

Abstract
Mice with functional genetic ablation of the Tacr1 (substance P-preferring receptor) gene (NK1R−/−) are hyperactive. Here, we investigated whether this is mimicked by NK1R antagonism and whether dopaminergic transmission is disrupted in brain regions that govern motor performance. The locomotor activity of NK1R−/− and wild-type mice was compared after treatment with an NK1R antagonist and/or psychostimulant (d-amphetamine or methylphenidate). The inactivation of NK1R (by gene mutation or receptor antagonism) induced hyperactivity in mice, which was prevented by both psychostimulants. Using in vivo microdialysis, we then compared the regulation of extracellular dopamine in the prefrontal cortex (PFC) and striatum in the two genotypes. A lack of functional NK1R reduced (>50%) spontaneous dopamine efflux in the prefrontal cortex and abolished the striatal dopamine response to d-amphetamine. These behavioural and neurochemical abnormalities in NK1R−/− mice, together with their atypical response to psychostimulants, echo attention deficit hyperactivity disorder (ADHD) in humans. These findings prompted genetic studies on the TACR1 gene (the human equivalent of NK1R) in ADHD patients in a case-control study of 450 ADHD patients and 600 screened supernormal controls. Four single-nucleotide polymorphisms (rs3771829, rs3771833, rs3771856, and rs1701137) at the TACR1 gene, previously known to be associated with bipolar disorder or alcoholism, were strongly associated with ADHD. In conclusion, our proposal that NK1R−/− mice offer a mouse model of ADHD was borne out by our human studies, which suggest that DNA sequence changes in and around the TACR1 gene increase susceptibility to this disorder.
Keywords: ADHD, d-amphetamine, dopamine, locomotor hyperactivity, methylphenidate, microdialysis, NK1R/TACR1, psychostimulant, single nucleotide polymorphism
Introduction
Substance P-preferring, neurokinin receptors (NK1R) are concentrated in brain regions that govern mood, cognition and motor performance, including the response to reward and stress (Rigby, et al., 2005). Mice with functional ablation of the NK1R gene (De Felipe, et al., 1998) (NK1R−/−) are hyperactive when compared with their wild-type (NK1R+/+) (Herpfer, et al., 2005; Fisher, et al., 2007) and their response to reward is disrupted. For instance, NK1R−/− mice develop conditioned place preference with cocaine, but not d-amphetamine or morphine (Murtra, et al., 2000; Gadd, et al., 2003). Further, they are more sensitive to the depressant effects of alcohol than their wild-type (George, et al., 2008), and they do not self-administer morphine (Ripley, et al., 2002) or develop locomotor sensitization when given this opiate (Ripley, et al., 2002; Gadd, et al., 2003).
There is evidence that α2A-autoreceptors are desensitized in the locus coeruleus of NK1R−/− mice (Herpfer, et al., 2005; Fisher, et al., 2007). This is consistent with reports that antagonism of NK1R increases burst-firing of noradrenergic neurones in this nucleus and release of noradrenaline from their terminals (Herpfer, et al., 2005; Fisher, et al., 2007; Gobbi, et al., 2007). Excessive noradrenergic transmission would disrupt attentional aspects of task performance (Aston-Jones and Cohen, 2005; Bouret and Sara, 2005) and augment release of serotonin in the forebrain (Froger, et al., 2001; Gobbi, et al., 2007); both these actions would impair response control (‘impulsivity’) (Winstanley, et al., 2006).
Hyperactivity, inattention, and impulsivity are core features of attention deficit hyperactivity disorder (ADHD), which affects between 2% and 5% of children in the UK and continues into adulthood. The disruption of the regulation of noradrenergic (reviewed by Brennan and Arnsten, 2008) and serotonergic (reviewed by Oades, 2008) transmission is thought to be amongst the neurobiological abnormalities underlying ADHD. To date, our findings suggest that all these behavioural and neurochemical changes can be secondary to blunted NK1R function. It follows that the disruption of NK1R could be a causal factor in ADHD.
Mounting evidence suggests that disruption of dopaminergic transmission in corticostriatal circuits of the brain is another prominent feature of ADHD (reviewed by Prince, 2008). The hyperactivity of NK1R−/− mice, together with their atypical response to certain rewarding stimuli, is consistent with this proposal. On this basis, the present experiments investigated whether dopaminergic transmission is abnormal in NK1R−/− mice. First, their locomotor response to the psychostimulants, methylphenidate (which blocks reuptake of dopamine) or d-amphetamine (a dopamine releasing agent), was compared with that of wild-type mice. Then, we used microdialysis to monitor extracellular dopamine in the dorsal striatum and prefrontal cortex (PFC): we compared both basal efflux and the effects of an acute challenge with an NK1R antagonist and/or d-amphetamine in the two genotypes. In both behavioural and neurochemical experiments, we tested whether the NK1R−/− mouse phenotype was mimicked by treating wild-types with an NK1R antagonist. The findings from all these experiments (described below) consolidated the status of NK1R−/− mice as a model of ADHD.
Prompted by the above evidence, we went on to genotype polymorphisms near or within the human TACR1 gene (the human equivalent of NK1R) in an ADHD case–control sample. However, ADHD is not a unitary disorder. In one subgroup, children also have conduct disorders and a greatly increased risk of criminality and alcoholism in adulthood. A second subgroup has been linked to parents with affective disorders: one study of parents with bipolar disorder (BPAD) found that a high proportion of their children were diagnosed as having ADHD (Hirshfeld-Becker, et al., 2006). Comorbidity between BPAD and alcoholism is also well established. Against this background, it is striking that the TACR1 gene, which is found on chromosome 2, is localised within a region linked to a combined alcoholism and conduct disorder phenotype (Dick, et al., 2004; Wiener, et al., 2005). The findings reported here strongly suggest that polymorphisms of TACR1 could distinguish a genetically defined subgroup of ADHD patients who have increased vulnerability to comorbid bipolar disorder and alcoholism.
We believe this is the first example of a mutant mouse phenotype leading to the discovery of a genetic susceptibility to a psychiatric disorder.
Materials and methods
Subjects
Mice
All procedures complied with the UK Animals (Scientific Procedures) Act, 1986. We used male mice (25–35 g) with a 129/Sv × C57BL/6 genetic background backcrossed with an outbred MF1 strain. The colony was maintained at UCL as described previously (Herpfer, et al., 2005). The genotype of each mouse was verified postmortem.
Humans
UK National Health Service (NHS) Multicenter Research Ethics Committee (MREC) and Local Research Ethics Committee (LREC) approval was obtained. All subjects, or their guardians or parents, signed an approved consent form after reading an information sheet. DNA samples were collected from 450 ADHD probands from NHS services in the UK.
ADHD patients
All were of white European origin. ADHD subjects were selected only if both parents were English, Scottish or Welsh. Subjects were referred for research assessment if experienced clinicians diagnosed ADHD (DSM-IV criteria) and an IQ of >70. Only individuals fulfilling the recruitment criteria after the completion of research assessments were included in the study. In the ADHD subsample, assembled at Cardiff University, parental information on the child's symptoms was obtained using the Child and Adolescent Psychiatric Assessment (CAPA) (Angold, et al., 1995). Data on the pervasiveness of symptoms were obtained from teachers, using the Child ADHD Teacher Telephone Interview (ChATTI) (Holmes, et al., 2004). For the second subsample, information on ADHD symptoms at school was obtained with the Conners Teacher questionnaire (long form) and scored as present if rated greater than one. HYPESCHEME, a computerized operational criteria checklist and diagnostic algorithm for DSM-IV and International Classification of Diseases (10th Revision), was used to confirm the diagnosis of the London-based subsample. HYPESCHEME data sheets were completed with data from the parent and school questionnaires together with the review of case notes. The HYPESCHEME diagnoses were checked against researcher-applied DSM-IV criteria and discrepancies were reviewed by two researchers. Situational pervasiveness was operationalized as the presence of one or more ADHD symptoms at home and at school, together with parental descriptions of impairment from ADHD symptoms, in more than one setting. If necessary, final consensus was agreed with a senior clinical researcher at a case conference.
