

Abstract
Parathyroid neoplasia is most commonly due to benign parathyroid adenoma but rarely can be caused by malignant parathyroid carcinoma. Evidence suggests that parathyroid carcinomas rarely, if ever, evolve through an identifiable benign intermediate, with the notable exception of carcinomas associated with the familial hyperparathyroidism-jaw tumor syndrome. Several genes have been directly implicated in the pathogenesis of typical sporadic parathyroid adenoma; somatic mutations in the MEN1 tumor suppressor gene are the most frequent finding, and alterations in the Cyclin D1/PRAD1 oncogene are also firmly established molecular drivers of sporadic adenomas. In addition, good evidence supports mutation in the CDKN1B/p27 cyclin-dependent kinase inhibitor (CDKI) gene, and in other CDKI genes as contributing to disease pathogenesis in this context. Somatic defects in additional genes, including β-catenin, POT1 and EZH2 may contribute to parathyroid adenoma formation but, for most, their ability to drive parathyroid tumorigenesis remains to be demonstrated experimentally. Further, genetic predisposition to sporadic presentations of parathyroid adenoma appears be conferred by rare, and probably low-penetrance, germline variants in CDKI genes and, perhaps, in other genes such as CASR and AIP. The HRPT2 tumor suppressor gene is commonly mutated in parathyroid carcinoma.
Keywords: Hyperparathyroidism, Parathyroid Adenoma, Parathyroid Carcinoma
1. Introduction
Primary hyperparathyroidism is a common endocrine disorder, and is almost always caused by parathyroid neoplasia. Benign parathyroid adenoma is the commonest form of sporadic parathyroid neoplasia, accounting for about 85% of all cases. Diffuse hypercellularity, classically termed hyperplasia, of multiple parathyroid glands is observed in approximately 15% of cases. Parathyroid carcinoma, in contrast, is a rare cause of primary hyperparathyroidism, accounting for less than 1% of cases, but is often associated with severe clinical manifestations and significant mortality.
Distinguishing between parathyroid adenoma and carcinoma is notoriously difficult on purely histopathologic grounds. In the absence of invasion of surrounding structures and/or metastasis, histopathologic features including fibrous bands, mitotic figures, and capsular invasion are strongly suggestive, but not pathognomonic of carcinoma. A definitive diagnosis of carcinoma depends upon the finding of regional or distant metastases or marked local invasion already present at the time of surgery, thereby lowering the likelihood for surgical cure of established parathyroid malignancy (Apel and Asa 2002). While there are a few reports of carcinoma occurring within (and apparently evolving from) an adenoma or a hyperplastic parathyroid gland (Murayama et al. 1977; Aldinger et al. 1982; Berland et al. 1982; Haghighi et al. 1983; Desch et al. 1984), the disproportionately overwhelming prevalence of typical sporadic parathyroid adenoma compared with carcinoma implies that progressive transformation from typical adenoma to carcinoma must be extremely rare. The genetic and epigenetic changes found in benign parathyroid adenomas and malignant parathyroid carcinomas will be discussed separately here.
2. Parathyroid Adenoma
Two genes, MEN1 and CCND1 (encoding cyclin D1), have been solidly established as genetic drivers of benign parathyroid adenomas, meeting the most stringent criteria of (a) the finding of recurrent, somatic mutations in typical, sporadically presenting human parathyroid adenomas and (b) their ability to drive parathyroid tumorigenesis in experimental animal models (or in humans, if strong/mendelian evidence of such a drive exists). CDKN1B and other CDKI genes appear to function rarely as genetic drivers of parathyroid tumorigenesis, and may more commonly be involved in predisposition to sporadically presenting (and familial) parathyroid tumors. CASR, encoding the calcium sensing receptor, also has a role, albeit very limited, in predisposition to parathyroid adenoma but not as a driver of sporadic tumors via somatic mutation. In this discussion and previously analyzed series, we have defined typical sporadic adenoma as cases with clinically determined single gland disease, occurring in adults with no personal/family history of primary hyperparathyroidism nor a personal/family history suggestive of multiple endocrine neoplasia or a related syndrome, and for which neither gross examination nor histopathology demonstrates atypical or malignant features (described below). Defining criteria for case ascertainment is important, since use of different (especially less stringent) criteria in some studies may well influence the apparent genetic contributors that emerge.
2.1 MEN1
The involvement of the MEN1 tumor suppressor gene in sporadic parathyroid adenomas was first suspected based on its involvement in the familial Multiple Endocrine Neoplasia Type 1 syndrome (Chandrasekharappa et al. 1997). MEN1 is discussed more thoroughly in another article in this issue [reference to follow] but its relevance to sporadic parathyroid tumors is detailed here.
Loss of heterozygosity of chromosome 11q, the genomic location of the MEN1 gene, is the most frequent genomic aberration found in parathyroid adenomas. Following positional cloning of the MEN1 in familial MEN1, somatic mutations of MEN1 were identified in sporadic parathyroid tumors, with biallelic inactivation of MEN1 occurring in 12-35% (Heppner et al. 1997; Carling et al. 1998a; Farnebo et al. 1998; Tanaka et al. 2002; Cromer et al. 2012; Newey et al. 2012). Rarely, patients presenting with apparently sporadic parathyroid adenoma will present with germline MEN1 mutations (Starker et al. 2012a).
Mouse models of MEN1 have been developed. Homozygous inactivation of Men1 is embryonic lethal and mice heterozygous null for Men1 develop a spectrum of tumors similar to the human syndrome, including parathyroid tumors (Crabtree et al. 2001; Harding et al. 2009). Additional conditional knockouts of Men1 have been developed using the Cre-LoxP system. Targeted inactivation of Men1 specifically to the parathyroid glands resulted in parathyroid neoplasia accompanied by hypercalcemic hyperparathyroidism (Libutti et al. 2003).
2.2 CCND1
The CCND1 (PRAD1) oncogene, encoding cyclin D1, was first identified as a human oncogene through its involvement in parathyroid tumorigenesis. In a subset of parathyroid adenomas, a peri-centromeric inversion of chromosome 11 results in the juxtaposition of the PTH 5’ regulatory region to the CCND1 coding region. This rearrangement places expression of CCND1 under the control of the PTH promoter/enhancer; this rearrangement is illustrated in Figure 1. Since PTH is normally expressed at high levels in parathyroid cells, the PTH-CCND1 rearrangement resulted in high-level overexpression of CCND1 (Motokura et al. 1991). While the PTH-CCND1 rearrangement is appears to occur in up to 8% of sporadic parathyroid adenomas (Westinet al. 2009), overexpression of cyclin D1 has been seen in 20-40% of these tumors (Hsi et al. 1996; Tominaga et al. 1999; Vasef et al. 1999). Subsequently, overexpression of cyclin D1 due to CCND1 DNA amplifications or gene rearrangements have been demonstrated in a variety of tumor types, confirming CCND1's importance as a bone fide human oncogene (Arnold 1995; Arnold and Papanikolaou 2005).
Fig. 1. Schematic diagram illustrating the DNA rearrangement involving the PTH gene and the CCND1 gene in a subset of parathyroid adenomas.

The chromosomal inversion event is deduced as the simplest cytogenetic event consistent with the molecular details of this DNA rearrangement.
A mouse model of the PTH-CCND1 rearrangement, similar to that found in human parathyroid tumors, has been developed (Imanishi et al. 2001). Cyclin D1 overexpression in the parathyroid glands of these transgenic mice is driven by the PTH regulatory region. These mice develop moderate chronic biochemical hyperparathyroidism and parathyroid gland hypercellularity, providing direct experimental evidence for cyclin D1's role as a driver of parathyroid neoplasia, and also establishing the mice as a model of human hyperparathyroidism.
2.3 CDKN1B/CDKIs
With the establishment of CyclinD1 as a bona fide parathyroid oncogene, its binding partners in cell cycle regulation, cyclin dependent inhibitors were also suspected to play a role in parathyroid tumorigenesis. Mutation of Cdkn1b, encoding the cyclin-dependent kinase inhibitor (CDKI) p27kip1, was later determined to cause a multiple endocrine neoplasia syndrome, including parathyroid tumors, in a spontaneous occurring rat model (Fritz et al. 2002; Pellegata et al. 2006). Patients with clinical criteria suggestive of MEN1 (e.g. multigland parathyroid hyperfunction or parathyroid plus another MEN1-related tumor), but lacking any detectable MEN1 mutation, were subsequently screened for sequence abnormalities in CDKN1B and other CDKI genes (reviewed in (Georgitsi)) and mutations of suspected pathologic significance were identified in CDKN1B and additional CDKI-encoding genes (CDKN1A, encoding p21Cip1, CDKN2B, encoding p15Ink4b and CDKN2C, encoding p18Ink4c) (Agarwal et al. 2009).The role of CDKI genes in familial endocrine tumors will be addressed elsewhere in this issue [reference to follow].
