

Abstract
Background
Atopic dermatitis (AD) is a common skin disease that is characterized by recurrent episodes of itching. Genetic variation associated with the persistence of AD has not been described for African-Americans.
Objective
To evaluate genetic variation of Filaggrin-2 (FLG2) in African-Americans with AD.
Methods
We evaluated a multiyear prospective cohort study of African-American children with AD with respect to FLG2 variation based on whole exome sequencing followed by a targeted analysis. We ultimately evaluated the association of rs rs12568784 and rs16833974 with the respect to the persistence of AD symptoms over time.
Results
Whole exome analysis was conducted on 60 subjects revealing premature stop codon in exon 3 at S2377X (rs12568784), X2392S (rs150529054), and a large exon 3 deletion mutation Q2053del224. Based on a priori criteria we then studied rs12568784, rs16833974 (H1249R) and Q2053del224. We noted that S2377X (OR = 0.44; 95% CI: 0.25, 0.46) and H1249R (0.23; 0.12, 0.46) were significantly less likely to be free of symptoms of AD and Q2053del224 (0.54; 0.16, 1.80) trended toward this outcome. S2377X and H1249R were in high linkage disequilibrium (D′=0.95).
Conclusions
In an African-American cohort with AD, FLG2 mutations were associated with more persistent AD. This is the first finding of genetic variation of a skin barrier protein in those of African ancestry with AD.
Clinical Implications
FLG2 variation is associated with more persistent AD in children of African ancestry.
Keywords: Atopic Dermatitis, Filaggrin, Epidemiology, Genetic Epidemiology, Atopy
Introduction
Atopic dermatitis (AD) is a common chronic relapsing disease that usually begins in childhood. It is found worldwide, although its prevalence varies by geographic location1;2. Recent studies have elegantly demonstrated that in some geographic locations more than 20% of individuals afflicted with AD have a genetic predisposition for this disorder3–9. The most common gene associated with AD is Filaggrin (FLG), which initially produces the profillagrin protein that is processed in the stratum granulosum into the FLG protein. FLG contributes to the barrier function of the stratum corneum of the skin. An intact skin barrier is presumed to protect the skin from penetration by environmental irritants and allergens. FLG loss-of-function mutations have been extensively described in people of European and Asian ancestry and these mutations vary by race 4;10. Among people of European ancestry, four FLG loss-of-function mutations have been consistently associated with the risk of developing AD or the risk of having persistent AD4;11. However, despite extensive evaluation, FLG variation has rarely been noted to be associated with AD in people of African ancestry4;10–12.
Why FLG loss-of-function mutations are not commonly found in people of African ancestry is currently unknown11–13. We recently noted that in a cohort of US children with AD, nearly 30% of the white children had a common European FLG loss-of-function mutation whereas the proportion was less than 6% in their African-American counterparts11. Further, attempts by our team and others to discover new FLG mutations in the African-American population have not been fruitful4;11;14. The FLG gene is in a technically difficult section of the genome to sequence because of highly redundant base sequences; so it is possible that important variations are still to be discovered15;16. However, it is also possible individuals of African ancestry with AD do not have inherited defects of skin barrier function due to FLG and that the defect is associated with another “skin barrier” gene.
Several other genes, identified through candidate gene and genome wide association studies, have been associated with AD; although many have not been replicated in additional investigations4;7;8;16–18. These discoveries include genes that are found in close proximity to FLG in a section of chromosome one (1q21) called the epidermal differentiation complex (EDC). The EDC contains several genes that encode proteins important for late epidermal differentiation. Six genes located within the EDC and close to FLG have structures similar to FLG15;19;20. Proteins coded for by these genes are characterized as S100-fused type proteins (SFTP) and contribute to skin barrier function in the stratum corneum15. The SFTPs include profilaggrin (FLG), hornerin (HRNR), filaggrin-2 (FLG2), repetin (RPTN), cornulin (CRNN), trichohyalin (TCHH), and trichohyalin-like 1(TCHHL1).