Controls
Screening of the controls for ancestry was carried out using a questionnaire to ensure that both parents were English, Scottish or Welsh with at least three grandparents having the same origins. One grandparent was allowed to be of another Caucasian European origin but not of Jewish or non-European Union (EU) ancestry (based on pre-2004 EU membership). All subjects were interviewed by a psychiatrist and recruited if there was no first-degree family history of any psychiatric disorder, according to self-report, including dissocial personality disorder, ADHD, schizophrenia, alcoholism, bipolar or unipolar disorder. Subjects were then interviewed with the SADS-L schedule and included only if there was no current or lifetime history of any research diagnostic criteria (RDC)-defined mental disorder (Spitzer and Endicott, 1977; 1978).
Mouse behaviour
Drug-naïve mice were placed individually in a light/dark exploration box (LDEB). They were allowed 90-min habituation in the dark zone (170 lux) as in the microdialysis studies, (below) (see Herpfer, et al., 2005; Fisher, et al., 2007). After 60 min, the mice were randomly assigned to groups given saline (10 mL/kg) or d-amphetamine (2.5 mg/kg, i.p.). After a further 30 min, they were transferred to a novel, light zone (350 lux) of the LDEB and allowed to commute between the compartments. Behaviour was recorded for 30 min and their locomotor activity (number of lines crossed) was scored blind. In a second experiment, methylphenidate (2.5 mg/kg; i.p.) replaced d-amphetamine but the protocol was otherwise the same.
When experiments involved two successive treatments, each mouse was placed in the LDEB, as before. After a further 30 min, they were randomly assigned to groups given vehicle (Tween 80 in 0.9% saline) or an NK1R antagonist: RP 67580 (5 or 10 mg/kg, i.p.). These test doses of RP 67580 are within the range of those reported to modify the behaviour of mice (Santarelli, et al., 2001; Yu, et al., 2002; Guiard, et al., 2004). After 30 min, pairs of subgroups were given either saline or d-amphetamine (2.5 mg/kg i.p.), and after a total of 90-min habituation, their locomotor activity was monitored. This experiment was repeated with another NK1R antagonist, L 733060, using the same doses (5 or 10 mg/kg, i.p.) so as to enable comparison of the effects of the two antagonists. Both compounds are believed to penetrate the blood-brain barrier (Bester, et al., 2001; Duffy, et al., 2002).
Microdialysis
A microdialysis probe (Cuprophan membrane, dialysis window of 1.0 mm) was implanted in halothane-anaesthetised mice in either the prefrontal cortex ((mm relative to Bregma): AP +2.1, ML+1.0, DV −2.0) or the dorsal striatum ((mm) AP + 1.10, ML+1.5, DV −3.3) (Paxinos and Franklin, 2001) (Figure 1). After overnight recovery, the probes were perfused with Ringer's solution (1.5 μL/min): stable (basal) efflux was confirmed in the home cage, before the mice were transferred to the LDEB where they were given either vehicle or an NK1R antagonist followed by saline or d-amphetamine at 30 and 60 min, respectively (see above). After 90 min, the mice were confined within the light zone of the LDEB (where they are most active).
Figure 1.
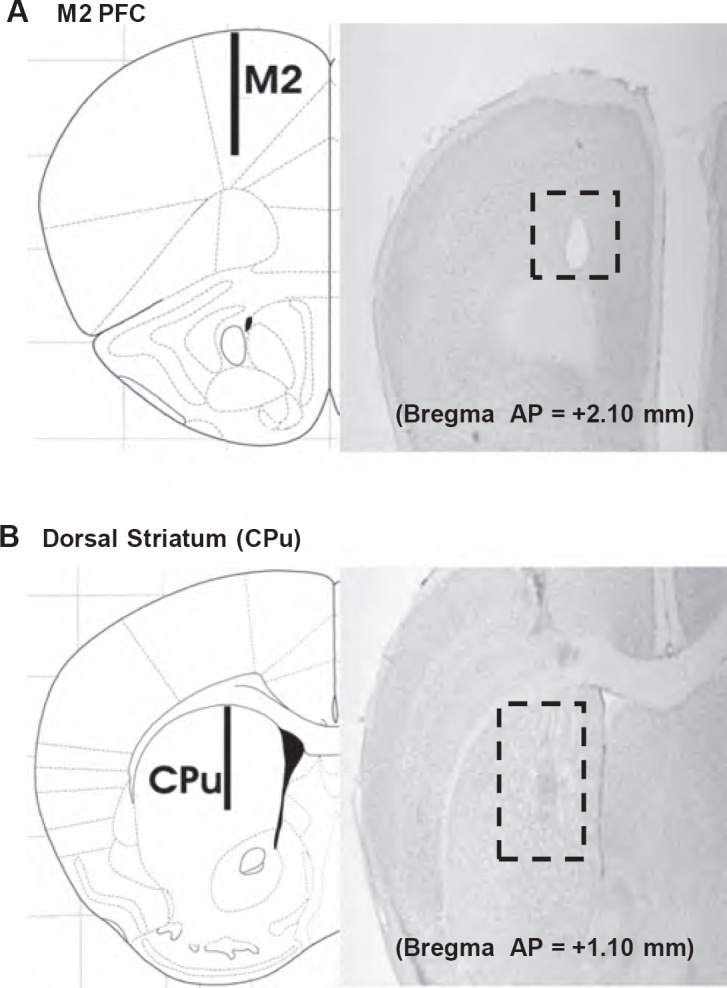
Schematic illustration (left) and photomicrograph (right) showing the typical location of the microdialysis probe track in the prefrontal cortex and dorsal striatum. Distances anterior to Bregma are indicated in mm (as defined in NK1(TACR1) gene mutation increases vulnerability to ADHD 29. Paxinos, G, Franklin, KBJ (2001). The Mouse Brain in Stereotaxic Coordinates. Second edition. London: Academic Press. Reproduced with permission from Elsevier)
Microdialysis samples were collected at 20-min intervals throughout the experiment and their dopamine content was analysed by high performance liquid chromatography coupled to an electrochemical detector (HPLC-ECD). The mobile phase comprised (mM) NaH2PO4 83, octanesulfonic acid 0.23, ethylenediaminetetraacetic acid disodium salt (EDTA) 0.84, and methanol 17% (adjusted to pH 4.0 with orthophosphoric acid) and was pumped through the HPLC system at 1 mL/min by a Shimadzu LC 6A isocratic dual-piston pump. Microdialysis samples were loaded into a rheodyne injection port fitted with a 50-μL loop and the solutes were separated on a 5-μM (4.6 × 250 mm) octadecylsilane column protected by a 7-μM aquapore guard column (4.6 × 30 mm). Dopamine content was measured with a Coulochem II detector with the conditioning electrode set at −280 mV and the measuring electrode set at +140 mV. A guard cell, set at +350 mV, was used to condition the mobile phase. The maximum sensitivity of the system was 5∼8 fmol (twice the baseline amplitude).