With the involvement of CDKNIB in familial/syndromic hyperparathyroidism having been established, a role for CDKN1B mutation in sporadic parathyroid tumors was sought. Non-synonymous, intragenic CDKN1B point mutations were identified in three patients with typically presenting sporadic parathyroid adenomas; in all three cases, the sequence variants were also present in the patients’ germline DNA. One additional adenoma contained a somatic frameshift mutation coupled with tumor-specific LOH at CDKN1B and the surrounding genomic region (Costa-Guda et al. 2011) and unpublished observations). Allelic loss at the CDKN1B locus is uncommon in parathyroid tumors (Tahara et al. 1996; Palanisamy et al. 1998), but decreased expression of p27 has been described at both the RNA (Buchwald et al. 2004) and protein levels (Erickson et al. 1998; Tokumoto et al. 2002). The finding of somatic mutation in sporadic human parathyroid adenomas coupled with the ability of Cdkn1B mutation to drive tumorigenesis in rats provides strong evidence supporting CDKN1B's candidacy as a genetic driver of benign parathyroid tumors. Future documentation of the recurrence of p27 mutation in sporadic cases would enable CDKN1B mutation to be solidly characterized as an established driver of typical sporadic parathyroid adenomas.
The remaining 6 CDKI genes, CDKN1A, CDKN1C, CDKN2A, CDKN2B, CDKN2C and CDKN2D, encoding p21, p57, p14ARF/p16, p15, p18, and p19 respectively, were subsequently examined for mutations in sporadic parathyroid adenomas. Non-synonymous, intragenic point mutations in CDKN1A, CDKN2B or CDKN2C were identified in five tumors; a single, somatic mutation was identified in CDKN2C, while the remaining mutations were germline or of undetermined germline/somatic status. No mutations of potential pathogenic significance were identified in CDKN1C, CDKN2A or CDKN2D. Functional evidence supporting the potential pathogenicity of observed sequence variants was demonstrated for 3 of the 5 variants, one in each gene. Hypermethylation of CDKN2A and CDKN2C has also been described in parathyroid tumors (Starker et al. 2011). Mice null for Ckdn2c, encoding p18, rarely develop parathyroid neoplasia but when crossed with Cdkn1b null mice, p18/p27 null animals demonstrate an increased incidence of parathyroid tumors (Franklin et al. 2000). p18 null/Men1 heterozygotes also demonstrate an increased incidence of parathyroid tumors as compared to single knockout of p18 or Men1 heterozygous littermates (Bai et al. 2007). Thus, the evidence that the mentioned mutations/variants CDKN1A, CDKN2B and CDKN2C may contribute to parathyroid tumorigenesis by somatic mutation or germline predisposition is strengthened their ability, at least in combination, to drive parathyroid tumorigenesis experimentally.
2.4 Calcium sensing receptor
The extracellular calcium level is monitored by the parathyroid glands via cell surface receptors and, at least when chronically low, can serve as an important regulator of parathyroid cell growth. The calcium sensing receptor (CaR) is encoded by the CASR gene. Calcium stimulates the CaR, a G-protein coupled receptor, which responds by activating phospholipase C, through Gq and G11, resulting in production of inositol triphosphate and release of calcium from intracellular stores. Diacylglycerol concentrations are also increased, stimulating protein kinase C, which phosphorylates the CaR, promoting β-arrestin binding and internalization of the CaR. CaR stimulation decreases PTH production and secretion. In the absence of a strong negative feedback stimulus from extracellular calcium, the CaR is relaxed and PTH secretion is relatively unrestrained. Over the short term, a parathyroid cell lacking CaR stimulation will secrete PTH from granules stored in the cytoplasm and begin to increase transcription of the PTH gene and protein production. A prolonged absence of stimulus from CaR will trigger the cell to grow and divide. Activating germline mutations of CASR are associated with a hypocalcemic, hypoparathyroid phenotype (Pearce et al. 1996) while inactivating mutations are seen in patients with familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT). Inactivating mutations result in reduced sensitivity to extracellular calcium, which alters the calcium-PTH set-point in parathyroid cells. An increased level of Ca2+ is required to suppress PTH release. Mutations in genes involved in CaR signaling, GNA11, which encodes Gα11, and AP2S1, which encodes AP2 σ, involved in CaR internalization, have also been demonstrated in FHH (Nesbit et al. 2013a; Nesbit et al. 2013b). Along with increased serum calcium and PTH, mild (FHH) to severe (NSHPT) parathyroid hyperplasia is seen in these patients, suggesting that CASR could function as a parathyroid tumor suppressor gene (Pollak et al. 1993). Parathyroid hyperplasia has also been demonstrated in homozygous Casr knockout mice, but not in heterozygous knockout mice (Ho et al. 1995). However, somatic, inactivating mutations have not been found in typical, sporadic parathyroid adenoma (Hosokawa et al. 1995; Cetani et al. 1999), and must be quite rare if they exist at all. Germline CASR mutations have been described in sporadically presenting hyperparathyroidism (Guarnieriet al. 2010; Starker et al. 2012a), although in only one case fitting our criteria for typically presenting, sporadic parathyroid adenoma (Guarnieri et al. 2010), underscoring the importance of case selection in genetic studies. Despite the rarity of mutations, aberrant CASR expression is more frequently seen in parathyroid tumors and may contribute at least to the hyperparathyroid biochemical phenotype, if not directly to parathyroid cell growth (Imanishi et al. 2001).
2.5 Additional Genetic Considerations
A number of additional genes have been reported as rare targets of somatic or germline mutation in benign, sporadic parathyroid adenomas but their ability to “drive” parathyroid tumorigenesis has yet to be established experimentally.
2.5.1 β-catenin
Several studies have examined the role of CTNNB1, encoding the oncogene β-catenin, in parathyroid adenomas. Phosphorylation of β-catenin by GSK3β normally leads to its proteosomal degradation (Clevers 2006); stabilization and accumulation of non-phosphorylated β-catenin, leading to increased activation of Wnt signaling, in human tumors can also be accomplished by mutation of the GSK3β recognition motif (encoded by exon 3) in the β-catenin gene CTNNB1 (Morin et al. 1997). Indeed, virtually all CTNNB1 mutations identified in human tumors are located in exon 3 and most affect serine-threonine phosphorylation sites or adjacent residues, making this a hotspot for mutational activation of CTNNB1 (Ilyas 2005).
Two studies of Swedish patients with parathyroid adenomas revealed an identical, somatic homozygous stabilizing mutation, encoding a serine to alanine change at amino acid 37 (S37A), in exon 3 of CTNNB1, in 9 of 124 tumors studied. Aberrant β-catentin staining was observed in all tumors analyzed immunohistochemically, regardless of mutation status(Bjorklund et al. 2007a; Bjorklund et al. 2008). Other groups have collectively interrogated nearly 600 additional parathyroid adenomas (including 98 from a distinct group of Swedish patients) for mutations of CTNNB1 exon 3 and S37A mutation has not been identified in any additional patients (Semba et al. 2000; Ikeda et al. 2002; Costa-Guda and Arnold 2007; Juhlin et al. 2009; Cetani et al. 2010; Haglund et al. 2010). However, a heterozygous, somatic mutation encoding a serine to cysteine change at amino acid 33 (S33C) has been identified in two patients from distinct cohorts (Guarnieri et al. 2012; Starker et al. 2012b), suggesting CTNNB's overall mutation frequency in parathyroid adenomas is likely less than 1.8% and perhaps as low as 0.3%. Beyond the noted discrepancies in reported frequencies of CTNNB1 mutations, it is difficult to explain why the S37A mutations reported by Bjorklund et al were uniformly homozygous, whereas the two S33C mutations reported by other groups were heterozygous; heterozygosity would be more consistent with CTNNB1's role as a direct-acting oncogene and the copy number of such mutations when observed in other types of human tumors. Also in contrast to the initial reports by Bjorklund et al, only two parathyroid adenomas of the 115 examined immunohistochemically in later studies, including only one of the two cases with S33C mutation, were reported to demonstrate abnormal β-catenin staining (Ikeda et al. 2002; Starker et al. 2012b), a value more consistent with the estimated mutation frequency. Aberrant splicing of the Wnt co-receptor LRP5, resulting in increased expression of β-catenin, has also been described in parathyroid adenoma (Bjorklund et al. 2007b). The role of β-catenin and other Wnt signaling pathway components in parathyroid tumorigenesis is an important issue that merits further investigation.
