LiPc) (Liu et al., 1993; Liu et al., 1995; Shen et al., 2009). LiPc has several desirable properties as a probe for cerebral pO2 including high sensitivity, resistance to chemical reactions, and high degree of inertness in biological systems for long- or short-term studies in vivo, in situ and in real time (Liu et al., 1995; Elas et al., 2003; Liu et al., 2004).
To further understand METH-induced effects on the brain we have investigated the local interstitial levels of pO2 in the striatum after METH administration using the novel technique of EPR oximetry and the spin probe, LiPc. Herein, we report that METH-treated mice experience decreased local interstitial levels of pO2 in the striatum. Additionally, the observed attenuation of brain pO2 is accompanied by a decrease in cerebral blood flow (CBF), indicating an ischemic condition and moreover, after single non-lethal dose of METH, brain tissue pO2 does not appear to fully recover to normal physiological levels.
Materials and Methods
Animals
The Laboratory Animal Care and Use Committee of the UNM HSC approved all experimental protocols. Male C57BL/6 mice, 16–20g, were obtained from Charles River Laboratory (Wilmington, MA, USA). Animals were maintained in a climate-controlled vivarium with a 12 h light–dark cycle and free access to food and water.
For all surgical stereotaxic LiPc implantation procedures, 4.0% isoflurane in N2O:O2 (70:30%) was used for anesthesia induction, and anesthesia was maintained with 1% isoflurane in mice. LiPc was a gift from Dr. Harold Swartz (NIH In vivo EPR Center, Dartmouth College, NH, USA). Animals were anesthetized throughout all EPR, MRI and Pulse Ox measurements with 1% isoflurane in N2O:O2 (70:30%) after induction at 4.0% isoflurane in N2O:O2 (70:30%). Physiological monitoring during all procedures comprised of measurement and maintenance of core (rectal) temperature at 37.5 ± 0.5°C using a heating pad, a heat lamp or a warm air heater in the MRI.
Drugs and Chemicals. d-Methamphetamine hydrochloride was purchased from Sigma (St. Louis, MO). Drugs were dissolved in 0.9% saline vehicle. Animals, under anesthesia, were given a single dose of METH (8 mg/kg i.v.) for 3 consecutive days. This dosage was selected based on previous studies in which similar single large doses of METH demonstrate persistent behavorial and neurochemical changes to assess its effects on the central nervous system of various mammalian species and to reflect METH overdoses or binging in human abusers (Cadet et al 2003). Control mice were injected with an equal volume of 0.9% saline vehicle.
Implantation of EPR Oximetry Probe LiPc
For every animal, correct assignment of the implantation site in the striatum was determined using the Mouse Brain Atlas (Paxinos and Franklin, 2001) and was confirmed by MRI. Under anesthesia, a pin hole on the parietal skull was made at the stereotaxic position of AP: +0.5 mm and L: +1.5 mm with respect to bregma. A small LiPc crystal (approximate diameter 0.2 mm) was placed at a depth of −3.5 mm using a microdialysis guide cannula with an inner diameter of 0.24 mm (CMA microdialysis, Stockholm, Sweden). LiPc crystal was placed in the striatum of the right hemisphere of mice. Mice were allowed to recover from implantation 48–72 h before further study.
Measurement of Cerebral pO2 by EPR Oximetry with LiPc
For non-invasive in vivo measurement of local cerebral pO2 in the anesthetized mouse before and directly after injection of METH, EPR oximetry was conducted according to previously described methods (Liu et al., 1993; Liu et al, 1995; Shen et al., 2009) with some modification. Briefly, an external loop resonator was placed over the position where LiPc was implanted, and an EPR spectrum was recorded using a Bruker EleXsys E540 EPR spectrometer equipped with an L-band bridge (Bruker Instruments, Billerica, MA, USA). The resonator has advanced automatching and autotuning capabilities that correct for any slight animal movements. The EPR spectrum was acquired with a scan time of 40s, and 5 scans were obtained and averaged to produce significant signal-to-noise ratio to allow accurate fitting. The peak-to-peak line width of the spectrum was obtained via computer line-fitting, and converted to pO2 values according to a calibration curve for LiPc as previously described (Liu et al., 1995; Liu et al., 2004; Shen et al., 2009). EPR acquisition parameters: microwave power of 18mW, a microwave frequency of 1.07 GHz, a center magnetic field strength of 380 G, a scan range of 1.0 G, with a modulation amplitude of less than one-third of the intrinsic EPR line-width.