Human DNA extraction and SNP characterization
Genomic DNA from all sources was extracted from whole blood samples using standard cell lysis, proteinase K digestion, and phenol/chloroform ethanol precipitation method (Sambrook, et al., 1989). Single nucleotide polymorphism (SNP) markers were genotyped using KASPar, which employs a universal fluorescent primer method of end-labelling amplified PCR product (KBioscience, Hoddesdon, UK), which was a modified Amplifluor SNP genotyping method. Seventeen percent of the samples from each microtitre plate were reduplicated to expose possible errors and to confirm the reproducibility of genotypes.
Statistics
Mouse
Behavioural data were analysed using the general linear model (SPSS (PC+)) with ‘genotype’ and ‘drug1’ and ‘drug2’ (when appropriate) as main factors in two-way or three-way ANOVA, followed by post hoc tests (LSD: see Herpfer, et al., 2005). Microdialysis data were analysed using repeated measures ANOVA (SPSS PC+) with ‘time’ as a within-subjects factor. Comparisons of blocks of data in different treatment groups were carried out on time-matched samples. When clusters of consecutive samples (‘bin’) within individual treatment groups were compared, bin was treated as a within-subjects factor. Raw data were analysed routinely but, for the dorsal striatum, the incremental (net) change in efflux was also analysed; this was calculated by the subtraction of mean efflux in the basal samples of each mouse from all samples in the series. A significant effect on main factor(s), or relevant interactions between them, was the criterion for progressing to post hoc comparisons of group means. The Greenhouse-Geisser ‘ε’ correction was applied to compensate for violation of sphericity of the variance-covariance matrix. Statistical significance was set at P ≤ 0.05.
Human
Human genetic data were first analysed to confirm Hardy-Weinberg equilibrium (HWE). Markers lacking HWE were repeated using an alternative Taqman method. Before any association analysis, the genotype data were assessed using the SCANGROUP program in GENECOUNTING (Zhao, et al., 2002). This program highlights potential genotyping errors by identifying significant differences in haplotypic frequencies between any single 96 well microtitre-plate when compared to haplotypic frequencies in all other plates combined.
Allelic association analysis with ADHD was carried out using a two-tailed chi-squared test for biallelic SNPs. The multilocus genotypes were then analysed for haplotypic association with ADHD using HAPLOVIEW, which estimates the maximum likelihood of haplotype frequencies from phase unknown case-control data. Monte Carlo significance of any haplotypic associations with ADHD was then obtained with a permutation test (Sham and Curtis, 1995). HAPLOVIEW was also used to calculate pair-wise tests of linkage disequilibrium with d′ and r2 for all markers visualised using Locus View 2.022.
Results
Psychostimulants prevent hyperactivity of NK1R−/− mice
The locomotor activity of NK1R−/− mice was more than 2-fold greater than that of their wild-type counterparts. An acute d-amphetamine challenge increased the activity of NK1R+/+ mice but reduced that of NK1R−/− mice (without inducing stereotypy [P < 0.05]: genotype × drug: F(1,36) = 14.9, P <0.001) (Figure 2A). The motor response to the dopamine reuptake inhibitor, methylphenidate, similarly depended on genotype [genotype × drug: F(1,43) = 11.1, P <0.01] (Figure 2A); the locomotor activity of NK1R+/+ mice was increased by this drug, but that of NK1R−/− mice did not differ from drug-free wild-types. Thus, the behaviour of NK1R−/− mice given either of these drugs did not differ from the NK1R+/+ phenotype (Figure 2A).
Figure 2.

Effects of psychostimulants and NK1R antagonists on the locomotor activity of NK1R+/+ and NK1R−/− mice. (A) Animals were given saline (‘Veh’: 10 mL/kg, i.p.), d-amphetamine (‘d-AMP’: 2.5 mg/kg, i.p.; N = 10) or methylphenidate (‘MPH’: 2.5 mg/kg, i.p.; N = 12–13). Bars show mean ± s.e. mean. The data were analysed by ANOVA with post hoc comparison of pairs of data (LSD test). Lines linking treatment groups indicate a statistically significant difference between the means at P ≤ 0.05 (or less). (B) Locomotor activity of NK1R+/+ mice and NK1R−/− mice following the administration of saline (Sal) or d-amphetamine (d-AMP: 2.5 mg/kg i.p.). These two treatments were assigned to mice that had been pretreated with either vehicle (Veh: Tween 80 in 0.9% saline) or an NK1R antagonist (RP 67580 (‘RP’) or L 733060 (‘L’)) at the doses indicated in parentheses (5 or 10 mg/kg i.p.). Bars show mean ± s.e. mean locomotor activity in the light zone of the LDEB per unit time. All the data from this experiment, which was fully randomised for each NK1R antagonist, were first analysed by multifactorial (three-way) ANOVA. When there was a difference in the main factors, or a relevant interaction between them, pairs of data were compared using a post hoc one-way ANOVA and LSD test. Lines linking pairs of treatment groups indicate differences between the means at P ≤ 0.05 (or less)
Behaviour of NK1R−/− mice resembles that of wild-type (NK1R+/+) mice given an NK1R antagonist
Next, we compared the effects of NK1R antagonists (RP 67580 or L 733060; 5 and 10 mg/kg, i.p.) on spontaneous and d-amphetamine-evoked locomotor activity in the two genotypes. There was an NK1R antagonist × d-amphetamine × genotype interaction with RP 67580 [F(2,33) = 6.5, P < 0.01] and L 733060 [F(2,24) = 3.3, P = 0.05] (Figure 2B). Both these compounds increased the locomotor activity of NK1R+/+ mice [P < 0.05, in both cases] but neither affected NK1R−/− mice. d-Amphetamine reduced the motor activity of NK1R−/− mice and that of NK1R+/+ mice pretreated with an NK1R antagonist [P < 0.05 in both cases].
Lack of functional NK1R disrupts regulation of dopamine efflux in the dorsal striatum and prefrontal cortex
Basal efflux of dopamine in the dorsal striatum did not differ in the two genotypes [NK1R+/+: 24.5 ± 0.7 fmol/20 min; NK1R−/−: 25.6 ± 3.2 fmol/20 min; N =16–17] and was unaffected by systemic administration of RP 67580 (Figure 3A). However, the dopamine response to d-amphetamine differed in NK1R−/− and NK1R+/+ mice [genotype × amphetamine: F(1,13) = 4.5, P = 0.05]. Specifically, d-amphetamine caused an incremental increase in dopamine efflux in NK1R+/+ mice [T80-T120 versus basal samples: F(1,3) = 14.7, P = 0.03] but not in NK1R−/− mice (Figure 3B). Furthermore, pretreatment with the NK1R antagonist prevented the dopamine response to d-amphetamine in the wild-types [Figure 3B: genotype × RP 67580: F(1,13) = 5.7, P < 0.05].
Figure 3.