2.5.2 mtDNA
Mitochondrial alterations, including mitochondrial DNA (mtDNA) mutations have been described in a variety of tumor types. It has been hypothesized that a selective advantage conferred by mtDNA mutation could in particular contribute to benign tumorigenesis of a slowly replicating tissue like the human parathyroid. Acquired mitochondrial DNA mutations were identified in a subset of parathyroid adenomas, particularly in those with an oxyphil cell phenotype (Costa-Guda et al. 2007). Oxyphil cells have a characteristic eosinophilic granular cytoplasm that is densely packed with mitochondria (Munger and Roth 1963; Apel and Asa 2002), as compared with the typical chief cell. While the exact mechanism remains controversial, mtDNA mutations may well contribute to the molecular pathogenesis of benign parathyroid tumors. Statistically significant differences in mutation prevalence in oxyphil vs. chief cell adenomas also suggest that mtDNA mutations may contribute to the oxyphil phenotype (Costa-Guda et al. 2007).
2.5.3 Next generation sequencing
Advances in sequencing technologies, allowing for analysis of virtually all transcribed exons throughout the entire genome, have recently been applied to sporadic parathyroid adenomas (Cromer et al. 2012; Newey et al. 2012). Interestingly, these two studies failed to identify any frequent genetic alterations in parathyroid adenomas or any alterations common to both studies (except MEN1), demonstrating the genetic heterogeneity of parathyroid adenomas. The most frequent, and the only recurrent, abnormality identified was mutation of the MEN1 gene which, as noted above, was already well-recognized as a key contributor to sporadic (and familial) parathyroid neoplasia. A heterozygous, somatic missense mutation of EZH2, an oncogenic contributor to multiple human tumor types, was identified in one of eight tumors subjected to next-generation sequencing and an identical mutation was found in one of 185 additional tumors examined by Sanger sequencing (Cromer et al. 2012). Identical mutation of EZH2 has previously been described in follicular lymphoma and diffuse large B-cell lymphoma and has been demonstrated to act as a dominant, gain-of-function mutation.(Yap et al. 2011). While EZH2 mutation has not yet been experimentally demonstrated to drive hyperparathyroidism, it is a strong candidate for rare involvement as a parathyroid oncogene. Mutations in a number of additional genes, such as POT1, were reported to affect single tumors, but could not be determined by Sanger sequencing of additional tumors to be recurrent. Further studies are required to determine the extent and nature of involvement of these additional genes in the pathogenesis of parathyroid adenomas.
2.5.4 Others
A number of additional candidate genes, whose involvement in parathyroid tumorigenesis has been suspected, have also been examined. Benign parathyroid tumors are found in patients with germline RET mutations (MEN2). However, somatic mutations of RET have not been identified in sporadic parathyroid adenomas (Pausova et al. 1996). It is unknown whether alterations in RET expression and/or function may contribute to the molecular pathogenesis of sporadic parathyroid tumors in some way.
Germline alterations of the AIP gene, located 2.6 megabases away from MEN1 on 11q13 and encoding the aryl hydrocarbon receptor interacting protein, predispose patients to pituitary tumors, often such heritable mutations have a sporadic presentation. A germline AIP mutation has also been described in a single patient with a pituitary tumor and parathyroid hyperplasia, who tested negative for MEN1 or CDKN1B gene mutations (Belar et al. 2012). A recent study examined a series of 132 sporadically presenting parathyroid adenomas for AIP mutations and identified germline mutation in 2 cases, accompanied by loss of the normal allele in one case. One of the two mutation-positive patients had persistent hypercalcemia/hyperparathyroidism following surgery, suggestive of multi-gland disease. The same c.911G>A (R304Q) mutation was identified in both, unrelated patients and has been previously seen in several familial isolated pituitary adenoma kindreds and sporadic pituitary tumors (Pardi et al. 2013). Homozygous inactivation of Aip in genetically engineered mice is embryonic lethal, and no parathyroid abnormalities were reported in Aip heterozygous knockout mice (Kang et al. 2011). Thus, a rare predisposition allele of AIP is a good candidate for linkage to occasional cases of sporadic hyperparathyroidism; in the absence of reported somatic mutations and direct experimental functional evidence it remains to be determined if such mutations can function as a genetic drivers of typical sporadic parathyroid adenomas.
Parathyroid cell proliferation is a normal response to vitamin D deficiency. Clinically, patients with vitamin D deficiency or suboptimal vitamin D nutrition have increased parathyroid gland weight (Silverberg et al. 1999). The PTH gene promoter contains a vitamin D responsive element (VDRE)(Demay et al. 1992) and 1,25(OH)2D3 (the active form of vitamin D)-VDR complex suppresses PTH transcription and secretion (Silver et al. 1999). Active vitamin D has been shown to suppress parathyroid cell growth both in vitro and in vivo (Cantley et al. 1985; Nygren et al. 1988). However, increased parathyroid cell proliferation seen in VDR-deficient mice can be rescued by a calcium and phosphate rich diet (Li et al. 1998), and mice with a parathyroid-specific VDR deletion do not demonstrate enlarged parathyroid glands (Meir et al. 2009), suggesting that parathyroid proliferation is primarily influenced by calcium levels. Vitamin D metabolism has been linked to tumorigenesis in various cell types. Active vitamin D can inhibit cell cycle progression, associated with decreased cyclin D1 and increased p21 and p27 levels, and functions in regulation of growth factors, angiogenesis, apoptosis and telomerase activity [reviewed in (Buchwald et al. 2005)]. Despite roles in both parathyroid cell growth and human tumorigenesis, and reduced expression in parathyroid adenomas (Carling et al. 1998b; Carling et al. 2000), VDR mutations have not been found in parathyroid tumors (Brown et al. 2000; Samander and Arnold 2006).
2.5 Epigenetics
The role of epigenetic alterations in parathyroid adenomas has not been extensively studied. A few studies, focusing on an individual gene or a small group of genes, have been performed. The Rb-interacting zinc finger gene, RIZ1, a tumor suppressor gene capable of driving tumor development in humans and experimental animals, was hypermethylated in 40% of the parathyroid adenomas studied. Further, hypermethylation was accompanied by loss of heterozygosity at 1p36, the genomic locus of RIZ1 (Carling et al. 2003). However, aberrant expression of RIZ1 has not been reported in parathyroid tumors and loss of RIZ1 does not appear to be able to drive parathyroid tumor development in genetically engineered mice (Steele-Perkins et al. 2001). Hypermethylation of APC, the gene responsible for familial adenomatous polyposis (FAP), occurs frequently in parathyroid adenomas (Juhlin et al. 2010). Parathyroid tumors have been reported in a few FAP patients (Sakai et al. 2002; Andreasson et al. 2012), but owing to the relatively high prevalence of hyperparathyroidism, this may be a chance occurence. Despite hypermethylation, aberrant APC expression has not been demonstrated in parathyroid adenomas (Juhlin et al. 2009; Svedlund et al. 2010), except in the setting of germline APC mutation (Andreasson et al. 2012). RASSF1A (Juhlin et al. 2010) and HIC1 (Svedlund et al. 2012), genes, frequently subject to epigenetic inactivation in human cancers, are also frequently hypermethylated in parathyroid adenomas. A comprehensive methylome analysis, including methylation sites of more than 14,000 genes, was performed on a series of benign and malignant parathyroid tumors. This study revealed aberrant methylation of 367 genes, including RIZ1, APC and RASSF1A, in parathyroid adenoma and 175 genes in parathyroid carcinoma, as compared to normal parathyroids, and methylation patterns of 263 genes differed between parathyroid adenoma and carcinoma (Starker et al. 2011). It remains unclear which of these aberrantly methylated genes may be important to the pathogenesis of parathyroid tumors and if any of them may eventually serve as the basis of novel therapeutic interventions.
3. Parathyroid Carcinoma
Parathyroid carcinoma is an exceedingly rare, but highly aggressive, form of primary hyperparathyroidism. The rarity of this tumor has made the study of its genetic basis more difficult. It has been controversial whether parathyroid cancers typically arise de novo or from preexisting benign adenomas through further accumulation of genetic abnormalities, akin to the colorectal cancer model (Vogelstein et al. 1989). While there are a few reports of carcinoma occurring within (and apparently evolving from) an adenoma or a hyperplastic parathyroid gland (Murayama et al. 1977; Aldinger et al. 1982; Berland et al. 1982; Haghighi et al. 1983; Desch et al. 1984), the disproportionately high prevalence of typical sporadic parathyroid adenoma compared with carcinoma implies that progressive transformation from typical adenoma to carcinoma must be extremely rare.