Confirmation of LiPc Implants and Measurement of CBF by MRI
Mice with LiPc implants were placed in a custom-built holder and moved to the isocenter of the magnet before obtaining MRI images. Throughout the imaging session, animals were anesthetized and monitored in real time. MR imaging was performed on a 4.7T Biospec® dedicated research MR scanner (Bruker Biospin, Billerica, MA), equipped with 500 mT/m (rise time 80–120 μs) gradient set (for performing small animal imaging) and a small bore linear RF coil (ID 72 mm). LiPc implant position was confirmed using T2-weighted 2D RARE (rapid acquisition with relaxation enhancement) imaging using the following parameters: TR/TE, 4000/65 ms; FOV, 2.5 cm ± 2.5 cm; slice thickness, 1.0 mm; slice gap, 0.5 mm; number of slices, 10; matrix, 256 × 128; number of averages, 20; receiver bandwidth, 50 kHz.
CBF in mice was measured on Day 1 before METH administration and on Day 3 after the last injection of METH. Mice were transferred to the MRI suite, placed in a dedicated holder and moved to the isocenter of the magnet prior to the imaging session. Animals were anesthetized and monitored during the entire duration of the study. Initial localizer images were acquired using the following parameters: 2D FLASH (Fast Low Angle SHot), TR/TE 10/3 ms, matrix 256 x 128, FOV 6.4 cm, 1 slice per orientation. After the localizer images were acquired, tissue perfusion was measured using non-invasive arterial spin labeling method (ASL). The sequence: Flow-sensitive Alternating Inversion Recovery Rapid Acquisition with Relaxation Enhancement (FAIR-RARE) was used to implement ASL with parameters: TE/TR, 46/16000ms; FOV, 2.5 cm x 2.1cm; slice thickness, 1mm; number of slices, 1; matrix = 128 x 128. Perfusion map was calculated using ASL_Perfusion_Processing macro in ParaVision 5.1 (Bruker Biospin MRI GmbH, Germany). The principle is as follows: Inversion recovery data from the imaging slice are acquired after selective inversion of the slice and after inversion of the slice including the surrounding tissue, containing the supplying arteries. The difference of the inverse of the apparent T1 images then yields a measure of the cerebral blood flow (Kim, 1995). The acquired data were transferred to a dedicated computer workstation for post processing.
Arterial Blood Gas Measurements
Arterial blood gas (SpO2) and other physiological parameters were measured using a Mouse Ox Plus (Starr Lab Science Corp, Oakmont, PA) with thigh sensor. Animals under anesthesia were given a single dose of METH (8 mg/kg) or 0.9% saline delivered i.v. and measurements were performed before and after injection for 3 consecutive days.
Statistical Analysis
Data are expressed as the Mean ± SEM. One-way ANOVA for repeated measures and Bonferroni’s modified t-test analysis were used for the data analyses. Significance was considered with P < 0.05.
Results
Cerebral pO2 in the Striatum After Acute METH Exposure
Our initial series of experiments were devoted to establishing whether EPR oximetry is capable of measuring changes in brain tissue pO2 levels were they to occur after mice were administered METH (8 mg/kg i.v. per day) (Figure 1A, 1B). For these experiments, a crystal of LiPc was implanted in the striatum of the brain. One of the major advantages of LiPc over other spin probes for EPR oximetry is its ability to obtain repetitive and accurate measurements at specific sites in brain tissue over a long period of time without frequent introduction of subsequent spin probe (Dunn and Swartz, 2003). Verification of the location of the LiPc crystal was obtained by MRI (Figure 1C). Under our experimental conditions, inflammation, tissue damage or reaction to the LiPc or the implantation procedure was not observed, similar to previous reports (Dunn and Swartz, 2003; Shen et al., 2009). The MRI technique greatly increases the apparent size of the crystal on the image (~1 mm) as the ability to visualize LiPc crystals on MR images under such conditions is based on the bulk magnetic susceptibility difference between the tissue and the crystal, which produces susceptibility-based signal attenuation around the crystal (Liu et al., 1995).
Figure 1. METH-induced percent change in interstitial pO2 levels and LiPc implant in the striatum of mice.