The structure and function of FLG2 protein has recently been described20;21. The FLG2 protein appears to have a role similar to FLG with respect to skin barrier function20;21. A previous study found that a FLG2 missense variant (rs16833974) was not associated with AD in Europeans 19;20. The goal of our study was to evaluate genetic variation of FLG2 in African-Americans with AD and then determine if FLG2 variation was associated with the persistence of AD.
Methods
Population
Subjects for this study were enrolled in the Pediatric Eczema Elective Registry (PEER), www.thepeerprogram.com, which is an ongoing prospective 10-year observational registry that is part of a post-marketing commitment originally by Novartis and now Valeant to the FDA and the European Medicines Agency. The enrollment criteria and goals of the PEER study have been described in detail elsewhere11;22. The diagnosis of AD for each child was made by the enrolling physicians, the majority of whom were pediatricians, allergists and dermatologists11;23. The diagnosis was confirmed based on the UK working party criteria22;24. Children/parents in the PEER study who also enrolled in our current study completed an additional informed consent approved by the Institutional Review Board of the University of Pennsylvania and provided a saliva sample from which DNA was extracted. Our study included subjects who self-described as African-American and provided saliva/DNA (N=380). A panel of ancestry informative markers (AIMS) was used to estimate genetic ancestry using clustering techniques as implemented in the program STRUCTURE11;25. Self-reported African ancestry was highly correlated with genetically derived ancestry based on these AIMS11. The percent African ancestry for subjects analyzed in this study was approximately 75% on average.
Outcome
We investigated the self-reported outcome of whether or not a child’s skin, without requiring the use of topical medication (e.g., steroids or calcineurin inhibitors to treat their AD), was AD symptom-free for the previous 6-months. Information about this outcome was collected longitudinally every six months by a survey thereby capturing the waxing and waning nature of AD.
Whole exome sequencing
We randomly selected 60 of the 380 eligible African-American individuals for whole exome sequencing. For this study, the primary goal of whole exome analysis was the discovery of new variants of FLG2 that were likely to be loss-of-function mutations in African-Americans with AD. The exome sequencing was conducted by Ambry Genetics (Aliso Viejo, California) utilizing whole exome target enrichment by Agilent SureSelect technology. The libraries were indexed 100 bp paired end and processed using Illumina HiSeq2000 at 100x coverage per exome. The data was then evaluated using two separate pipelines; a proprietary pipeline established by Ambry and a pipeline created at the University of Pennsylvania based on the best practices guidelines of the Broad Institute. The SFTP genes, including FLG, have three exons15;21;26. The tiny first exon is non-coding whereas the small second exon is responsible for initiating translation. The large third exon contains sequence information for the precursor protein15;21;26. It has been hypothesized that stop-gain (nonsense) mutations of SFTP genes in exon 3 are loss-of-function mutations12;15;16;19;20;27. Because of the structural similarity between FLG and FLG2, we targeted stop-gain variants and insertion/deletion variants of exon 3 of FLG2 present in more than one subject 4;16;19;20. Further, we required these variants to be present in more than one individual, in order to minimize false discovery, to avoid further study of sporadic mutations, and to achieve our goal of investigating more common variants (minor allele frequency (MAF) >0.05). We also decided a priori to pursue rs12568784, which was a nonsense mutation of exon 3 previously studied in European subjects19.
Targeted analysis
As noted in the Results section, three variants were analyzed further. As appropriate, SNPs were genotyped via TaqMan® assay using TaqMan® 5′ nuclease PCR primers and probes designed by Life Technologies Corporation Assay-By-DesignSM custom oligonucleotide reagent service (Life Technologies, Carlsbad, CA). Each probe consisted of an oligonucleotide with a fluorescent reporter dye, a non-fluorescent quencher and minor groove binder. Allele-specific cleavage of probes was detected using different reporter dyes for each probe (6FAM and VIC fluorophores for each allele) and evaluated using an ABI 7900 HT Sequence Detection System. PCR end-point fluorescence levels were measured automatically by using the SDS 2.3 manufacturer’s custom software (Life Technologies). Allelic discrimination results were then graphed on a scatter plot contrasting reporter dye florescence (i.e., Allele A vs. Allele a).