Substantial evidence for a progression model has been demonstrated in colon cancer and other solid tumors, with normal tissue advancing through hyperplastic/dysplastic and benign neoplasia stages, via incremental accumulation of acquired genetic abnormalities, before becoming malignant. In a progression model, genetic alterations already present in early/benign disease are found at equal or greater frequencies in advanced/malignant disease, and additional alterations (that were important for progression) are present selectively in the malignant tumors. For this progression model to be generally true for parathyroid cancer, the same genetic alterations already present in parathyroid adenomas should be at least equally well represented in parathyroid carcinoma, along with additional acquired genomic changes found in carcinomas. The most common (and most informative) alterations in benign parathyroid tumors, loss of 11q and mutation of MEN1, occur in 35% of parathyroid adenomas (Tahara et al. 1996; Agarwal et al. 1998; Farnebo et al. 1999; Hunt et al. 2005; Cromer et al. 2012; Newey et al. 2012; Costa-Guda et al. 2013a). A progression model would predict that 11q loss and MEN1 mutation would be found in at least 35% of carcinomas; however, these changes are rarely, if ever, seen in parathyroid cancer (Agarwal et al. 1998; Farnebo et al. 1999; Kytola et al. 2000; Costa-Guda et al. 2013a). These observations suggest that parathyroid cancer generally arises de novo, rather than evolving from a preexisting typical benign adenoma.
3.1 HRPT2
An exception to the above-noted predominant process of de novo parathyroid carcinomagenesis appears to occur in patients with germline HRPT2 mutations. These patients do indeed appear to develop parathyroid carcinomas that evolve from preexisting benign or atypical adenomas, and might explain those rare reports of apparent progression. HRPT2 mutation is responsible for Hyperparathyroidism Jaw-Tumor syndrome (HPT-JT), an autosomal dominant disorder predisposing to multiple benign and/or malignant tumors in the parathyroid glands, kidneys and uterus and benign tumors of the jaw bones, typically classified as ossifying and/or cementifying fibroma. Germline HRPT2 mutations are also found in a subset of families with Familial Isolated Hyperparathyroidism.
Studies of sporadic parathyroid tumors demonstrated somatic, intragenic, inactivating mutations of HRPT2 in a large percentage of malignant parathyroid tumors (Shattuck et al. 2003b), but they very rarely occur in sporadically presenting benign parathyroid adenomas (Howell et al. 2003; Krebs et al. 2005; Bradley et al. 2006). Expression of parafibromin, the protein product of HRPT2, is also lost in the majority of parathyroid carcinomas but retained in adenomas. Owing to its specificity for parathyroid cancer, except in the setting of germline mutation, parafibromin immunohistochemistry has been proposed as an aid to diagnosis of parathyroid cancer in clinically equivocal cases (Tan et al. 2004; Gill et al. 2006; Cetani et al. 2007) but parafibromin staining alone may not be sufficient to serve as a diagnostic marker of parathyroid cancer (DeLellis 2011).
Unexpectedly, a substantial minority of patients with seemingly sporadic parathyroid cancer possess germline HRPT2 mutations, suggesting they may represent novel cases of HPT-JT or a phenotypic variant, and having important implications for long term management of the patients and their families (Shattuck et al. 2003b; Cetani et al. 2004; Guarnieri et al. 2006; Arnold and Marx 2008). A summary of HRPT2 mutations in both familial and sporadic hyperparathyroidism is shown in Figure 2. Identified mutations are scattered throughout the 1593 coding base pairs of the 17 exon gene, with an over-representation of mutations in exons 1, 2 and 7. As expected for a classical tumor suppressor gene, in many tumors biallelic inactivation of the gene can be demonstrated, through mutation accompanied by LOH or through independent mutations in both alleles (Howell et al. 2003; Shattuck et al. 2003b; Cetani et al. 2004). Inactivation of HRPT2 through gross deletion of the gene has also been observed (Cascon et al. 2011; Domingues et al. 2012; Bricaire et al. 2013).
Fig. 2. Schematic diagram of known mutations and polymorphisms in HRPT2 (CDC73).
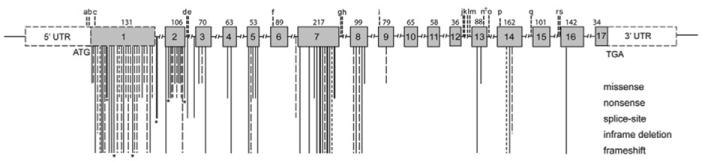
The HRPT2 gene consists of 17 exons (boxes), the coding region is indicated by shaded boxes and the untranslated regions are unshaded. The sites of known mutations are indicated by vertical lines below the gene. The length of line indicates mutation type as illustrated. Germline mutations (solid lines), somatic mutations (wide interrupted lines), and undefined mutations (narrow interrupted) are indicated . Figure originally published in (Newey et al. 2010).
Parafibromin is a 531 amino acid, ubiquitously expressed and evolutionarily conserved protein with a nuclear localization signal and no homology to any known protein functional domains. The C-terminal portion contains sequence similarity to yeast cell-division protein Cdc73p, a component of the yeast polymerase-associated factor 1 complex (Paf1c), which associates with RNA polymerase II during transcriptional initiation and elongation. Additional evidence suggests Paf1c is involved in histone modification and posttranscriptional events, including modification of the poly (a) tail. The human PAF1 complex (hPAF1C) includes homologs most of the same subunits as the yeast Paf1c and shares similar functions.
Studies in Drosophila have demonstrated that Hyrax, the Drosophila homolog of parafibromin, is involved in canonical Wnt/Wingless signaling (Mosimann et al. 2006), a central regulator of development and proliferation, thereby providing one potential mechanism for parafibromin's role in tumorigenesis. Activation of canonical Wnt signaling leads to activation of gene transcription by β-catenin; many targets of Wnt signaling promote cell proliferation. Expression of cyclin D1, an oncogene capable of driving parathyroid neoplasia, is regulated in part by Wnt signaling (Shtutman et al. 1999; Tetsu and McCormick 1999) and Wnt pathway abnormalities have been well documented in various types of human tumors (reviewed in (Karim et al. 2004)). Loss of Wnt pathway components APC and GSK3β (Juhlin et al. 2009) and accumulation of β-catenin have also been described in parathyroid cancer (Svedlund et al. 2010). Parafibromin has been demonstrated to inhibit cancer cell growth and cause G1 phase arrest in vitro, in part through regulation of Cyclin D1 (Woodard et al. 2005; Lin et al. 2008).
Conventional and conditional transgenic mouse knockouts of Hrpt2 have been developed to elucidate the in vivo function of parafibromin. Homozygous deletion of Hrpt2 is embryonic lethal by embryonic day 6.5 and controlled germline deletion of Hrpt2 at later stages of development led to growth retardation, severe cachexia and death within 20 days (Wang et al. 2008). These results indicate that Hrpt2 expression is important for both embryonic development and survival of adult mice. It remains to be determined how loss of Hrpt2 expression promotes tumorigenesis in tissues such as parathyroid and kidney in humans and whether this phenotype can be recapitulated in the setting of a model organism.
3.2 Additional Genetic Considerations
Studies of parathyroid cancer have focused primarily on identifying locations of allelic imbalance and interrogating candidate genes for sequence and/or expression abnormalities. Using comparative genomic hybridization and molecular allelotyping, recurrent regions of loss likely to contain a key tumor suppressor gene(s) have been localized to chromosomes 1p, 3, 13q and 14. Recurrent regions of gains likely to contain driver oncogene(s) are located on chromosomes 1q and 16 (Agarwal et al. 1998; Farnebo et al. 1999; Kytola et al. 2000; Hunt et al. 2005; Sulaiman et al. 2012; Costa-Guda et al. 2013a). Mutations in important human tumor suppressor genes such as p53 (Cryns et al. 1994; Hakim and Levine 1994), pRb and BRCA2 (Shattuck et al. 2003a) have been sought but none have been identified.