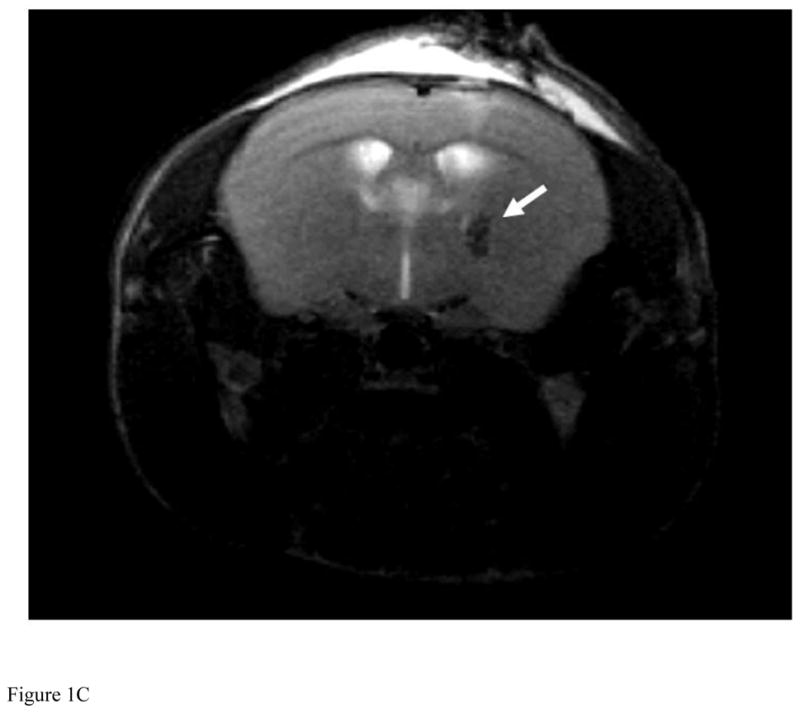
For all experiments, LiPc crystal was implanted in the mouse brain 48 hr before EPR measurement and EPR spectra were collected each day before and after injection of METH (8 mg/kg i.v. per day) or vehicle (0.9% saline i.v.) from mice breathing 21% oxygen and converted to pO2. The 0 time point is time of injection. (A) Percent change in interstitial pO2 levels in the striatum of mice injected with METH for 3 days (*P < 0.05 vs Day 1 and Day 2 at the same time point). (B) Percent change in interstitial pO2 levels in the striatum of vehicle mice were injected with vehicle for 3 days as a control. Data are expressed as the Mean ± SEM (N=6 for each group). (C) A representative in vivo MR image of a mouse implanted with LiPc crystal (arrow) confirmed using T2-weighted 2D RARE imaging.
For measurement of cerebral pO2, the peak-to-peak line width of the EPR spectrum for LiPc in mice was recorded and these data were converted to pO2 according to a calibration curve for this oximetry probe, as previously described (Liu et al., 1993; Liu et al., 1995; Liu et al., 2004; Shen et al., 2009). Using this approach, levels of cerebral pO2 in vehicle (0.9% saline, i.v.) and METH treated mice were recorded every 5 min for 60 min on 3 consecutive days. It was found that after a single injection of METH, a significant decrease in brain pO2 in the striatum was observed from the pre-injection level. For ease of illustration, we converted these data to % cerebral pO2 change as a function of time (Figure 1A and 1B). Maximal decrease in striatal pO2 was achieved roughly 20 min after injection from 33.5 ± 1.0 mmHg to 23.9 ± 2.0 mmHg (a decrease of 28.7%) and was sustained for the duration of the experiment, e.g., up to 60 min. Next, we examined what effect a single injection of METH per day for 3 consecutive days would have on brain pO2 levels, as a mimic of chronic exposure to METH. Brain pO2 decreased overall from 33.5 ± 1.0 mmHg (pre-exposure to METH on Day 1) to 21.6 ± 2.6 mmHg (a decrease of 35.5% in pO2; post-exposure to METH on Day 3) over the duration of 3 days METH exposure (Figure 1A). Again, a maximal daily decrease was achieved roughly 20 min after injection on each day and brain pO2 levels did not fully recover to control level before each mouse received another dose of METH. In essence, pre-injection values of striatal pO2 were consistently lower than the previous day’s baseline (pre-injection) measurement. In fact, a sustained decrease in brain pO2, only recovering partially to 96% on Day 2 and then to 86% on Day 3, from the observed decreases on Days 1 and 2 of 71% and 70%, respectively, at 60 min post injection. as Injection of 0.9% saline to vehicle mice for 3 consecutive days showed no change in brain pO2 demonstrating that decreases were a consequence of METH exposure (Figure 1B). These results indicate that acute exposure to METH leads to reduced brain tissue oxygenation, and that chronic exposure may decrease the brain pO2 to a hypoxia level, rendering the brain to potential ischemic damage.