The FLG2 deletion mutation was assayed by fragment analysis on ABI 3130XL capillary sequencer. The primers for a PCR product covering the deletion region were designed by Primer 3 software. For generation of fluorescent PCR products the forward primer was labeled in the 5′ end with VIC dye. The primer sequences and the PCR protocol are available on request. The PCR products were run on 3130XL using LIZ 600 as size standard and analyzed by GeneMapper v4.1.
Individuals in this study were followed longitudinally and surveyed every six months. Hence, we assessed the statistical association between the repeated measures of the binary outcome (presence or absence of AD) and each variant using a mixed-effects framework called a generalized linear latent and mixed model (GLLAMM) which accounts for the correlation of the outcomes within patient over time. All analyses assumed an additive genetic model. 28;29; However, we have also presented results from a dominant model wherever appropriate.
Results
Characteristics of the study subjects
Out of 380 African-American subjects who provided DNA, 81 were excluded from further analysis due to insufficient DNA. The median age (in years) at enrollment into PEER and that of AD onset were 6.8 (interquartile range (IQR) = 4.1, 10.2) and 0.8 (IQR = 0.3, 3) respectively. Male subjects accounted for 56.5% (n = 169) of the cohort. At enrollment, 47.5% (N=142) had limited or uncontrolled AD, 55.5% (N=166) had a history of asthma or wheezing, 68.2% (N=204) had a history of seasonal allergies, and 25.4% reported food allergies (N=76). The mothers of 21.7% (N=65) had a history of eczema and the fathers of 15.4% (N=46) had eczema. At enrollment, no child had experienced skin free of symptoms of AD while not using topical medications in the last 6 months.
FLG2 exome analysis to identify potential loss of function mutations (N=60)
A premature stop codon was noted in exon 3 at S2377X (rs12568784) and another at X2392S (rs150529054). Rs12568784 was found in 16 subjects and rs150529054 was noted in only one subject (and therefore not studied further). A deletion mutation was also noted at Q2053del224 in 4 subjects. By exome sequencing, all variants were heterozygotes. As per our a priori decision to evaluate premature stop codon and deletion variants that occurred in more than 1 subject, S2377X and Q2053del224 were further followed up in the full cohort. H1249R (rs16833974) on exon 3, which was present in 14 subjects, was also included in the next phase of the study because it was a nonsynonymous variant of prior interest (Table 1).
Table 1.
General information about FLG2 variants in African-Americans noted in a whole exome analysis (n=60).
| Type of variation | Location | dbSNP | Variant Allele |
|---|---|---|---|
| Stop-gain | NM_001014342:exon3:c.C7130A:p.S2377X | rs12568784 | G>T |
| Nonsynonymous | NM_001014342:exon3:c.A3746G:p.H1249R | rs16833974 | T>C |
| Deletion mutation | NM_001014342:exon3:c.5937_6161delp.A1979_Q2053del | N/A | Deletion of 224 base pairs |
FLG2 loss-of-function mutations in the African-American PEER cohort (N=299)
The minor allele frequencies (MAFs) and results from association testing of rs12568784, rs16833974 and Q2053del224 are presented in Tables 2 and 3 and Figures 1 and 2. Rs12568784, which is a single base pair substitution of T for G resulting in a premature stop codon, fulfilled the previously defined criteria for FLG by Sandilands et al as a loss-of-function mutation16. Children with this variant, which had a MAF of 0.22, were more likely to have persistent symptoms of AD requiring topical therapy (OR = 0.44; 95% CI: 0.25, 0.46) (Table 3). Rs16833974, is a single base pair substitution of T for C, had a MAF of 0.24, and was also associated with the outcome (0.23 (0.12, 0.46)) (Table 3 & Figure 1). These two loci were highly concordant (kappa=0.68, R=0.77, and 82.1% agreement) and were in high linkage disequilibrium (LD) (D′=0.95) (As represented in HapMap see Figure 230). Q2053del224, with a MAF of 0.04, was not statistically associated but did have a trend toward increased persistence (0.54 (0.16, 1.80)). None of the studied variants were associated with an increased risk of asthma, seasonal allergies, or food allergies (Table 3).