Recently, a single parathyroid carcinoma and a recurrence from the same patient were subjected to whole genome, next-generation sequence analysis. Somatic point mutations were detected in 23 genes; of these, 15 were detected in both the primary tumor and the recurrence, 7 were found only in the recurrence and one (PIK3CA) was found only in the primary tumor. Of particular interest, mutations were identified in MLL2, a putative tumor suppressor gene known to interact with MEN1; mTOR, a gene upstream of the parathyroid oncogene Cyclin D1; and THRAP3, a member of the SNARP complex that can regulate Cyclin D1 expression. A mutation was also identified inCDKN2C/p18; interestingly affecting the same amino acid residue as a mutation previously identified in a parathyroid adenoma (Costa-Guda et al. 2013b). Two inter-chromosomal translocations and one intra-chromosomal inversion, expected to result in the formation of fusion proteins, were also identified; it is unclear however if any of these fusion proteins were actually expressed in the tumor (Kasaian et al. 2013). Since these results are from a single patient, it is impossible to predict which of the mutated genes identified in this study will emerge as important driver genes in parathyroid cancer. However, the identification of such mutations is a potentially important step in expanding our understanding of the molecular pathogenesis of parathyroid cancer.
4. Conclusions
Several important advances have been made towards the goal of understanding the molecular basis of sporadic parathyroid tumors. Mutations in the MEN1 tumor suppressor gene are the most common finding in benign parathyroid adenomas. Mutations in CDKN1B, encoding p27, and other CDKI genes are found at a low frequency in parathyroid adenomas and germline variants may function as predisposition alleles in seemingly sporadic cases. The cyclin D1/PRAD1 oncogene has been identified as a parathyroid oncogene, is overexpressed in 20–40% of parathyroid adenomas, and is also involved in the development of many additional tumor types. Alterations in additional genes such as those encoded by the mitochondrial genome, β-catenin, and others identified by next-generation sequencing methods, including POT1 and EZH2, may contribute to parathyroid adenoma formation but their ability to drive parathyroid tumorigenesis remains to be demonstrated experimentally. Somatic mutations in the RET gene, the causal defect in MEN2, plus CASR and VDR, appear to contribute rarely if ever to the development of sporadic parathyroid tumors. The identification of the tumor suppressor parafibromin, encoded by the HRPT2 gene, has provided important insight into parathyroid carcinoma. Observations from comparing regions of allelic imbalance as well as known gene mutations suggest that parathyroid cancer generally arises de novo, rather than evolving from a preexisting typical benign adenoma. Additional genes important to the development of parathyroid tumors are likely to be identified by next-generation sequence analysis and the extent and nature of their involvement will need to be carefully examined and validated with genetic and experimental-functional approaches.
Highlights.
Parathyroid adenoma is the most common parathyroid tumor; parathyroid carcinoma is rare.
Parathyroid carcinoma does not evolve through a benign intermediate, except in HPT-JT.
Somatic MEN1 mutations are the most frequent finding in parathyroid adenoma.
Cyclin D1/PRAD1 is also an established driver of sporadic adenomas.
Mutation in CDKN1B/p27 and other CDKI also appears to contribute to parathyroid adenoma development.
Genetic predisposition to sporadic parathyroid adenoma may be conferred by rare, germline variants in CDKI genes, CASR and AIP.
The HRPT2 tumor suppressor gene is commonly mutated in parathyroid carcinoma.
Acknowledgements
This work was supported in part by the Murray-Heilig Fund in Molecular Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94(5):1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Schrock E, Kester MB, Burns AL, Heffess CS, Ried T, Marx SJ. Comparative genomic hybridization analysis of human parathyroid tumors. Cancer Genet Cytogenet. 1998;106(1):30–36. doi: 10.1016/s0165-4608(98)00049-1. [DOI] [PubMed] [Google Scholar]
- Aldinger KA, Hickey RC, Ibanez ML, Samaan NA. Parathyroid carcinoma: a clinical study of seven cases of functioning and two cases of nonfunctioning parathyroid cancer. Cancer. 1982;49(2):388–397. doi: 10.1002/1097-0142(19820115)49:2<388::aid-cncr2820490230>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Andreasson A, Sulaiman L, do Vale S, Martins JM, Ferreira F, Miltenberger-Miltenyi G, Batista L, Haglund F, Bjorck E, Nilsson IL, Hoog A, Larsson C, Juhlin CC. Molecular characterization of parathyroid tumors from two patients with hereditary colorectal cancer syndromes. Fam Cancer. 2012;11(3):355–362. doi: 10.1007/s10689-012-9520-z. [DOI] [PubMed] [Google Scholar]
- Apel RL, Asa SL. The Parathyroid Glands. Endocrine Pathology. V. LiVolsi and S. Asa. Philadelphia, Churchill Livingstone. 2002:103–147. [Google Scholar]
- Arnold A. The cyclin D1/PRAD1 oncogene in human neoplasia. J Investig Med. 1995;43(6):543–549. [PubMed] [Google Scholar]
- Arnold A, Marx SJ. Familial Hyperparathyroidism. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 2008:361–366. [Google Scholar]
- Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23(18):4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Bai F, Pei XH, Nishikawa T, Smith MD, Xiong Y. p18Ink4c, but not p27Kip1, collaborates with Men1 to suppress neuroendocrine organ tumors. Mol Cell Biol. 2007;27(4):1495–1504. doi: 10.1128/MCB.01764-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belar O, De La Hoz C, Perez-Nanclares G, Castano L, Gaztambide S. Novel mutations in MEN1, CDKN1B and AIP genes in patients with multiple endocrine neoplasia type 1 syndrome in Spain. Clin Endocrinol (Oxf) 2012;76(5):719–724. doi: 10.1111/j.1365-2265.2011.04269.x. [DOI] [PubMed] [Google Scholar]
- Berland Y, Olmer M, Lebreuil G, Grisoli J. Parathyroid carcinoma, adenoma and hyperplasia in a case of chronic renal insufficiency on dialysis. Clin Nephrol. 1982;18(3):154–158. [PubMed] [Google Scholar]
- Bjorklund P, Akerstrom G, Westin G. Accumulation of nonphosphorylated beta-catenin and c-myc in primary and uremic secondary hyperparathyroid tumors. J Clin Endocrinol Metab. 2007a;92(1):338–344. doi: 10.1210/jc.2006-1197. [DOI] [PubMed] [Google Scholar]
- Bjorklund P, Akerstrom G, Westin G. An LRP5 receptor with internal deletion in hyperparathyroid tumors with implications for deregulated WNT/beta-catenin signaling. PLoS Med. 2007b;4(11):e328. doi: 10.1371/journal.pmed.0040328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund P, Lindberg D, Akerstrom G, Westin G. Stabilizing mutation of CTNNB1/beta-catenin and protein accumulation analyzed in a large series of parathyroid tumors of Swedish patients. Mol Cancer. 2008;7:53. doi: 10.1186/1476-4598-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, Besser GM, Conceicao Pereira M, Davie MW, Dudley N, Leite V, Sadler GP, Seller A, Thakker RV. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf) 2006;64(3):299–306. doi: 10.1111/j.1365-2265.2006.02460.x. [DOI] [PubMed] [Google Scholar]
- Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, Vezzosi D, Kuhn JM, Murat A, Caron P, Sadoul JL, Silve C, Chanson P, Barlier A, Clauser E, Porchet N, Groussin L. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98(2):E403–408. doi: 10.1210/jc.2012-2789. [DOI] [PubMed] [Google Scholar]
- Brown SB, Brierley TT, Palanisamy N, Salusky IB, Goodman W, Brandi ML, Drueke TB, Sarfati E, Urena P, Chaganti RS, Pike JW, Arnold A. Vitamin D receptor as a candidate tumor-suppressor gene in severe hyperparathyroidism of uremia. J Clin Endocrinol Metab. 2000;85(2):868–872. doi: 10.1210/jcem.85.2.6426. [DOI] [PubMed] [Google Scholar]