Effect of d-amphetamine on dopamine efflux in the dorsal striatum of NK1R+/+ and NK1R−/− mice. Stable basal efflux was first established in the home cage before the transfer of the mice to the dark zone of the LDEB for 90 min where they were given a first injection (at 30 min) of either vehicle (Veh: Tween 80 in 0.9% saline) or RP 67580 (‘RP’, 5 mg/kg. i.p.) followed by a second injection (at 60 min) of either (A) saline (sal) or (B) d-amphetamine (‘d-AMP’: 2.5 mg/kg i.p.), as indicated by the arrows. The mice were then transferred to the light zone of the LDEB. Points show mean ± s.e. mean efflux of the incremental increase (‘net’) from baseline: Net efflux was calculated, as described in Methods. The experiment was fully randomised across the eight groups but the data are illustrated in two graphs for clarity. Results were first analysed by multifactorial (three-way) ANOVA. When there was a difference in the main factors, or a relevant interaction between them, time-matched bins of data were compared using (post hoc) two-way repeated measures ANOVA (N = 3 or 4 per cell). Bins of data that differ by P ≤ 0.05 (or less) are indicated by the solid bar. (A) There was no difference in basal efflux in saline injected mice after treatment with RP 67580. (B) Comparison of efflux in the sample bin, T80-T120 (i.e. after treatment with d-amphetamine) with that of the three basal samples revealed an interaction between bin × genotype × RP/vehicle ($: F1,13 = 11.7; P < 0.01). Further, within the sample bin, T80-T120, there was a genotype × RP/vehicle interaction (#: F1,13 = 5.7; P < 0.05)
In the PFC, dopamine efflux in NK1R−/− mice was less than half that of their wild-type [Figure 4A and 4B: F(1,18) = 182.2, P < 0.001]. Moreover, efflux in NK1R+/+ mice given RP 67580 progressively declined to that of NK1R−/− mice (Figure 4A). d-Amphetamine did not affect dopamine efflux in vehicle-pretreated mice of either genotype (Figure 4B) or that in NK1R+/+ mice given RP 67580 (Figure 4A and 4B).
Figure 4.

Effect of d-amphetamine on dopamine efflux in the PFC of NK1R+/+ and NK1R−/− mice. Mice were given vehicle (Veh: Tween 80 in 0.9% saline) or RP 67580 (‘RP’, 5 mg/kg. i.p.) followed by a second injection of (A) saline (Sal) or (B) d-amphetamine (d-AMP: 2.5 mg/kg i.p.), as indicated by the arrows (see legend to Figure 3 for further details). Points show mean ± s.e. mean dopamine efflux in the LDEB (N = 3 or 4 per cell). Across the eight groups, there was a main effect of genotype in the basal samples (*: F1,18 = 181.2; P < 0.001). When comparing efflux in the final bin of three samples with that in the basal samples, there was a bin × genotype × RP/vehicle interaction ($: F1,18 = 6.7; P = 0.02). There was also an interaction (#) between genotype × RP/ vehicle in the final bin (F1,18 = 5.9; P = 0.03)
SNPs in TACR1 (human NK1R) gene are associated with ADHD
In an earlier unpublished study, which used seven SNP ‘tagging’ markers that capture most of the haplotypic variation in the region of the TACR1 gene, we identified a single SNP (rs3771856) that was associated with alcoholism. More recently, in part of a genome-wide association study (Sklar, et al., 2008), we observed an association between a further 18 SNPs at, or within, 20 kb of TACR1 and bipolar disorder, including polymorphisms of TACR1. In view of the well established comorbidity of ADHD with conduct disorder and alcoholism, we decided to explore allelic and haplotypic association between genetic markers at, or within, 20 kb of the TACR1 gene with ADHD. We focussed on eight SNPs that had already been shown to be significantly associated with bipolar disorder or alcoholism or had shown a strong trend towards significance (see Table 1).
Table 1.
Tests of allelic association between genetic markers at TACR1 with ADHD in the samples from the University of Cardiff and Institute of Psychiatry, London
| # | SNP Code | Associated Allele | Case allele counts | Control allele counts | Case allele frequencies | Control allele frequencies | HW Cases | HW Controls | Allelic χ2 | 2 sided P value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs3886110 | A | 367 | 457 | 437 | 583 | 0.445 | 0.428 | 0.884 | 0.637 | 0.533 | 0.4654 |
| 2 | rs13012537 | G | 355 | 459 | 431 | 589 | 0.436 | 0.423 | 0.905 | 0.461 | 0.34 | 0.5596 |
| 3 | rs3771829 | C | 89 | 735 | 64 | 942 | 0.108 | 0.064 | 0.356 | 0.979 | 11.651 | 0.0006 |
| 4 | rs3771833 | A | 105 | 693 | 76 | 928 | 0.132 | 0.076 | 0.203 | 0.176 | 15.366 | 0.00008 |
| 5 | rs10203484 | A | 296 | 514 | 351 | 669 | 0.365 | 0.344 | 0.382 | 0.389 | 0.897 | 0.3435 |
| 6 | rs12477554 | A | 339 | 461 | 431 | 589 | 0.424 | 0.423 | 0.709 | 0.848 | 0.003 | 0.9589 |
| 7 | rs3771856 | G | 431 | 321 | 604 | 572 | 0.573 | 0.514 | 0.273 | 0.462 | 6.538 | 0.0106 |
| 8 | rs17011370 | G | 781 | 33 | 946 | 70 | 0.959 | 0.931 | 0.673 | 0.776 | 6.842 | 0.0089 |
SNPs numbered 3, 4, 7, 8 show allelic association with ADHD. All markers remained significant after correcting for the use of eight SNPs.
Four of these SNPs showed allelic or haplotypic association with ADHD (rs3771829: P = 0.0006; rs3771833: P = 0.00008; rs3771856: P = 0.0106; and rs1701137: P = 0.0089). Robust evidence for allelic association with ADHD remained after the correction for multiple testing (Table 1 and Figure 5). We then tested for haplotypic association and found a single haplotype that was associated with ADHD. This consisted of the alleles C and A of the SNPs rs3771829 and rs3771833, respectively, which are tagging SNPs for haplotype block 2 of the TACR1 gene (Table 2). The statistical significance of the association with ADHD remained after empirical tests of significance were applied, empirical P = 0.0028. Output from the Human Genome Browser (http://genome.ucsc.edu/) (March 2006 release) in Figure 5 shows the distances between markers, vis à vis, TACR1 together with the linkage disequilibrium relationships between adjacent markers in the ADHD case-control sample. As shown in Figure 5, the associated SNPs are within the first intron of TACR1 or in the 5′ promoter region.
Figure 5.

Schematic diagram of NK1R/TACR1 gene structure and associated SNPs on human chromosome 2p12. Exons and introns of the TACR1 gene are shown according to the March 2006 release of the Human Genome Browser (http://genome.ucsc.edu). The gene is transcribed from the right of the diagram (5′) to the left (3′). The names and positions of the four SNP markers, found to show association with ADHD, are shown relative to the structure of the genes in the region. Three of the four SNPs were within the first intron of TACR1
Table 2.