Table 2.
Minor allele and genotype frequencies for the African-American PEER cohort.
| Variant | Minor allele frequency | Genotype frequency | ||
|---|---|---|---|---|
| African - American PEER Cohort (n = 299) | AA | Aa | aa | |
| S2377X (rs12568784) | 0.22 | 0.55 | 0.39 | 0.06 |
| H1249R (rs16833974) | 0.24 | 0.58 | 0.37 | 0.05 |
| Q2053del224 | 0.04 | 0.92 | 0.08 | 0 |
Table 3.
Association of FLG2 variants in the African-American PEER cohort with the persistence of AD, asthma, seasonal allergy and food allergy at study entry. OR = odds ratio; CI = confidence intervals
| Variant | OR (95% CI) ADD p-value | ||||
|---|---|---|---|---|---|
| Additive genetic model * | Dominant genetic model* | Asthma+ | Seasonal allergy+ | Food allergy+ | |
| S2377X (rs12568784) | 0.44 (0.25, 0.77) p=0.004 |
0.43 (0.21, 0.88) p=0.022 |
1.05 (0.70,1.57) p=0.8010 |
1.01 (0.66,1.56) p=0.625 |
0.96 (0.84, 1.11) p=0.928 |
| H1249R (rs16833974) | 0.23 (0.12, 0.46) p=0.00002 |
0.23 (0.11, 0.46) p=0.00004 |
1.12 (0.66,1.90) p=0.677 |
0.89 (0.48,1.44) p=0.391 |
0.79 (0.42, 1.47) p=0.451 |
| Q2053del224 | 0.54 (0.16, 1.80) p=0.234 |
---- | 0.88 (0.34,2.32) p=0.804 |
0.58 (0.23, 1.52) p=0.220 |
1.42 (0.71, 2.81) p=0.319 |
GLLAMM model;
Logistic regression model
Figure 1.
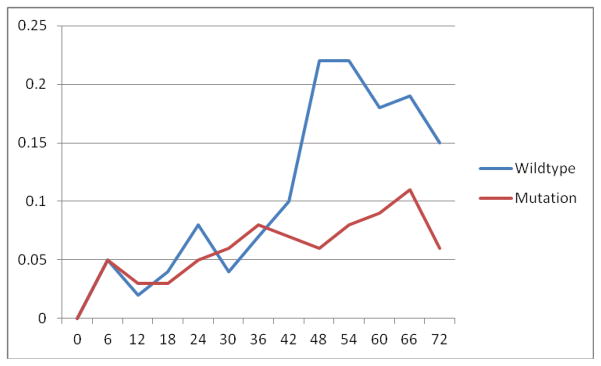
Percent individuals reporting skin symptom free off of medication by rs16833974 over the first 6 years of follow up
Figure 2.


LD plots for rs16833974 (A.) and rs12568784 (B.) utilizing the ASW population demonstrating LD relationships (brightness and size of diamond is proportional to R-squared) for these variants with respect to the FLG2 as well as other nearby genes of the EDC.