- Buchwald PC, Akerstrom G, Westin G. Reduced p18INK4c, p21CIP1/WAF1 and p27KIP1 mRNA levels in tumours of primary and secondary hyperparathyroidism. Clin Endocrinol (Oxf) 2004;60(3):389–393. doi: 10.1111/j.1365-2265.2004.01995.x. [DOI] [PubMed] [Google Scholar]
- Buchwald PC, Westin G, Akerstrom G. Vitamin D in normal and pathological parathyroid glands: new prospects for treating hyperparathyroidism (review). Int J Mol Med. 2005;15(4):701–706. [PubMed] [Google Scholar]
- Cantley LK, Russell J, Lettieri D, Sherwood LM. 1,25-Dihydroxyvitamin D3 suppresses parathyroid hormone secretion from bovine parathyroid cells in tissue culture. Endocrinology. 1985;117(5):2114–2119. doi: 10.1210/endo-117-5-2114. [DOI] [PubMed] [Google Scholar]
- Carling T, Correa P, Hessman O, Hedberg J, Skogseid B, Lindberg D, Rastad J, Westin G, Akerstrom G. Parathyroid MEN1 gene mutations in relation to clinical characteristics of nonfamilial primary hyperparathyroidism. J Clin Endocrinol Metab. 1998a;83(8):2960–2963. doi: 10.1210/jcem.83.8.4977. [DOI] [PubMed] [Google Scholar]
- Carling T, Du Y, Fang W, Correa P, Huang S. Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery. 2003;134(6):932–939. doi: 10.1016/s0039-6060(03)00422-7. discussion 939-940. [DOI] [PubMed] [Google Scholar]
- Carling T, Rastad J, Akerstrom G, Westin G. Vitamin D receptor (VDR) and parathyroid hormone messenger ribonucleic acid levels correspond to polymorphic VDR alleles in human parathyroid tumors. J Clin Endocrinol Metab. 1998b;83(7):2255–2259. doi: 10.1210/jcem.83.7.4862. [DOI] [PubMed] [Google Scholar]
- Carling T, Rastad J, Szabo E, Westin G, Akerstrom G. Reduced parathyroid vitamin D receptor messenger ribonucleic acid levels in primary and secondary hyperparathyroidism. J Clin Endocrinol Metab. 2000;85(5):2000–2003. doi: 10.1210/jcem.85.5.6607. [DOI] [PubMed] [Google Scholar]
- Cascon A, Huarte-Mendicoa CV, Javier Leandro-Garcia L, Leton R, Suela J, Santana A, Costa MB, Comino-Mendez I, Landa I, Sanchez L, Rodriguez-Antona C, Cigudosa JC, Robledo M. Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer. 2011;50(11):922–929. doi: 10.1002/gcc.20911. [DOI] [PubMed] [Google Scholar]
- Cetani F, Ambrogini E, Viacava P, Pardi E, Fanelli G, Naccarato AG, Borsari S, Lemmi M, Berti P, Miccoli P, Pinchera A, Marcocci C. Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol. 2007;156(5):547–554. doi: 10.1530/EJE-06-0720. [DOI] [PubMed] [Google Scholar]
- Cetani F, Pardi E, Banti C, Collecchi P, Viacava P, Borsari S, Fanelli G, Naccarato AG, Saponaro F, Berti P, Miccoli P, Pinchera A, Marcocci C. Beta-catenin activation is not involved in sporadic parathyroid carcinomas and adenomas. Endocr Relat Cancer. 2010;17(1):1–6. doi: 10.1677/ERC-09-0147. [DOI] [PubMed] [Google Scholar]
- Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, Ambrogini E, Gazzerro E, Colussi G, Berti P, Miccoli P, Pinchera A, Marcocci C. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. 2004;89(11):5583–5591. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- Cetani F, Pinchera A, Pardi E, Cianferotti L, Vignali E, Picone A, Miccoli P, Viacava P, Marcocci C. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Miner Res. 1999;14(6):878–882. doi: 10.1359/jbmr.1999.14.6.878. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-Catenin Signaling in Development and Disease. Cell. 2006;127(3):469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Costa-Guda J, Arnold A. Absence of stabilizing mutations of beta-catenin encoded by CTNNB1 exon 3 in a large series of sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2007;92(4):1564–1566. doi: 10.1210/jc.2006-2554. [DOI] [PubMed] [Google Scholar]
- Costa-Guda J, Imanishi Y, Palanisamy N, Kawamata N, Phillip Koeffler H, Chaganti RS, Arnold A. Allelic imbalance in sporadic parathyroid carcinoma and evidence for its de novo origins. Endocrine. 2013a doi: 10.1007/s12020-013-9903-4. 10.1007/s12020-013-9903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011;96(4):E701–706. doi: 10.1210/jc.2010-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Guda J, Soong CP, Parekh VI, Agarwal SK, Arnold A. Germline and Somatic Mutations in Cyclin-Dependent Kinase Inhibitor Genes CDKN1A, CDKN2B, and CDKN2C in Sporadic Parathyroid Adenomas. Horm Cancer. 2013b doi: 10.1007/s12672-013-0147-9. 10.1007/s12672-013-0147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Guda J, Tokura T, Roth SI, Arnold A. Mitochondrial DNA mutations in oxyphilic and chief cell parathyroid adenomas. BMC Endocr Disord. 2007;7:8. doi: 10.1186/1472-6823-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98(3):1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, Carling T. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J Clin Endocrinol Metab. 2012;97(9):E1774–1781. doi: 10.1210/jc.2012-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns VL, Rubio MP, Thor AD, Louis DN, Arnold A. p53 abnormalities in human parathyroid carcinoma. J Clin Endocrinol Metab. 1994;78(6):1320–1324. doi: 10.1210/jcem.78.6.8200932. [DOI] [PubMed] [Google Scholar]
- DeLellis RA. Parathyroid tumors and related disorders. Mod Pathol. 2011;24(Suppl 2):S78–93. doi: 10.1038/modpathol.2010.132. [DOI] [PubMed] [Google Scholar]
- Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM. Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1992;89(17):8097–8101. doi: 10.1073/pnas.89.17.8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desch CE, Arsensis G, Woolf PD, May AG, Amatruda JM. Parathyroid hyperplasia and carcinoma within one gland. Am J Med. 1984;77(1):131–134. doi: 10.1016/0002-9343(84)90447-9. [DOI] [PubMed] [Google Scholar]
- Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, Cavaco BM. Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin Endocrinol (Oxf) 2012;76(1):33–38. doi: 10.1111/j.1365-2265.2011.04184.x. [DOI] [PubMed] [Google Scholar]
- Erickson LA, Jin L, Wollan PC, Thompson GB, van Heerden J, Lloyd RV. Expression of p27kip1 and Ki-67 in benign and malignant thyroid tumors. Mod Pathol. 1998;11(2):169–174. [PubMed] [Google Scholar]
- Farnebo F, Kytola S, Teh BT, Dwight T, Wong FK, Hoog A, Elvius M, Wassif WS, Thompson NW, Farnebo LO, Sandelin K, Larsson C. Alternative genetic pathways in parathyroid tumorigenesis. J Clin Endocrinol Metab. 1999;84(10):3775–3780. doi: 10.1210/jcem.84.10.6057. [DOI] [PubMed] [Google Scholar]
- Farnebo F, Teh BT, Kytola S, Svensson A, Phelan C, Sandelin K, Thompson NW, Hoog A, Weber G, Farnebo LO, Larsson C. Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab. 1998;83(8):2627–2630. doi: 10.1210/jcem.83.8.4846. [DOI] [PubMed] [Google Scholar]
- Franklin DS, Godfrey VL, O'Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20(16):6147–6158. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz A, Walch A, Piotrowska K, Rosemann M, Schaffer E, Weber K, Timper A, Wildner G, Graw J, Hofler H, Atkinson MJ. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62(11):3048–3051. [PubMed] [Google Scholar]
- Georgitsi M. MEN-4 and other multiple endocrine neoplasias due to cyclin-dependent kinase inhibitors (p27(Kip1) and p18(INK4C)) mutations. Best Pract Res Clin Endocrinol Metab. 24(3):425–437. doi: 10.1016/j.beem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Gill AJ, Clarkson A, Gimm O, Keil J, Dralle H, Howell VM, Marsh DJ. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism-jaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol. 2006;30(9):1140–1149. doi: 10.1097/01.pas.0000209827.39477.4f. [DOI] [PubMed] [Google Scholar]
- Guarnieri V, Baorda F, Battista C, Bisceglia M, Balsamo T, Gruppioni E, Fiorentino M, Muscarella LA, Coco M, Barbano R, Corbetta S, Spada A, Cole DE, Canaff L, Hendy GN, Carella M, Scillitani A. A rare S33C mutation of CTNNB1 encoding beta-catenin in a parathyroid adenoma found in an Italian primary hyperparathyroid cohort. Endocrine. 2012;41(1):152–155. doi: 10.1007/s12020-011-9558-y. [DOI] [PubMed] [Google Scholar]