Haplotype analysis of SNP data from ADHD cases and controls at the TACR1 locus
| Block and SNPs | Haplotype | Freq. | Counts | Frequencies | χ2 | Asymptotic P Value | χ2 | Permutation P Value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Control | Case | Control | ||||||||||
| Block 1 | |||||||||||||
| rs3886110 | C | A | 0.556 | 453.7 | 378.3 | 575.9 | 444.1 | 0.545 | 0.565 | 0.69 | 0.4062 | 0.69 | 0.9899 |
| & | A | G | 0.421 | 356.5 | 475.5 | 423.9 | 596.1 | 0.429 | 0.416 | 0.316 | 0.574 | 0.316 | 0.9989 |
| rs13012537 | A | A | 0.015 | 15.5 | 816.5 | 13.1 | 1007 | 0.019 | 0.013 | 0.985 | 0.321 | 0.985 | 0.9517 |
| Block 2 | |||||||||||||
| rs3771829 | G | G | 0.893 | 716.3 | 113.7 | 935.7 | 84.3 | 0.863 | 0.917 | 14.147 | 2.00E-04 | 14.147 | 0.0008 |
| & | C | A | 0.083 | 89.2 | 740.8 | 63.6 | 956.4 | 0.107 | 0.062 | 12.281 | 5.00E-04 | 12.281 | 0.0028 |
| rs3771833 | G | A | 0.023 | 22.3 | 807.7 | 19.5 | 1001 | 0.027 | 0.019 | 1.267 | 0.2604 | 1.267 | 0.9166 |
| Block 3 | |||||||||||||
| rs10203484 | G | G | 0.575 | 470.8 | 353.2 | 589 | 431 | 0.571 | 0.577 | 0.07 | 0.792 | 0.07 | 1 |
| & | A | A | 0.354 | 301.8 | 522.2 | 351 | 669 | 0.366 | 0.344 | 0.98 | 0.3221 | 0.98 | 0.9526 |
| rs12477554 | G | A | 0.071 | 51.4 | 772.6 | 80 | 940 | 0.062 | 0.078 | 1.779 | 0.1823 | 1.779 | 0.813 |
SNP markers are combined into haplotypes using the program HAPLOVIEW. Two haplotypes show association with ADHD but only one was positively associated with ADHD and this remained significant after an empirical test of association (9,999 iterations). The associated haplotype comprised allele C from SNP rs3771829 and allele A from rs3771833.
Discussion
The hyperactivity of NK1R−/− mice and its prevention by d-amphetamine or methylphenidate together, with disrupted monoamine transmission, all echo features of ADHD (see Di Michele, et al., 2005). Crucially, the prevention of hyperactivity by methylphenidate distinguishes these mutant mice from the spontaneously hypertensive rat (Amini, et al., 2004), which is the most widely studied model of this disorder. Both the hyperactivity and the atypical effects of d-amphetamine in NK1R−/− mice rest on the lack of functional NK1R rather than extraneous (e.g. developmental) changes. This is confirmed by our finding that NK1R antagonists (RP 67580 or L 733060) increased the locomotor activity of NK1R+/+ mice and that this was prevented by d-amphetamine. Neither of these antagonists affected the locomotor hyperactivity of the mutant mice and so their NK1R selectivity is assured.
We then used in vivo microdialysis to investigate whether the disruption of dopaminergic transmission in NK1R−/−mice could underlie their abnormal behaviour. Striatal dopaminergic transmission has long been linked with the modulation of locomotor activity. There is also evidence that disruption of mesocorticolimbic dopaminergic transmission gives rise to a lack of tolerance of delayed reward and contributes to impulsivity, a prominent feature of ADHD (Santarelli, et al., 2001; Oades, et al., 2005; Sagvolden, et al., 2005; Dalley, et al., 2007).
First, we compared dopamine efflux in the dorsal striatum. We studied this region because the activation of NK1R in either the substantia nigra (Reid, et al., 1991) or the striatum (Tremblay, et al., 1992; Krolewski, et al., 2005) increases striatal dopamine efflux. Further, although antagonism of NK1R within the striatum has no effect on substance P mRNA expression or spontaneous locomotor activity, it prevents a d-amphetamine-induced increase in both these measures (Gonzales-Nicolini and McGinty, 2002). NK1R antagonism in the striatum also prevents hyperactivity mediated by the activation of dopamine D1 receptors (Krolewski, et al., 2005). Against this background, we reasoned that basal dopamine efflux in the striatum would be unaffected by functional ablation of the NK1R gene, or NK1R antagonism, but that the dopamine response to d-amphetamine would be diminished.
As predicted, basal dopamine efflux did not differ in the two genotypes and was unaffected by NK1R antagonism. Evidently, spontaneous dopamine efflux in the dorsal striatum of these mice does not depend on tonic activation of NK1R. This is consistent with their low density on dopaminergic neurones in the substantia nigra, which project to the dorsal striatum.
Most, if not all, NK1R in the dorsal striatum are expressed by cholinergic interneurones (Gerfen, 1991). Recent evidence suggests that these interneurones influence the activity of medium-spiny striatal output neurones (Wang, et al., 2006), and as a consequence, motor activity (Pisani, et al., 2007). Striatal acetylcholine release is increased by the activation of dopamine D1-like receptors (Acquas and di Chiara, 1999) and this depends on functional NK1R (Steinberg, et al., 1996). Since cholinergic (M1) receptor knock-out mice are hyperactive (Gerber, et al., 2001), one explanation for the hyperactivity of NK1R−/− mice could be that their lack of functional NK1R impairs acetylcholine release mediated by D1 receptor activation (Anderson, et al., 1994). However, this is unlikely because basal dopamine efflux did not differ in the two genotypes and local activation of DRD1-like receptors induces hyperactivity, rather than reduces it (Krolewski, et al., 2005). An alternative explanation focuses on glutamatergic afferents from the cortex and thalamus, which converge on the cholinergic interneurones and medium-spiny output neurones. Glutamate-evoked acetylcholine release in the striatum is similarly facilitated by NK1R (Kemel, et al., 2002). It follows that disruption of glutamate/NK1R/acetylcholine coupling could contribute to the hyperactivity of NK1R−/− mice. In either case, impaired NK1R function could account for hyperactivity in ADHD.
The administration of d-amphetamine increased extracellular dopamine in NK1R+/+ mice, but not in NK1R−/− mice or NK1R+/+ mice treated with RP 67580. It seems that the striatal dopamine response to d-amphetamine depends on functional NK1R. This is supported by evidence that antagonism of striatal NK1R prevents an increase in dopamine efflux following the administration of cocaine, another psychostimulant (Loonam, et al., 2003). This difference in the dopamine response to d-amphetamine in the two genotypes could be explained by the absence of NK1R on noradrenergic neurones that project to the substantia nigra. This follows from reports that local infusion of substance P in the region of the locus coeruleus (Cheesman, et al., 1983) activates noradrenergic neurones and that the dopamine response to d-amphetamine in the dorsal striatum is attenuated in DβH−/− mice, which cannot synthesize noradrenaline (Schanks, et al., 2006). The prevention of hyperactivity of NK1R−/− mice by d-amphetamine is harder to explain but the answer to this question could hold the key to its therapeutic effects in ADHD. We are currently testing our prediction, based on reports outlined above, that d-amphetamine increases acetylcholine efflux in the dorsal striatum of wild-type mice but not in mice lacking functional NK1R.
Next, we monitored dopamine efflux in the M2 subregion of the PFC. We studied this subregion for several reasons. First, in rats, the equivalent (Fr2) region has reciprocal connections with the locus coeruleus, forming part of a circuit that determines focussed attention/associative learning (Jones and Yang, 1985; Jodo, et al., 1998; Bouret and Sara, 2005). Secondly, all noradrenergic neurones in the locus coeruleus express NK1R (Chen, et al., 2000). Thirdly, ectopic uptake and release of dopamine by noradrenergic neurones in the PFC (Devoto, et al., 2005) indicate functional coupling between these two groups of neurones.