Discussion
Mutations in the FLG gene have been associated with both risk of developing AD and persistence of AD in European and Asian populations4. However, FLG mutations have not been consistently or commonly found in African-Americans with AD4;11;12. Investigators have postulated that other proteins involved in skin barrier function could, like FLG, be associated with AD15;19;20. However, this hypothesis, which has been tested mainly in Europeans, has not been fully substantiated15;19–21. Like FLG, FLG2 is located within the EDC15;19;20. To the best of our knowledge, our study is the first report showing that a common loss-of-function mutation of FLG2 is associated with the persistence of AD in African-American children. Children with the FLG2 loss-of-function mutation, rs12568784, or the nonsynonymous variant rs16833974, which was in high LD with rs12568784, were more than 50% more likely to have persistent AD as compared to those without these variants. The magnitude of this finding is similar to previous findings in Europeans with FLG loss-of-function mutations 11. The presence of FLG2 variants was not associated with asthma, seasonal allergies, or food allergies. Finally, the presence of a large deletion mutation of FLG2 showed a trend toward an association with persistent AD.
We did not have direct access to study participants because of the design of the PEER study. Previous studies have shown that the FLG2 protein, which resides in the stratum corneum, is an important component of skin barrier function, and is diminished as a consequence of skin inflammation15;19–21. Thus it has been conjectured, but not proven, that diminished FLG2 production could be associated with the risk of developing AD in a fashion similar to diminished or absent production of FLG15;19–21. We hypothesize that rs12568784 variant is a loss-of-function mutation, because like the FLG loss-of-function mutations, rs12568784 represents a stop codon in exon 3. If true, children with this variant are unable to produce or produce diminished amounts of FLG24;16. This is consistent with current knowledge of the pathophysiology of AD. However, the two reported FLG2 variants are in high LD with each other and other variants of the gene (see Figure 2) so even if they are not loss-of-function mutations it is likely that another variant will be.
The presumption is that FLG2 protein and the other SFTPs, like FLG, ultimately contribute to skin barrier function as part of skin natural moisturizing factor9. A recent study examined expression of structural proteins and regulatory molecules in the stratum corneum after experimental disruption of skin barrier in human volunteers with AD and psoriasis and demonstrated that both FLG and FLG2 levels were decreased as a consequence of barrier disruption31. The production of both FLG and FLG2 have also been shown to be diminished in lesional as well as non-lesional skin of AD patients15;20. This reduction, which was more pronounced in the lesional skin and occurred independent of FLG genotypes, was presumably due to the influence of pro-inflammatory cytokines. This study failed to find a link between FLG null mutations and reduction in protein production31. However, it is to be noted that the study investigated only the 4 common mutations found in Europeans; therefore the possibility of a less common, non-genotyped mutation or copy number variation within the FLG gene having influenced the results cannot be excluded.
Alterations in the bacterial flora of children with AD have been associated with flares of AD32. Recent studies have implied that FLG, FLG2, and, another SFTP, HRNR may function as part of cutaneous antimicrobial defenses as they are further processed in the stratum corneum to become peptides that make the skin surface inhospitable for pathogenic bacteria32–38. Further, as reported by Broccardo et al. skin barrier proteins including FLG2 can act as powerful antimicrobial agents against Staphylococcus aureus, which is known to be associated with exacerbations of AD32;39. It is now well accepted that AD is a disorder resulting at least partially from immune dysfunction, with TH2 cells playing a bigger part during the acute manifestation of the disease whereas TH1 cells assume greater importance during the chronic phase40–42. The TH1 cells secrete several cytokines including gamma interferon and IL-1241–43. Bacterial colonization propagates immune dysfunction and stimulates TH1 cells to secrete gamma interferon, which eventually leads to macrophage activation thus perpetuating the chronic nature of the disease. Deficiency of FLG2, therefore, could encourage bacterial colonization, which could in turn lead to persistence of AD.
Most genetic studies of AD to date have evaluated the risk of developing AD; however, very few have studied the persistence of AD as an outcome. Often when persistence was measured, it was measured as time to when the child first noted that he/she was symptom free44. This might not be the appropriate definition of persistence for a chronic waxing and waning illness like AD. In our study as well as in a previous report, we focused on the persistence of AD over years of observation, which mimics the natural history of AD 11. In fact, we believe that our definition of outcome is clinically more meaningful since it takes into account the chronicity as well as the intermittently remitting and relapsing character of AD 11;44.