Effect of METH on CBF by Diffusion MRI
To understand the potential causes of the observed reduction in brain pO2, we further investigated the METH-induced alteration in O2 delivery and other physiological parameters. Tissue O2 delivery is highly dependent on the regulation of CBF to control normal tissue oxygenation, thus the brain regulates regional and total blood flows (Zauner et al., 2002). A review of the pertinent literature would suggest that METH’s effect on CBF, both focal and global, is controversial dependent on the drug exposure and various methods of measurement (Polesskaya et al., 2011). To determine whether METH-induced reduction in cerebral pO2 is due, at least in part, to altered CBF in mice over the same 3-day time course, we measured CBF changes in the striatum by diffusion MRI and ASL methods (Figures 2A). After a baseline CBF reading from day 1, mice were injected with METH for 3 days and CBF was measured again on Day 3 after the final injection. CBF was observed to decrease significantly from Day 1 pre-injection to Day 3 post-injections in the striatum (Figure 2A), corresponding with a decrease in brain tissue pO2 as measured by EPR over the same 3 days (Figure 1A). CBF in mice was measured approximately 20–30 min after the 3rd injection to allow for insertion of the mouse into the MR imager and acquisition of images and to assess CBF when maximal decrease of cerebral pO2 was also observed by EPR. Although the focus of our study is on the striatum, the effects of METH on the brain are global; therefore CBF measurements were also undertaken in the whole brain and a similar significant decrease over the same 3 days was observed (Figure 2B). Of note, whole brain CBF data should be taken with consideration as this decrease incorporates the decrease in CBF observed in the striatum and the change in other areas of brain are unclear. Injection of 0.9% saline to vehicle mice for 3 days caused no change in CBF in the whole brain or striatum under the same conditions (Figure 2) indicating that alterations of CBF were a consequence of acute METH exposure. These results suggest that METH-induced hypoxia in the brain tissue is due to, at least in part, decreased CBF.
Figure 2. Alteration of CBF from acute exposure to METH.


Tissue perfusion was measured using non-invasive ASL MRI. “Vehicle” and “Pre-METH” represents the control measurement before mice received any injections of METH (8 mg/kg i.v.) or vehicle (0.9% saline i.v.). Measurements shown as “Vehicle Day 3” and “Post-METH Day 3” reflect CBF changes after exposure to METH or vehicle given for 3 consecutive days. (A) Decrease in CBF in the striatum of mice injected with METH for 3 consecutive days (*P < 0.05 vs Vehicle, pre-METH, and Vehicle Day 3). (B) Decrease in CBF in the whole brain of mice injected with METH for 3 consecutive days (*P < 0.05 vs Vehicle, pre-METH, and Vehicle Day 3). Data are expressed as the Mean ± SEM (N=5 for each group).
Effect of METH on Arterial Blood Gas (SpO2) and Other Physiological Parameters
In addition to CBF, METH-induced hypoxia could also be contributed by altered physiological parameters. Under normal physiological conditions, there is a linear relationship between arterial pO2 and brain pO2. After insults such as TBI and ischemia, CBF is often compromised and O2 delivery is decreased, which can cause brain pO2 to decline to lower levels (Zauner et al., 2002). Given that METH causes a sustained decrease in both CBF (Figure 2) and brain pO2 (Figure 1), we measured the effect of METH on SpO2 and several other physiological parameters, heart rate (HR) and breath rate (BR), known to be compromised by METH (Cruickshank and Dyer, 2009; Polesskaya et al., 2011). Measurements were taken by pulse oximetry before and after METH administration for 3 consecutive days (Figures 3A–3B). Day 1 measurements were taken before mice were injected with METH as a control. This was followed by the first injection of METH and results presented for DAY 2 were obtained 24 hr after the exposure to METH on Day 1 followed by the 2nd exposure to METH. This was repeated for Day 3 and measurements taken on Day 4 were done to determine the change in these physiological parameters from Day 3 exposure. As observed with cerebral pO2 and CBF (Figure 1A and Figure 2), SpO2 decreased daily and levels did not recover fully from the previous day’s exposure to METH or to the baseline measurement of SpO2 on Day 1 (Figure 3A). Therefore, this daily decrease in SpO2 paralleled EPR brain pO2 data. Interestingly, an increase in HR from Day 1 to Day 2 was observed and then remained at this level for the duration of the experiment (Figures 3B). Specifically, after the baseline measurement for HR on Day 1 of 392 ± 5.7 bpm HR increased to an average of 442 ± 2.1 bpm for the duration of the experiment. BR remained unchanged at the same time points and control animals treated with 0.9% saline showed no significant change in SpO2 or HR from the baseline measurements indicating that physiological changes observed were a result of METH exposure.