Extracellular dopamine in the PFC of NK1R−/− mice was more than two-fold lower than that in the wild-type. This finding supports the view that ADHD involves a deficit in dopaminergic transmission in the PFC: ‘hypofrontality’ (Sagvolden, et al., 2005; Pliszka, 2005). The deficit in dopamine efflux was due to a lack of functional NK1R, rather than an extraneous compensatory response, because efflux in NK1R+/+ mice given RP 67580 progressively declined to that of NK1R−/−mice. In the rat (Seabrook, et al., 1995) and mouse (our unpublished data), dopaminergic neurones in the ventral tegmental area (which project to the PFC) express few, if any, NK1R and so these receptors are unlikely to have a direct effect on dopaminergic transmission in the PFC. Instead, the increased serotonin and/or noradrenaline transmission in NK1R−/− mice (Fisher, et al., 2007; Gobbi, et al., 2007) could indirectly blunt transmitter release from dopaminergic neurones in the VTA (through activation of somatodendritic- or heteroceptors) (Gresch, et al., 1995; Pan, et al., 1996). Whatever the explanation, the deficit in dopaminergic transmission in the prefrontal cortex could contribute to the hyperactivity in NK1R−/− mice and ADHD patients by disinhibiting glutamatergic (pyramidal cell) efferents that project from the PFC to the striatum (Le Moal and Simon, 1991; Ventura, et al., 2004). The increased serotonergic and noradrenergic transmission in NK1R−/−mice would also activate α1-adrenoceptors and 5-HT2A receptors, which could further contribute to locomotor hyperactivity (Auclair, et al., 2004).
d-Amphetamine did not affect dopamine efflux in the PFC of either genotype and so we infer that the difference in their behavioural response to d-amphetamine does not depend on its effects on extracellular dopamine in this region. Nevertheless, our findings indicate that functional ablation of the NK1R gene disrupts dopaminergic transmission in frontostriatal circuits, as well as that of noradrenergic and serotonergic neurones, all of which feature prominently in ADHD and are targeted by all the drugs that are currently used to treat this disorder.
Studies of the genetic susceptibility of humans similarly point to abnormal monoamine transmission in ADHD. So far, findings include genetic effects from the dopamine transporter (SLC6A3) gene, dopamine D4 (DRD4), and dopamine D5 receptor (DRD5) genes. It is interesting that DRD5 are expressed by cholinergic neurones in the dorsal striatum and the PFC (Berlanga, et al., 2005) where TACR1 (NK1R) is expressed. Weaker, albeit replicated, genetic evidence also supports the association between ADHD with the gene encoding synaptosomal-associated protein 25 kDa (SNAP-25) and the serotonin transporter (SLC6A4) gene (Asherson, 2004; Faraone, 2004; Thapar, et al., 2005). However, the effect size of any of these variants is small and compatible with the known genetic heterogeneity of ADHD established by previous linkage studies.
It has already been suggested that it is the disruption of the interactions between monoamine transmitters that holds the key to this disorder (Oades, et al., 2005). Our evidence suggests that impaired NK1R function causes such an imbalance. Furthermore, the NK1R−/− mouse phenotype could incorporate functional abnormalities in gene products already associated with ADHD, either directly (e.g. by modifying the activity of noradrenergic projections to the prefrontal cortex and dorsal raphe nuclei) or indirectly (e.g. via cholinergic neurones in the striatum, which co-express dopamine D5 receptors and NK1R).
These findings prompted us to carry out genetic association studies to test for abnormalities in the TACR1 gene of patients with a diagnosis of ADHD. Our results revealed four SNPs that are positively associated with ADHD. This suggests that there are genetic variants, mutations or aetiological base-pair changes that alter the expression or function of the TACR1 receptor in ways that predispose to a subtype of ADHD. Because TACR1 SNPs are also associated with bipolar disorder, it is tempting to speculate that they genetically define a new clinical subtype of ADHD, which is more related to mood disorder than to conduct disorder. Three scenarios can be envisaged. The first is that different mutations in TACR1 could be responsible for ADHD and for bipolar disorder. The second is that the same mutations are responsible for bipolar disorder and ADHD, and therefore, there is a ‘bipolar’ subtype of ADHD, which is expressed in childhood as ADHD and later in life as bipolar disorder. Thirdly, it is possible that the same or another set of mutations in TACR1 predisposes to conduct disorder and explains the strongly established comorbidity between alcoholism and conduct disorder. The fact that TACR1 is associated with bipolar disorder both in our study and the study by Perlis, et al., (2008) seems to strengthen the first explanation and the likelihood that the association with alcoholism is mediated by TACR1 associated affective disorders, which are also strongly associated with alcoholism. These issues will best be resolved by further study of the TACR1 gene association in carefully clinically characterized subgroups of alcoholism with ADHD and subgroups of ADHD associated with dissocial personality or conduct disorder and by direct genomic resequencing of TACR1 in each of these TACR1 associated subgroups.
In conclusion, functional ablation of the NK1R gene causes neurochemical, pharmacodynamic, and behavioural changes in mice that resemble abnormalities in humans with ADHD and to a lesser extent resemble bipolar affective disorder. These findings provide compelling evidence that the SNPs in the TACR1 gene locus increase susceptibility to ADHD although replication in other samples is needed. New diagnostic, treatment and preventive strategies could emerge from further research of the molecular pathology of NK1R/TACR1 gene expression and the function of this receptor.
Acknowledgements
This work was funded by the Clinical Research and Development Committee UCL (SNP genotyping (HG)), the Wellcome Trust (AT, Michael O'Donovan, Michael Owen (Cardiff University)), UCL Business PLC (support for the mouse colony), and the Edendale Fund (SCS). We wish to thank all human subjects for their co-operation, Kate Langley for collation of information on the patients' ADHD status (Cardiff samples) and Evangelia Stergyiakouli for the preparation and dispatch of human DNA (Cardiff) samples.
Author contributions
TCY carried out the experiments on mice and analysed the behavioural and neurochemical data. AM and HG carried out the genetic studies on bipolar disorder and the genotyping of the ADHD samples provided by AT (Cardiff) and PA (London). SPH generated the NK1R−/− mice and carried out the cytochemistry. SCS initiated the study, designed the experiments using mice (in collaboration with SPH and TCY), supervised the preclinical data analysis, and drafted the manuscript, with inputs from all co-authors.
Competing interests statement
UCL Business PLC has filed patents for this murine model of ADHD and genetic markers for diagnosis and prognosis of ADHD. HG, SCS, and SPH are named inventors.
Contributor Information
TC Yan, Department of Neuroscience, Physiology and Pharmacology, University College London, London, UK.
A McQuillin, Molecular Psychiatry Laboratory, Department of Mental Health Sciences, Royal Free & UCL School of Medicine, London, UK.
A Thapar, Department of Psychological Medicine, School of Medicine, Cardiff University, Cardiff, Wales, UK.
P Asherson, ADHD genetics group, MRC Social Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King's College London, London, UK.
SP Hunt, Department of Cell and Developmental Biology, University College London, London, UK.
SC Stanford, Department of Neuroscience, Physiology and Pharmacology, University College London, London, UK.
H Gurling, Molecular Psychiatry Laboratory, Department of Mental Health Sciences, Royal Free & UCL School of Medicine, London, UK.