The two FLG2 mutations associated with the persistence of AD in African-Americans are either absent or only infrequently found in people of European origin. The variant allele of rs16833974 is not found in the HapMap database of Utah residents with Northern and Western European Ancestry (CEU) (as reference MAF in American of African Ancestry in the South West (ASW) = 0.26; browser. 1000genomes.org). Why FLG loss-of-function mutations that are prevalent in whites are not found consistently in people of African ancestry and while the opposite appears true with respect to FLG2 is not clear. However, all these mutations appear to be functionally similar; i.e., they diminish or stop the production of skin barrier proteins irrespective of their location in the EDC. It is tempting to speculate that these mutations arose in different populations during different time periods and were propagated selectively due to a multitude of factors that could include natural selection, genetic drift or a population bottleneck effect45. Whether these mutations confer any survival advantage is a matter of ongoing debate.
An important limitation of our study was that we were not able to evaluate the presence or absence of FLG2 protein in the skin of our study subjects. As a result, we can only hypothesize (as others have also done) that based on previous reports of a structurally related FLG, the mutations noted in our study would diminish the production of FLG215;19;21;26. Also, it is important to note that our study was limited to subjects with mild to moderate AD, which is the approved patient population to receive pimecrolimus. Our results are also not directly comparable to other studies that have focused on genetic factors that are associated with the onset of AD. Our study design did not allow us to explore whether children with rs12568784 or rs16833974 were at increased risk for developing AD.
In conclusion, we found that two mutations in FLG2, rs12568784 and rs16833974, were associated with more persistent AD in African-American children. This is the first report of this finding. Of particular interest is that rs12568784 is a stop-gain mutation in exon 3 and is in a high degree of LD with rs16833974. Ultimately functional studies will be needed to confirm that these mutations could lead to absence of or diminished ability to produce FLG2. While mutations in FLG that are common in Europeans and Asians have not been consistently found in individuals of African ancestry, the results of our study indicate that in African-Americans, AD could be associated with mutations in another skin barrier protein like FLG2.
Acknowledgments
This study was funded by R01-AR0056755 from the National Institute of Arthritis Musculoskeletal and Skin Diseases and from a grant from Valeant Pharmaceuticals for the PEER study.
Abbreviations
- AD
Atopic Dermatitis
- AIMS
Ancestral informative markers
- CI
Confidence interval
- CRNN
Cornulin
- EDC
Epidermal Differentiation Complex
- FLG
Filaggrin
- FLG2
Filaggrin-2
- GLLAMM
Generalized linear latent and mixed models
- HRNR
Hornerin
- MAF
Minor allele frequency
- OR
Odds ratio
- PEER
Pediatric Eczema Elective Registry
- RPTN
Repetin
- SFTP
S100-fused type protein
- TCHH
Trichohyalin
- TCHHL1
Trichohyalin-like 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Williams HC. Epidemiology of atopic dermatitis. Clinical & Experimental Dermatology. 2000;25:522–9. doi: 10.1046/j.1365-2230.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 2.Asher MI, Montefort S, Bjorksten B, et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet. 2006;368:733–43. doi: 10.1016/S0140-6736(06)69283-0. [DOI] [PubMed] [Google Scholar]
- 3.Brown SJ, Relton CL, Liao H, et al. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. British Journal of Dermatology. 2009;161:884–9. doi: 10.1111/j.1365-2133.2009.09339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown SJ, McLean WH. One remarkable molecule: filaggrin. Journal of Investigative Dermatology. 2012;132:751–62. doi: 10.1038/jid.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marenholz I, Nickel R, Ruschendorf F, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. Journal of Allergy & Clinical Immunology. 2006;118:866–71. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 6.Sandilands A, O’Regan GM, Liao H, et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. Journal of Investigative Dermatology. 2006;126:1770–5. doi: 10.1038/sj.jid.5700459. [DOI] [PubMed] [Google Scholar]