Figure 3. Acute METH treatment modulates SpO2, heart rate (HR) and breath rate (BR).


For all experiments, the Day 1 represents the control measurement by pulse oximetry before mice received any injections of METH (8 mg/kg i.v.) or vehicle (0.9% saline i.v.). Measurements shown on Days 2, 3 and 4 reflect changes in SpO2 and HR measured at 24hr after exposure to METH or vehicle given on Days 1, 2, and 3, respectively. (A) Results represent the decrease in SpO2 of mice injected with METH for 3 consecutive days (*P < 0.05 vs Vehicle). (B) Results represent the increase in HR of mice injected with METH for 3 consecutive days (*P < 0.05 vs Vehicle). Data are expressed as the Mean ± SEM (N=6 for each group).
As observed in Figure 1A, maximal decrease in brain pO2 was observed 20 min post METH administration on each day. Therefore, physiological measurements were also taken 20 min after METH exposure on each day to determine if there were further changes to these parameters directly after injection compared to the 24 hr time point results shown in Figures 3A and B. Ironically, fluctuations in SpO2 and HR observed at 20 min (data not shown) were comparable to values given in Figures 3A and 3B. Therefore, only day to day values are reported which reflect the overall changes from acute exposure over a 3 day period. Contrary to measurements taken 24 hrs after exposure, BR did increase significantly to 173.8 ± 4.3 brpm (P < 0.05, t-test) 20 min directly after exposure to METH from the daily baseline average of 93.7 ± 1.4 brpm, but returned to a control levels 24 hrs later. Together, these results show that METH administration causes reduced SpO2 but increased heart rate and parallels previous results including the increase in BR directly after injection (Cruickshank and Dyer, 2009; Polesskaya et al., 2011).
Discussion
The mechanisms underlying METH-induced neurotoxicity are not fully understood, but several animal and clinical studies have suggested that changes in brain O2 may contribute to METH-induced neurotoxicity, leading to vascular events including stroke (Perez et al., 1999; Wang et al., 2001; Ho et al., 2009). O2 is the terminal electron acceptor for many enzymes that are important in brain function, but the impact of METH on brain tissue pO2 in vivo, in situ and real time remains largely uncharacterized even though much is known about the effects of METH on the brain. Therefore, using the unique capability of EPR oximetry, this is the first study to provide direct evidence in vivo and in real time that once daily exposure of mice to METH (8 mg/kg, i.v.) for 3 consecutive days induces a sustained and significant attenuation of brain tissue pO2. As illustrated in Figure 1A, striatal pO2 decreased 35% with consecutive daily acute METH exposure compared to pre-treatment values and control mice. More importantly, pO2 did not recover fully to control levels even 24 hr after exposure to a single non-lethal dose of METH and continual exposure exacerbated the condition (Figure 1A).