References
- Acquas E, di Chiara G, (1999). Dopamine D1 receptor-mediated control of striatal acetylcholine release by endogenous dopamine. Eur J Pharmacol 383: 121–127 [DOI] [PubMed] [Google Scholar]
- Amini B, Yang PB, Swann AC, Dafny N, (2004). Differential locomotor responses in male rats from three strains to acute methylphenidate. Int J Neurosci 4: 1063–1084 [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Kuo S, Chase TN, Engber TM, (1994). Dopamine D1 receptor-stimulated release of acetylcholine in rat striatum is mediated indirectly by activation of striatal neurokinin1 receptors. J Pharmacol Exp Ther 269: 1144–1151 [PubMed] [Google Scholar]
- Angold A, Prendergast M, Cox A, Harrington R, Simonoff E, Rutter M, (1995). The child and adolescent psychiatric assessment. Psychol Med 25: 739–753 [DOI] [PubMed] [Google Scholar]
- Asherson P, (2004). Attention-deficit hyperactivity disorder in the post-genomic era. Eur Child Adolesc Psychiatry 13 (Suppl. 1): 150–170 [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD, (2005). Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J Comp Neurol 493: 99–110 [DOI] [PubMed] [Google Scholar]
- Auclair A, Crouin C, Cotecchia S, Glowinski J, Tassin J-P, (2004). 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioral sensitization to opiates and psychostimulants. Eur J Neurosci 20: 3073–3084 [DOI] [PubMed] [Google Scholar]
- Berlanga ML, Simpson TK, Alcantara AA, (2005). Dopamine D5 receptor localization on cholinergic neurones of the rat forebrain and diencephalon: A potential neuroanatomical substrate involved in mediating dopaminergic influences on acetylcholine release. J Comp Neurol 492:34–49 [DOI] [PubMed] [Google Scholar]
- Bester H, de Felipe C, Hunt SP, (2001). The NK1 receptor is essential for the full expression of noxious inhibitory controls in the mouse. J Neurosci 21: 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret S, Sara SJ, (2005). Network reset: A simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci 28: 574–582 [DOI] [PubMed] [Google Scholar]
- Brennan AR, Arnsten AF, (2008). Neuronal mechanisms underlying attention deficit hyperactivity disorder: The influence of arousal on prefrontal cortical function. Ann NY Acad Sci 1129: 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheesman HJ, Pinnock RD, Henderson G, (1983). Substance P excitation of rat locus coeruleus neurones. Eur J Pharmacol 94:93–99 [DOI] [PubMed] [Google Scholar]
- Chen LW, Wei LC, Liu HL, Rao ZR, (2000). Noradrenergic neurones expressing substance P receptor (NK1) in the locus coeruleus complex: A double immunofluorescence study in the rat. Brain Res 873: 155–159 [DOI] [PubMed] [Google Scholar]
- Dalley JW, Fryer TD, Brichard L, Robinson ES, Theobald DE, Laane K, et al. , (2007). Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science 315: 1267–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe C, Herrero JF, O'Brien JA, Palmer JA, Doyle CA, Smith A, et al. , (1998). Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 392: 394–397 [DOI] [PubMed] [Google Scholar]
- Devoto P, Flore G, Saba P, Fa M, Gessa GL, (2005). Stimulation of the locus coeuleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J Neurochem 92: 368–374 [DOI] [PubMed] [Google Scholar]
- Di Michele F, Prichep L, Joh ER, Chabot RJ, (2005). The neurophysiology of attention-deficit/hyperactivity disorder. Int J Psychophysiol 58:81–93 [DOI] [PubMed] [Google Scholar]
- Dick DM, Lim TK, Edenberg HJ, Hesselbrock V, Kramer J, Kuperman S, et al. , (2004). A genome-wide screen for genes influencing conduct disorder. Mol Psychiatry 9: 81–86 [DOI] [PubMed] [Google Scholar]
- Duffy RA, Varty GB, Morgan CA, Lachowic JE, (2002). Correlation of neurokinin (NK)1 receptor occupancy in gerbil striatum with behavioral effects of NK1 antagonists. J Pharmacol Exp Ther 301: 536–542 [DOI] [PubMed] [Google Scholar]
- Faraone SV, (2004). Genetics of adult attention-deficit/hyperactivity disorder. Psychiatr Clin North Am 27: 303–321 [DOI] [PubMed] [Google Scholar]
- Fisher AS, Stewart RJ, Yan TC, Hunt SP, Stanford SC, (2007). Disruption of noradrenergic transmission and the behavioural response to a novel environment in NK1R −/− mice. Eur J Neurosci 25: 1195–1204 [DOI] [PubMed] [Google Scholar]
- Froger N, Gardier AM, Moratalia R, Alberti I, Lena I, Boni C, et al. , (2001). 5 Hydroxytryptamine (5-HT)1A autoreceptor adaptive changes in Substance P: (Neurokinin 1) receptor knock-out mice mimic antidepressant induced desensitization. J Neurosci 21: 8188–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd CA, Murtra P, de Felipe C, Hunt SP, (2003). Neurokinin-1 receptor-expressing neurones in the amygdala modulate morphine reward and anxiety behaviors in the mouse. J Neurosci 23: 8271–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, et al. , (2008). Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science 319: 1536–1539 [DOI] [PubMed] [Google Scholar]
- Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S, (2001). Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc Natl Acad Sci USA 98: 15312–15317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, (1991). Substance P (neurokinin-1) receptor mRNA is selectively expressed in cholinergic neurones in the striatum and basal forebrain. Brain Res 556: 165–170 [DOI] [PubMed] [Google Scholar]
- Gobbi G, Cassano T, Radja F, Morgese MG, Cuomo V, Santarelli L, et al. , (2007). Neurokinin1 receptor antagonism requires norepinephrine to increase serotonin function. Eur Neuropsychopharmacol 17: 328–338 [DOI] [PubMed] [Google Scholar]
- Gonzales-Nicolini V, McGinty JF, (2002). NK-1 receptor blockade decreases amphetamine-induced behavior and neuropeptide mRNA expression in the striatum. Brain Res 931: 41–49 [DOI] [PubMed] [Google Scholar]
- Gresch PJ, Sved AF, Zigmond MJ, Finlay JM, (1995). Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J Neurochem 65: 111–116 [DOI] [PubMed] [Google Scholar]
- Guiard BP, Przybylwski C, Guilloux J-P, Seif I, Froger N, de Felipe C, et al. , (2004). Blockade of substance P (neurokinin 1) receptors enhances extracellular serotonin when combined with a selective serotonin reuptake inhibitor: An in vivo microdialysis study in mice. J Neurochem 89: 54–63 [DOI] [PubMed] [Google Scholar]
- Herpfer I, Hunt SP, Stanford SC, (2005). A comparison of neurokinin 1 receptor knock-out (NK1−/−) and wild type mice: Exploratory behaviour and extracellular noradrenaline concentration in the cerebral cortex of anaesthetised subjects. Neuropharmacology 48: 706–719 [DOI] [PubMed] [Google Scholar]
- Hirshfeld-Becker DR, Biederman J, Henin A, Faraone SV, Dowd ST, De Petrillo LA, et al. , (2006). Psychopathology in the young offspring of parents with bipolar disorder: A controlled pilot study. Psychiatry Res 145: 155–167 [DOI] [PubMed] [Google Scholar]
- Holmes J, Lawson D, Langley K, Fitzpatrick H, Trumper A, Pay H, et al. , (2004). The child attention-deficit hyperactivity disorder teacher telephone interview (CHATTI): Reliability and validity. Br J Psychiatry 184: 74–78 [DOI] [PubMed] [Google Scholar]
- Jodo E, Chiang C, Aston-Jones G, (1998). Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurones. Neurosci 83: 63–79 [DOI] [PubMed] [Google Scholar]
- Jones BJ, Yang T-Z, (1985). The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J Comp Neurol 242: 56–92 [DOI] [PubMed] [Google Scholar]
- Kemel M-L, Perez S, Godeheu G, Soubrie P, Glowinski J, (2002). Facilitation by endogenous tachykinins of the NMDA-evoked release of acetylcholine after acute and chronic suppression of dopaminergic transmission in the matrix of the rat striatum. J Neurosci 22: 1929–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolewski DM, Bishop C, Walker PD, (2005). Intrastriatal dopamine D1 receptor agonist-mediated motor behavior is reduced by local neurokinin 1 receptor antagonism. Synapse 57: 1–7 [DOI] [PubMed] [Google Scholar]
- Le Moal M, Simon H, (1991). Mesocorticolimbic dopaminergic network: Functional and regulatory roles. Physiol Rev 71: 155–234 [DOI] [PubMed] [Google Scholar]
- Loonam TM, Noailles PAH, Yu J, Zhu JPQ, Angulo JA, (2003). Substance P and cholecystokinin regulate neurochemical responses to cocaine and methamphetamine in the striatum. Life Sci 73: 727–739 [DOI] [PubMed] [Google Scholar]
- Murtra P, Sheasby A, Hunt SP, de Felipe C, (2000) Loss of rewarding effects of morphine and amphetamine but not cocaine, in mice with disruption of the substance P receptor (NK1) gene. Soc Neurosci Abstracts 658.5.