- Guarnieri V, Canaff L, Yun FH, Scillitani A, Battista C, Muscarella LA, Wong BY, Notarangelo A, D'Agruma L, Sacco M, Cole DE, Hendy GN. Calcium-sensing receptor (CASR) mutations in hypercalcemic states: studies from a single endocrine clinic over three years. J Clin Endocrinol Metab. 2010;95(4):1819–1829. doi: 10.1210/jc.2008-2430. [DOI] [PubMed] [Google Scholar]
- Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, Minisola S, Mascia ML, D'Agruma L, Cole DE. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab. 2006;91(8):2827–2832. doi: 10.1210/jc.2005-1239. [DOI] [PubMed] [Google Scholar]
- Haghighi P, Astarita RW, Wepsic HT, Wolf PL. Concurrent primary parathyroid hyperplasia and parathyroid carcinoma. Arch Pathol Lab Med. 1983;107(7):349–350. [PubMed] [Google Scholar]
- Haglund F, Andreasson A, Nilsson IL, Hoog A, Larsson C, Juhlin CC. Lack of S37A CTNNB1/beta-catenin mutations in a Swedish cohort of 98 parathyroid adenomas. Clin Endocrinol (Oxf) 2010;73(4):552–553. doi: 10.1111/j.1365-2265.2010.03830.x. [DOI] [PubMed] [Google Scholar]
- Hakim JP, Levine MA. Absence of p53 point mutations in parathyroid adenoma and carcinoma. J Clin Endocrinol Metab. 1994;78(1):103–106. doi: 10.1210/jcem.78.1.8288693. [DOI] [PubMed] [Google Scholar]
- Harding B, Lemos MC, Reed AA, Walls GV, Jeyabalan J, Bowl MR, Tateossian H, Sullivan N, Hough T, Fraser WD, Ansorge O, Cheeseman MT, Thakker RV. Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia. Endocr Relat Cancer. 2009;16(4):1313–1327. doi: 10.1677/ERC-09-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi SE, Skarulis MC, Doppman JL, Alexander RH, Kim YS, Saggar SK, Lubensky IA, Zhuang Z, Liotta LA, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997;16(4):375–378. doi: 10.1038/ng0897-375. [DOI] [PubMed] [Google Scholar]
- Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet. 1995;11(4):389–394. doi: 10.1038/ng1295-389. [DOI] [PubMed] [Google Scholar]
- Hosokawa Y, Pollak MR, Brown EM, Arnold A. Mutational analysis of the extracellular Ca(2+)-sensing receptor gene in human parathyroid tumors. J Clin Endocrinol Metab. 1995;80(11):3107–3110. doi: 10.1210/jcem.80.11.7593409. [DOI] [PubMed] [Google Scholar]
- Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, Nelson AE, Krause U, Hammje K, Dralle H, Hoang-Vu C, Gimm O, Marsh DJ, Morreau H, Teh BT. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40(9):657–663. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsi ED, Zukerberg LR, Yang WI, Arnold A. Cyclin D1/PRAD1 expression in parathyroid adenomas: an immunohistochemical study. J Clin Endocrinol Metab. 1996;81(5):1736–1739. doi: 10.1210/jcem.81.5.8626826. [DOI] [PubMed] [Google Scholar]
- Hunt JL, Carty SE, Yim JH, Murphy J, Barnes L. Allelic loss in parathyroid neoplasia can help characterize malignancy. Am J Surg Pathol. 2005;29(8):1049–1055. [PubMed] [Google Scholar]
- Ikeda S, Ishizaki Y, Shimizu Y, Fujimori M, Ojima Y, Okajima M, Sugino K, Asahara T. Immunohistochemistry of cyclin D1 and beta-catenin, and mutational analysis of exon 3 of beta-catenin gene in parathyroid adenomas. Int J Oncol. 2002;20(3):463–466. [PubMed] [Google Scholar]
- Ilyas M. Wnt signalling and the mechanistic basis of tumour development. J Pathol. 2005;205(2):130–144. doi: 10.1002/path.1692. [DOI] [PubMed] [Google Scholar]
- Imanishi Y, Hosokawa Y, Yoshimoto K, Schipani E, Mallya S, Papanikolaou A, Kifor O, Tokura T, Sablosky M, Ledgard F, Gronowicz G, Wang TC, Schmidt EV, Hall C, Brown EM, Bronson R, Arnold A. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J Clin Invest. 2001;107(9):1093–1102. doi: 10.1172/JCI10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhlin CC, Haglund F, Villablanca A, Forsberg L, Sandelin K, Branstrom R, Larsson C, Hoog A. Loss of expression for the Wnt pathway components adenomatous polyposis coli and glycogen synthase kinase 3-beta in parathyroid carcinomas. Int J Oncol. 2009;34(2):481–492. [PubMed] [Google Scholar]
- Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenstrom J, Hoog A, Larsson C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One. 2010;5(3):e9472. doi: 10.1371/journal.pone.0009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BH, Xia F, Pop R, Dohi T, Socolovsky M, Altieri DC. Developmental control of apoptosis by the immunophilin aryl hydrocarbon receptor-interacting protein (AIP) involves mitochondrial import of the survivin protein. J Biol Chem. 2011;286(19):16758–16767. doi: 10.1074/jbc.M110.210120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim R, Tse G, Putti T, Scolyer R, Lee S. The significance of the Wnt pathway in the pathology of human cancers. Pathology. 2004;36(2):120–128. doi: 10.1080/00313020410001671957. [DOI] [PubMed] [Google Scholar]
- Kasaian K, Wiseman SM, Thiessen N, Mungall KL, Corbett RD, Qian JQ, Nip KM, He A, Tse K, Chuah E, Varhol RJ, Pandoh P, McDonald H, Zeng T, Tam A, Schein J, Birol I, Mungall AJ, Moore RA, Zhao Y, Hirst M, Marra MA, Walker BA, Jones SJ. Complete genomic landscape of a recurring sporadic parathyroid carcinoma. J Pathol. 2013;230(3):249–260. doi: 10.1002/path.4203. [DOI] [PubMed] [Google Scholar]
- Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2005;90(9):5015–5017. doi: 10.1210/jc.2005-0717. [DOI] [PubMed] [Google Scholar]
- Kytola S, Farnebo F, Obara T, Isola J, Grimelius L, Farnebo LO, Sandelin K, Larsson C. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol. 2000;157(2):579–586. doi: 10.1016/S0002-9440(10)64568-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139(10):4391–4396. doi: 10.1210/endo.139.10.6262. [DOI] [PubMed] [Google Scholar]
- Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O'Connor S, Ward JM, Emmert-Buck MR, Remaley A, Miller M, Turner E, Alexander HR, Arnold A, Marx SJ, Collins FS, Spiegel AM. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 2003;63(22):8022–8028. [PubMed] [Google Scholar]
- Lin L, Zhang JH, Panicker LM, Simonds WF. The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-myc proto-oncogene. Proc Natl Acad Sci U S A. 2008;105(45):17420–17425. doi: 10.1073/pnas.0710725105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir T, Levi R, Lieben L, Libutti S, Carmeliet G, Bouillon R, Silver J, Naveh-Many T. Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am J Physiol Renal Physiol. 2009;297(5):F1192–1198. doi: 10.1152/ajprenal.00360.2009. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Mosimann C, Hausmann G, Basler K. Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/Armadillo. Cell. 2006;125(2):327–341. doi: 10.1016/j.cell.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350(6318):512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- Munger BL, Roth SI. The cytology of the normal parathyroid glands of man and Virginia deer; a light and electron microscopic study with morphologic evidence of secretory activity. J Cell Biol. 1963;16:379–400. doi: 10.1083/jcb.16.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama T, Kawabe K, Tagami M. A case of parathyroid carcinoma concurred with hyperplasia: an electron microscopic study. J Urol. 1977;118(1 Pt 1):126–127. doi: 10.1016/s0022-5347(17)57920-3. [DOI] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, Rust N, Hobbs MR, Heath H, 3rd, Thakker RV. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013a;368(26):2476–2486. doi: 10.1056/NEJMoa1300253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, Gregory L, Rimmer AJ, Rust N, Graham U, Morrison PJ, Hunter SJ, Whyte MP, McVean G, Buck D, Thakker RV. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013b;45(1):93–97. doi: 10.1038/ng.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat. 2010;31(3):295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT, Gorvin CM, Stechman M, Gregory L, Mihai R, Sadler G, McVean G, Buck D, Thakker RV. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. J Clin Endocrinol Metab. 2012;97(10):E1995–2005. doi: 10.1210/jc.2012-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren P, Larsson R, Johansson H, Ljunghall S, Rastad J, Akerstrom G. 1,25(OH)2D3 inhibits hormone secretion and proliferation but not functional dedifferentiation of cultured bovine parathyroid cells. Calcif Tissue Int. 1988;43(4):213–218. doi: 10.1007/BF02555137. [DOI] [PubMed] [Google Scholar]