METH abuse has also been associated with both transient and permanent alterations in CBF. Although the effect of METH exposure on CBF changes is controversial (Polesskaya et al., 2011), our findings show a decrease in CBF in the striatum after injections with METH, as compared to the saline control (Figure 2). Decreased CBF may lead to metabolically limiting tissue hypoxia, deprivations in O2 and glucose delivery, as well as a build-up of potentially toxic compounds (Bramlett and Dietrich, 2004; Polesskaya et al 2011). Therefore, we suggest that METH-induced decrease in brain pO2 may be associated with a reduction in CBF, although some residual flow persists, indicating incomplete ischemia. Incomplete ischemia, if sufficiently severe and/or prolonged, can still cause irreversible cell damage, due in part to the initial period of hypoxia and the secondary period of compromised energy metabolism, which continues after adequate oxygenation is restored. Blood flow and O2 delivery are vital for the maintenance of normal brain function and tissue viability; therefore, tissue O2 delivery to the brain is highly dependent on the regulation CBF, and the brain will attempt to extract a larger fraction of O2 from arterial blood, but the compensating mechanism is limited (Zauner et al., 2002). Also, during ischemia the O2 demand of the brain increases resulting in a decrease in oxygenation saturation. Thus, a decrease in CBF and striatal pO2 was accompanied by decreases in SpO2 (Figure 3A). The minor decrease in SpO2 is expected with METH-mediated increases in both cerebral and peripheral metabolic activation and increased O2 demand in the brain. Of note, SpO2 remained within physiological levels (Lee at el., 2009; Kutscher et al 2013), indicating that it has limited contribution to the observed changes in brain tissue pO2 or CBF. Consistent with prior reports (Cruickshank and Dyer, 2009; Polesskaya et al., 2011), we also observed an increase in HR, which was sustained for the duration of the experiments, and an increase in BR directly after injection (up to 20 min) which returned to control levels within 24 hr.
It has also been suggested that METH promotes blood acidosis and alkalosis supported by findings of METH-induced vasoconstriction of blood vessels, decrease in CBF, and an increase in respiration potentially leading to hypocapnia (Polesskaya et al., 2011). Our findings, including the increase in BR, support an association of METH with induced hypocapnia. Furthermore, decreased cerebral pO2 and CBF also support concerns that prolonged hypocapnia may lead to cerebral ischemia (Laffey and Kavanagh, 2002; Polesskaya et al., 2011). The ischemic condition is characterized by (i) cellular energy failure and brain edema, (ii) excess neurotransmitter release (particularly the excitatory amino acid neurotransmitters such as glutamate) and uptake inhibition, (iii) oxidative stress with subsequent lipid peroxidation, and (iv) disturbances in autoregulation of CBF levels (Calvert and Zhang 2005; Busl and Greer, 2010). In our study, we find that local striatum brain tissue pO2 decreases using EPR oximetry with particulate LiPc for single site measurement which suggests that ischemic-like changes occur in the striatum. However, we do not know if pO2 changes are global or localized based on our data from a single site. Although whole brain decrease in CBF is noteworthy, data should be taken with consideration as the measurement includes that observed in the striatum and it is still not clear how other regions of the brain may contribute. Given that METH neurotoxicity affects different parts of the brain, we speculate METH-induced hypoxia might occur heterogeneously throughout the brain tissue, which may lead to heterogeneous hypoxic brain injury and thus play a significant role in oxidative mechanisms associated with METH neurotoxicity (Yamamoto and Zhu, 1998; Shiba et al., 2001; Cadet et al 2003; Pubill et al., 2005). Further studies are warranted to test this hypothesis with the continued development of EPR imaging to measure heterogeneous changes in brain tissue pO2 (Shen et al., 2009).
A review of literature suggests METH neurotoxicity is a characterized by a variety of neuropathological changes, including degeneration of monoaminergic terminals, dysregulation of energy metabolism as well as neuronal apoptosis in various regions of the brain (Cadet et al 2003; Krasnova and Cadet, 2009). The impact of differential pO2 or CBF on the mechanistic significance of METH neurotoxicity, focally or globally, remains to be seen as neurotoxicity may be modulated based on metabolic demand mediated via dopaminergic and serotonergic pathways. Similarly, many questions remain to be answered with respect to METH neurotoxicity in monoaminergic terminals and striatal neurons (Cadet et al 2003). Additional studies are ongoing to determine the impact differential pO2 may have on the neurotoxicity of METH in the striatum or other regions of the brain. Likewise, alternative routes of administration and dosage schemes have been used to study METH abuse and it will be of interest to determine if comparable results are observed under various methodologies. Furthermore, the adverse neurologic consequences of METH-induced neurotoxicity would stem from the restoration of normal tissue perfusion similar to those observed in oxidative reperfusion injury and an imbalance between O2 supply and demand as METH’s effect diminishes over time with discontinued use. Results herein show an initial decrease in brain pO2 that is partially recovered to near normal after 24 hr and this phenomenon is repeated with continued exposure. Further studies are also needed to test this hypothesis, but our findings support a concern for METH-induced oxidative reperfusion injury.