- Oades RD, (2008). Dopamine-serotonin interactions in attention-deficit hyperactivity disorder (ADHD). Prog Brain Res 172: 543–565 [DOI] [PubMed] [Google Scholar]
- Oades RD, Sadile AG, Sagvolden T, Viggiano D, Zuddas A, Devoto P, et al. , (2005). The control of responsiveness in ADHD by catecholamines: Evidence for dopaminergic, noradrenergic and interactive roles. Dev Sci 8: 122–131 [DOI] [PubMed] [Google Scholar]
- Pan WHT, Sung J-C, Fuh SMR, (1996). Local application of amphetamine into the ventral tegmental area enhances dopamine release in the nucleus accumbens and the medial prefrontal cortex through noradrenergic neurotransmission. J Pharmacol Exp Ther 278: 725–781 [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ, (2001) The Mouse Brain in Stereotaxic Coordinates. second ed. London: Academic Press [Google Scholar]
- Perlis RH, Purcell S, Fagerness J, Kirby A, Petryshen TL, et al. , (2008). Family-based association study of lithium-related and other candidate genes in bipolar disorder. Arch Gen Psychiatry 65: 53–61 [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ, (2007). Re-emergence of striatal interneurones in movement disorders. Trends Neurosci 30: 545–553 [DOI] [PubMed] [Google Scholar]
- Pliszka SR, (2005). The neuropsychopharmacology of attention-deficit hyperactivity disorder. Biol Psychiatry 57: 1385–1390 [DOI] [PubMed] [Google Scholar]
- Prince J, (2008). Catecholamine dysfunction in attention-deficit-hyperactivity disorder. J Clin Psychopharmacol 28 (Suppl. 2): S39–S45 [DOI] [PubMed] [Google Scholar]
- Reid MS, Herrera-Marchitz M, Ungerstedt U, (1991). Effects of intranigral substance P and neurokinin A injections on extracellular dopamine levels measured with microdialysis in the striatum and frontoparietal cortex of rats. J Neurochem 57: 970–974 [DOI] [PubMed] [Google Scholar]
- Rigby M, O'Donnell R, Rupniak NMJ, (2005). Species differences in tachykinin receptor distribution: Further evidence that the substance P (NK1) receptor predominates in human brain. J Comp Neurol 490: 335–353 [DOI] [PubMed] [Google Scholar]
- Ripley TL, Gadd CA, de Felipe C, Hunt SP, Stephens DN, (2002). Lack of self-stimulation and behavioural sensitization to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology 43: 1258–1268 [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Aase H, Johansen EB, Russell VA, (2005). A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive combined subtypes. Behav Brain Sci 28: 397–168 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T, (1989) In: Molecular Cloning: A Laboratory Manual. Vol. 1, 2, 3 New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Santarelli L, Gobbi G, Debs PC, Sibille EL, Blier P, Hen R, et al. , (2001). Genetic and pharmacological disruption of neurokinin 1 receptor function decreases anxiety-related behaviours and increases serotonin function. Proc Natl Acad Sci USA 98: 1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanks JR, Ventura R, Puglisi-Allegra S, Alcare A, Cole CD, Liles LC, et al. , (2006). Dopamine-β-hydroxylase knock-out mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacol 31: 2221–2230 [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Bowery BJ, Hill RG, (1995). Pharmacology of tachykinin receptors on neurones in the ventral tegmental area of rat brain slices. Eur J Pharmacol 273: 113–119 [DOI] [PubMed] [Google Scholar]
- Sham PC, Curtis D, (1995). Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet 59: 97–105 [DOI] [PubMed] [Google Scholar]
- Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K, et al. , (2008). Whole-genome association study of bipolar disorder. Mol Psychiatry 13: 558–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer RL, Endicott J, (1977) The Schedule for Affective Disorders and Schizophrenia. Lifetime Version, third ed. New York State Psychiatric Institute; New York [Google Scholar]
- Spitzer RL, Endicott J, (1978) Schedule for Affective Disorders and Schizophrenia, third ed Biometrics Research; New York: [DOI] [PubMed] [Google Scholar]
- Steinberg R, Rodier D, Souilhac I, Bougault I, Emonds-Alt X, Soubrie P, et al. , (1996). Pharmacological characterization of tachykinin receptors controlling acetylcholine release from rat striatum: An in vivo microdialysis study. J Neurochem 65: 2543–2548 [DOI] [PubMed] [Google Scholar]
- Thapar A, O'Donovan M, Owen MJ, (2005). The genetics of attention deficit hyperactivity disorder. Hum Mol Genet 14: R275–R282 [DOI] [PubMed] [Google Scholar]
- Tremblay L, Keme M-L, Desban M, Gauchy C, Glowinski J, (1992). Distinct presynaptic control of dopamine release in striosomal and matrix enriched areas of rat striatum by selective agonists of NK1, NK2 and NK3 tachykinin receptors. Proc Natl Acad Sci USA 89: 11214–11218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura R, Alcaro A, Cabib S, Conversi D, Mandolesi L, Puglisi-Allegra S, (2004). Dopamine in the medial prefrontal cortex controls genotype-dependent effects of amphetamine on mesoaccumbens dopamine release and locomotion. Neuropsychopharmacol 29: 72–80 [DOI] [PubMed] [Google Scholar]
- Wang A, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. , (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurones is mediated by cholinergic transmission. Neuron 50: 443–452 [DOI] [PubMed] [Google Scholar]
- Wiener HW, Go RC, Tiwari H, George V, Page GP, (2005). COGA phenotypes and linkages on chromosome 2. BMC Genet 6 (Suppl. 1): S125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Eagle DM, Robbins TW, (2006). Behavioral models of impulsivity in relation to ADHD: Translation between clinical and preclinical studies. Clin Psychol Rev 26: 379–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Cadet JL, Angulo JA, (2002). Neurokinin-1 (NK-1) receptor antagonists abrogate methamphetamine-induced striatal dopaminergic neurotoxicity in the murine brain. J Neurochem 83: 613–622 [DOI] [PubMed] [Google Scholar]
- Zhao JH, Lissarraguem S, Essioux L, Sham PC, (2002). Genecounting: Haplotype analysis with missing genotypes. Bioinformatics 18: 1694–1695 [DOI] [PubMed] [Google Scholar]