- Palanisamy N, Imanishi Y, Rao PH, Tahara H, Chaganti RS, Arnold A. Novel chromosomal abnormalities identified by comparative genomic hybridization in parathyroid adenomas. J Clin Endocrinol Metab. 1998;83(5):1766–1770. doi: 10.1210/jcem.83.5.4806. [DOI] [PubMed] [Google Scholar]
- Pardi E, Marcocci C, Borsari S, Saponaro F, Torregrossa L, Tancredi M, Raspini B, Basolo F, Cetani F. Aryl Hydrocarbon Receptor Interacting Protein (AIP) Mutations Occur Rarely in Sporadic Parathyroid Adenomas. J Clin Endocrinol Metab. 2013;98(7):2800–2810. doi: 10.1210/jc.2012-4029. [DOI] [PubMed] [Google Scholar]
- Pausova Z, Soliman E, Amizuka N, Janicic N, Konrad EM, Arnold A, Goltzman D, Hendy GN. Role of the RET proto-oncogene in sporadic hyperparathyroidism and in hyperparathyroidism of multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 1996;81(7):2711–2718. doi: 10.1210/jcem.81.7.8675600. [DOI] [PubMed] [Google Scholar]
- Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335(15):1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42):15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75(7):1297–1303. doi: 10.1016/0092-8674(93)90617-y. [DOI] [PubMed] [Google Scholar]
- Sakai Y, Koizumi K, Sugitani I, Nakagawa K, Arai M, Utsunomiya J, Muto T, Fujita R, Kato Y. Familial adenomatous polyposis associated with multiple endocrine neoplasia type 1-related tumors and thyroid carcinoma: a case report with clinicopathologic and molecular analyses. Am J Surg Pathol. 2002;26(1):103–110. doi: 10.1097/00000478-200201000-00014. [DOI] [PubMed] [Google Scholar]
- Samander EH, Arnold A. Mutational analysis of the vitamin D receptor does not support its candidacy as a tumor suppressor gene in parathyroid adenomas. J Clin Endocrinol Metab. 2006;91(12):5019–5021. doi: 10.1210/jc.2006-1543. [DOI] [PubMed] [Google Scholar]
- Semba S, Kusumi R, Moriya T, Sasano H. Nuclear Accumulation of B-Catenin in Human Endocrine Tumors: Association with Ki-67 (MIB-1) Proliferative Activity. Endocr Pathol. 2000;11(3):243–250. doi: 10.1385/ep:11:3:243. [DOI] [PubMed] [Google Scholar]
- Shattuck TM, Kim TS, Costa J, Yandell DW, Imanishi Y, Palanisamy N, Gaz RD, Shoback D, Clark OH, Monchik JM, Wierman ME, Hollenberg A, Tojo K, Chaganti RS, Arnold A. Mutational analyses of RB and BRCA2 as candidate tumour suppressor genes in parathyroid carcinoma. Clin Endocrinol (Oxf) 2003a;59(2):180–189. doi: 10.1046/j.1365-2265.2003.01814.x. [DOI] [PubMed] [Google Scholar]
- Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, Tojo K, Robbins CM, Carpten JD, Farnebo LO, Larsson C, Arnold A. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003b;349(18):1722–1729. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Yalcindag C, Sela-Brown A, Kilav R, Naveh-Many T. Regulation of the parathyroid hormone gene by vitamin D, calcium and phosphate. Kidney Int Suppl. 1999;73:S2–7. doi: 10.1046/j.1523-1755.1999.07310.x. [DOI] [PubMed] [Google Scholar]
- Silverberg SJ, Shane E, Dempster DW, Bilezikian JP. The effects of vitamin D insufficiency in patients with primary hyperparathyroidism. Am J Med. 1999;107(6):561–567. doi: 10.1016/s0002-9343(99)00294-6. [DOI] [PubMed] [Google Scholar]
- Starker LF, Akerstrom T, Long WD, Delgado-Verdugo A, Donovan P, Udelsman R, Lifton RP, Carling T. Frequent germ-line mutations of the MEN1, CASR, and HRPT2/CDC73 genes in young patients with clinically non-familial primary hyperparathyroidism. Horm Cancer. 2012a;3(1-2):44–51. doi: 10.1007/s12672-011-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starker LF, Fonseca A, Akerstrom G, Bjorklund P, Westin G, Carling T. Evidence of a stabilizing mutation of beta-catenin encoded by CTNNB1 exon 3 in a large series of sporadic parathyroid adenomas. Endocrine. 2012b;42(3):612–615. doi: 10.1007/s12020-012-9690-3. [DOI] [PubMed] [Google Scholar]
- Starker LF, Svedlund J, Udelsman R, Dralle H, Akerstrom G, Westin G, Lifton RP, Bjorklund P, Carling T. The DNA methylome of benign and malignant parathyroid tumors. Genes Chromosomes Cancer. 2011;50(9):735–745. doi: 10.1002/gcc.20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, Buyse IM, Fletcher JA, Liu J, Bronson R, Chadwick RB, de la Chapelle A, Zhang X, Speleman F, Huang S. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15(17):2250–2262. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman L, Haglund F, Hashemi J, Obara T, Nordenstrom J, Larsson C, Juhlin CC. Genome-wide and locus specific alterations in CDC73/HRPT2-mutated parathyroid tumors. PLoS One. 2012;7(9):e46325. doi: 10.1371/journal.pone.0046325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund J, Auren M, Sundstrom M, Dralle H, Akerstrom G, Bjorklund P, Westin G. Aberrant WNT/beta-catenin signaling in parathyroid carcinoma. Mol Cancer. 2010;9:294. doi: 10.1186/1476-4598-9-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund J, Koskinen Edblom S, Marquez VE, Akerstrom G, Bjorklund P, Westin G. Hypermethylated in cancer 1 (HIC1), a tumor suppressor gene epigenetically deregulated in hyperparathyroid tumors by histone H3 lysine modification. J Clin Endocrinol Metab. 2012;97(7):E1307–1315. doi: 10.1210/jc.2011-3136. [DOI] [PubMed] [Google Scholar]
- Tahara H, Smith AP, Gas RD, Cryns VL, Arnold A. Genomic localization of novel candidate tumor suppressor gene loci in human parathyroid adenomas. Cancer Res. 1996;56(3):599–605. [PubMed] [Google Scholar]
- Tan MH, Morrison C, Wang P, Yang X, Haven CJ, Zhang C, Zhao P, Tretiakova MS, Korpi-Hyovalti E, Burgess JR, Soo KC, Cheah WK, Cao B, Resau J, Morreau H, Teh BT. Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin Cancer Res. 2004;10(19):6629–6637. doi: 10.1158/1078-0432.CCR-04-0493. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Uchino S, Noguchi S, Nishioka T, Yamasaki H, Hashimoto K, Yoshimoto K. Biallelic inactivation by somatic mutations of the MEN1 gene in sporadic parathyroid tumors. Cancer Lett. 2002;175(2):175–179. doi: 10.1016/s0304-3835(01)00729-7. [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Tokumoto M, Tsuruya K, Fukuda K, Kanai H, Kuroki S, Hirakata H. Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism. Kidney Int. 2002;62(4):1196–1207. doi: 10.1111/j.1523-1755.2002.kid585.x. [DOI] [PubMed] [Google Scholar]
- Tominaga Y, Tsuzuki T, Uchida K, Haba T, Otsuka S, Ichimori T, Yamada K, Numano M, Tanaka Y, Takagi H. Expression of PRAD1/cyclin D1, retinoblastoma gene products, and Ki67 in parathyroid hyperplasia caused by chronic renal failure versus primary adenoma. Kidney Int. 1999;55(4):1375–1383. doi: 10.1046/j.1523-1755.1999.00396.x. [DOI] [PubMed] [Google Scholar]
- Vasef MA, Brynes RK, Sturm M, Bromley C, Robinson RA. Expression of cyclin D1 in parathyroid carcinomas, adenomas, and hyperplasias: a paraffin immunohistochemical study. Mod Pathol. 1999;12(4):412–416. [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, White R. Allelotype of colorectal carcinomas. Science. 1989;244(4901):207–211. doi: 10.1126/science.2565047. [DOI] [PubMed] [Google Scholar]
- Wang P, Bowl MR, Bender S, Peng J, Farber L, Chen J, Ali A, Zhang Z, Alberts AS, Thakker RV, Shilatifard A, Williams BO, Teh BT. Parafibromin, a component of the human PAF complex, regulates growth factors and is required for embryonic development and survival in adult mice. Mol Cell Biol. 2008;28(9):2930–2940. doi: 10.1128/MCB.00654-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin G, Bjorklund P, Akerstrom G. Molecular genetics of parathyroid disease. World J Surg. 2009;33(11):2224–2233. doi: 10.1007/s00268-009-0022-6. [DOI] [PubMed] [Google Scholar]
- Woodard GE, Lin L, Zhang JH, Agarwal SK, Marx SJ, Simonds WF. Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene. 2005;24(7):1272–1276. doi: 10.1038/sj.onc.1208274. [DOI] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]