In conclusion, recent advancements have established EPR oximetry, as a versatile method for highly sensitive and repetitive measurements of pO2 in the brain related to METH abuse. We demonstrate that METH can attenuate brain tissue pO2 in the striatum and acute METH exposure leads to a sustained and significant decrease in striatal pO2 that is not fully recovered. These findings suggest that even a one-time exposure to METH may promote ischemic insult and further support findings of potentially deleterious consequences to brain function and a risk factor for the development of stroke in young adults.
Highlights.
Investigated striatal tissue pO2 changes following methamphetamine administration in vivo using EPR oximetry.
Striatal pO2 was reduced by 81% after a single injection of methamphetamine and 64% after exposure for 3 consecutive days.
pO2 did not recover fully to control levels even 24 hrs after a single dose.
Decrease in brain tissue pO2 may be associated with a decrease in CBF.
Administration of methamphetamine may lead to hypoxic insult.
Acknowledgments
This work was supported in part by grants from National Institute of Health [P30GM103400, R01AG031725, R21DA023473 and 8UL1TR000041]
Abbreviations
- METH
methamphetamine
- pO2
partial pressure of oxygen
- EPR
electron paramagnetic resonance
- SpO2
arterial blood gas
- HR
heart rate
- BR
breath rate
- CBF
cerebral blood flow
- LiPc
lithium phthalocyanine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad R, Kuppusamy P. Theory, instrumentation, and applications of electron paramagnetic resonance oximetry. Chem Rev. 2010;110:3212–3236. doi: 10.1021/cr900396q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudelet C, Gallez B. How does blood oxygen level-dependent (BOLD) contrast correlate with oxygen partial pressure (pO2) insider tumors. Magn Reson Med. 2002;48:980–986. doi: 10.1002/mrm.10318. [DOI] [PubMed] [Google Scholar]
- Busl KM, Greer DM. Hypoxic-ischemic brain injury: pathophysiology, neuropathology and mechanisms. Neuro Rehabilitation. 2010;26:5–13. doi: 10.3233/NRE-2010-0531. [DOI] [PubMed] [Google Scholar]
- BramLett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. JCBFM. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Brannock C. Free radicals and the pathobiology of brain dopamine systems. Neurochem Int. 1998;32:117–131. doi: 10.1016/s0197-0186(97)00031-4. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Jayanthi S, Deng X. Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB. 2003;17:1775–1788. doi: 10.1096/fj.03-0073rev. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Zhang JH. Pathophysiology of an hypoxic-ischemic insult during the perinatal period. Neurological Research. 2005;27:246–260. doi: 10.1179/016164105X25216. [DOI] [PubMed] [Google Scholar]
- Clemens KJ, Cornish JL, Hunt GE, McGregor IS. Repeated weekly exposure to MDMA, methamphetamine or their combination: long-term behavioural and neurochemical effects in rats. Drug Alcohol Depend. 2007;86:183–190. doi: 10.1016/j.drugalcdep.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Cruickshank CC, Dyer KR. A review of the clinical pharmacology of methamphetamine. Addiction. 2009;104:1085–1099. doi: 10.1111/j.1360-0443.2009.02564.x. [DOI] [PubMed] [Google Scholar]
- Dunn JF, Swartz HM. In vivo electron paramagnetic resonance oximetry with particulate materials. Methods. 2003;30:159–166. doi: 10.1016/s1046-2023(03)00077-x. [DOI] [PubMed] [Google Scholar]
- Elas E, Williams BB, Parasca A, Mailer C, Pelizzari CA, Lewis MA, River JN, Karczmar GS, Barth ED, Halpern HJ. Quantitative tumor oxymetric images from 4D electron paramagnetic resonance imaging (EPRI): Methodology and comparison with blood oxygen level-dependent (BOLD) MRI. Magn Reson Med. 2003;49:682–691. doi: 10.1002/mrm.10408. [DOI] [PubMed] [Google Scholar]
- Ho EL, Josephson A, Lee HS, Smith WS. Cerebrovascular complications of methamphetamine abuse. Neurocrit Care. 2009;10:295–305. doi: 10.1007/s12028-008-9177-5. [DOI] [PubMed] [Google Scholar]
- Kim SG. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (FAIR) technique: Application to functional mapping. MRM. 1995;34:293–301. doi: 10.1002/mrm.1910340303. [DOI] [PubMed] [Google Scholar]