- 7.Paternoster L, Standl M, Chen CM, et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nature Genetics. 2012;44:187–92. doi: 10.1038/ng.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez E, Baurecht H, Herberich E, et al. Meta-analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. Journal of Allergy & Clinical Immunology. 2009;123:1361–70. doi: 10.1016/j.jaci.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 9.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. New England Journal of Medicine. 2011;365:1315–27. doi: 10.1056/NEJMra1011040. [DOI] [PubMed] [Google Scholar]
- 10.Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. British Journal of Dermatology. 2010;162:472–7. doi: 10.1111/j.1365-2133.2009.09582.x. [DOI] [PubMed] [Google Scholar]
- 11.Margolis DJ, Apter AJ, Gupta J, et al. The persistence of atopic dermatitis and Filaggrin mutations in a US longitudinal cohort. Journal of Allergy & Clinical Immunology. 2012;130:912–7. doi: 10.1016/j.jaci.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winge MC, Bilcha KD, Lieden A, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. British Journal of Dermatology. 2011;165:1074–80. doi: 10.1111/j.1365-2133.2011.10475.x. [DOI] [PubMed] [Google Scholar]
- 13.Gao PS, Rafaels NM, Hand T, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. Journal of Allergy & Clinical Immunology. 2009;124:507–13. doi: 10.1016/j.jaci.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winge MC, Bilcha KD, Lieden A, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. British Journal of Dermatology. 2011;165:1074–80. doi: 10.1111/j.1365-2133.2011.10475.x. [DOI] [PubMed] [Google Scholar]
- 15.Henry J, Toulza E, Hsu CY, et al. Update on the epidermal differentiation complex. Frontiers in Bioscience. 2012;17:1517–32. doi: 10.2741/4001. [DOI] [PubMed] [Google Scholar]
- 16.Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nature Genetics. 2007;39:650–4. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 17.Weidinger S, O’Sullivan M, Illig T, et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. Journal of Allergy & Clinical Immunology. 2008;121:1203–9. doi: 10.1016/j.jaci.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. Journal of Allergy & Clinical Immunology. 2010;125:16–29. doi: 10.1016/j.jaci.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marenholz I, Rivera VA, Esparza-Gordillo J, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. Journal of Investigative Dermatology. 2011;131:1644–9. doi: 10.1038/jid.2011.90. [DOI] [PubMed] [Google Scholar]
- 20.Pellerin L, Henry J, Hsu CY, et al. Defects in filaggrin-like proteins in both lesional and nonlesional atopic skin. Journal of Allergy & Clinical Immunology. 2013;131:1094–102. doi: 10.1016/j.jaci.2012.12.1566. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z, Hansmann B, Meyer-Hoffert U, et al. Molecular identification and expression analysis of filaggrin-2, a member of the S100 fused-type protein family. PLoS ONE [Electronic Resource] 2009;4:e5227. doi: 10.1371/journal.pone.0005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapoor R, Hoffstad O, Bilker W, et al. The frequency and intensity of topical pimecrolimus treatment in children with physician-confirmed mild to moderate atopic dermatitis. Pediatric Dermatology. 2009;26:682–7. doi: 10.1111/j.1525-1470.2009.01013.x. [DOI] [PubMed] [Google Scholar]
- 23.Margolis DJ, Papadopoulos M, Apter AJ, et al. Obtaining DNA in the mail from a national sample of children with a chronic non-fatal illness. Journal of Investigative Dermatology. 2011;131:1765–7. doi: 10.1038/jid.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams HC, Burney PG, Hay RJ, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. I. Derivation of a minimum set of discriminators for atopic dermatitis. British Journal of Dermatology. 1994;131:383–96. doi: 10.1111/j.1365-2133.1994.tb08530.x. [DOI] [PubMed] [Google Scholar]