- Kousik SM, Graves SM, Napier TC, Zhao C, Carvey PM. Methamphetamine-induced vascular changes lead to striatal hypoxia and dopamine reduction. Neuro Report. 2011;22:923–928. doi: 10.1097/WNR.0b013e32834d0bc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutscher HL, Gao D, Li S, Massa CB, Cervelli J, Deshmukh M, Joseph LB, Laskin DL, Sinko PJ. Toxicodynamics of rigid polystyrene microparticles on pulmonary gas exchange in mice: Implications for microemboli-based drug delivery systems. Toxicol Appl Pharmcol. 2013;266:214–223. doi: 10.1016/j.taap.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffey JG, Kavanagh BP. Hypocapnia. N Engl J Med. 2002;347:43–53. doi: 10.1056/NEJMra012457. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Woodske ME, Zou B, O’Donnell CP. Dynamic arterial blood gas analysis in conscious unrestrained C57BL/6J mice during exposure to intermitten hypoxia. J Appl Phsiol. 2009;107:290–294. doi: 10.1152/japplphysiol.91255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Bacić G, Hoppes PJ, Jang J, Du H, Ou LC, Dunn JF, Swartz HM. Assessment of cerebral pO2 by EPR oximetry in rodents: effects of anesthesia, ischemia, and breathing gas. Brain Res. 1995;685:91–98. doi: 10.1016/0006-8993(95)00413-k. [DOI] [PubMed] [Google Scholar]
- Liu KJ, Gast P, Moussavi M, Norby SW, Vahidi N, Walczak T, Wu M, Swartz HM. Lithium phthalocyanine: a probe for electron paramagnetic resonance oximetry in viable biological systems. Proc Natl Acad Sci USA. 1993;90:5438–5442. doi: 10.1073/pnas.90.12.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Rosenberg GA. Matrix matalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Liu S, Timmins GS, Shi H, Gasparovic CM, Liu KJ. Application of in vivo EPR in brain research: monitoring tissue oxygenation, blood flow, and oxidative stress. NMR Biomed. 2004;17:327–334. doi: 10.1002/nbm.899. [DOI] [PubMed] [Google Scholar]
- Love S. Oxidative stress in brain ischemia. Brain Pathol. 1999;9:119–131. doi: 10.1111/j.1750-3639.1999.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. 2. Academic Press; San Diego: 2001. [Google Scholar]
- Perez JA, Jr, Arsura EL, Strategos S. Methamphetamine-related stroke: four cases. J Emerg Med. 1999;17:469–471. doi: 10.1016/s0736-4679(99)00009-8. [DOI] [PubMed] [Google Scholar]
- Polesskaya O, Silva J, Sanfilippo C, Desrosiers T, Sun A, Shen J, Feng C, Polesskiy A, Deane R, Zlokovic B, Kasischke K, Dewhurst S. Methamphetamine causes sustained depression in cerebral blood flow. Brain Res. 2011;1373:91–100. doi: 10.1016/j.brainres.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pubill D, Chipnan C, Camins A, Pallás M, Camarasa J, Escubedo E. Free radical production induced by methamphetamine in rat striatal synaptosomes. Toxicol and App Pharmaco. 2005;204 :57–68. doi: 10.1016/j.taap.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Riddle EL, Fleckenstein AE, Hanson GR. Mechanisms of METH-induced dopaminergic neurotoxicity. AAPS J. 2006;8:E413–E418. doi: 10.1007/BF02854914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Sood R, Weaver J, Timmins GS, Schnell A, Miyake M, Kao JPY, Rosen GM, Liu KJ. Direct visualization of mouse brain oxygen distribution by electron paramagnetic resonance imaging: Application to focal cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1695–1703. doi: 10.1038/jcbfm.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba T, Yamato M, Kudo W, Watanabe T, Utsumi H, Yamada KI. In vivo imaging of mitochondrial function in methamphetamine-treated rats. Neuroimage. 2001;57:866–872. doi: 10.1016/j.neuroimage.2011.05.041. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hayashi T, Chang C-F, Chiang Y-H, Tsao L-I, Su T-P, Borlongan C, Lin S-Z. Methamphetamine potentiates ischemia/reperfusion insults after transient middle cerebral artery ligation. Stroke. 2001;32:775–782. doi: 10.1161/01.str.32.3.775. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Zauner A, Daugherty WP, Bullock MR, Warner DA. Brain oxygenation and energy metabolism: part 1- biological function and pathophysiology. Neurosurgery. 2002;51:289–302. [PubMed] [Google Scholar]