- 25.Pritchard JK, Stephens M, Rosenberg NA, et al. Association mapping in structured populations. American Journal of Human Genetics. 2000;67:170–81. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandilands A, Smith FJ, Irvine AD, et al. Filaggrin’s fuller figure: A glimpse into the genetic architecture of atopic dermatitis. Journal of Investigative Dermatology. 2007;127:1282–4. doi: 10.1038/sj.jid.5700876. [DOI] [PubMed] [Google Scholar]
- 27.Smith FJ, Irvine AD, Terron-Kwiatkowski A, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nature Genetics. 2006;38:337–42. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- 28.Rabe-Hesketh S, Pickles A, Skrondal A. GLLAMM Manual. 2001/01, 1–138. 10-28-2001. Institute of Psychiatry, Kiing’s College, University of London; [Google Scholar]
- 29.Rabe-Hesketh S, Skrondal A, Pickles A. Reliable estimation of generalized linear mixed models using adaptive quadrature. The Stata Journal. 2002;2:1–21. [Google Scholar]
- 30.Johnson AD, Handsaker RE, Pulit S, et al. SNAP: A web based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–9. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Koning HD, van den Bogaard EH, Bergboer JG, et al. Expression profile of cornified envelope structural proteins and keratinocyte differentiation-regulating proteins during skin barrier repair. British Journal of Dermatology. 2012;166:1245–54. doi: 10.1111/j.1365-2133.2012.10885.x. [DOI] [PubMed] [Google Scholar]
- 32.Kong HH, Oh J, Deming C, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Research. 2012;22:850–9. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glaser R, Harder J, Lange H, et al. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nature Immunology. 2004;6:57–64. doi: 10.1038/ni1142. [DOI] [PubMed] [Google Scholar]
- 34.Harder J, Schroder JM, Glaser R. The skin surface as antimicrobial barrier:present concepts and future outlooks. Experimental Dermatology. 2012;22:5. doi: 10.1111/exd.12046. [DOI] [PubMed] [Google Scholar]
- 35.Donovan M, Thomas-Colligon A, Rain J, et al. The N terminal S100 domain of filaggrin activates the skin aspartic acid protease in the epidermis of human skin, in vitro. Journal of Investigative Dermatology. 2013;133:S109. [Google Scholar]
- 36.Hsu C, Mechin M, Raymond A, et al. Hornerin is a new target of calpain-1. Journal of Investigative Dermatology. 2013;133:S124. doi: 10.1016/j.jid.2016.09.030. [DOI] [PubMed] [Google Scholar]
- 37.Natsuga K, Cipolat S, Watt F. Skin barrier defect allows more abundent microbiota in skin and induces skin inflammation and the expression of antibmicrobial peptides independently of microbiota. Journal of Investigative Dermatology. 2013;133:S192. [Google Scholar]
- 38.McAleer M, Irvine A. The multifunctional role of filaggrin in allergic skin disease. Journal of Allergy & Clinical Immunology. 2013;131:280–91. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- 39.Broccardo CJ, Mahaffey S, Schwarz J, et al. Comparative proteomic profiling of patients with atopic dermatitis based on a history of eczema herpecticum infection and Staphylococcus aureus colonization. J Allergy Clin Immunol. 2011;127:186–93. doi: 10.1016/j.jaci.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunological Reviews. 2011;242:233–46. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandt EB, Sivaprasad U. Th2 cytokines and atopic dermatitis. J Clin Cell Immunol. 2011;2:110. doi: 10.4172/2155-9899.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bieber T. Atopic Dermatitis. New England Journal of Medicine. 2008;358:1483–94. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 43.Rahman RS, Collins M, Williams CM, et al. The pathology and immunology of atopic dermatitis. Inflamm Allergy Drug Targets. 2011;10:486–96. doi: 10.2174/187152811798104935. [DOI] [PubMed] [Google Scholar]
- 44.Henderson J, Northstone K, Lee SP, et al. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. Journal of Allergy & Clinical Immunology. 2008;121:872–7. doi: 10.1016/j.jaci.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 45.Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]