

Abstract
Efficient correlative imaging of small targets within large fields is a central problem in cell biology. Here, we demonstrate a series of technical advances in focused ion beam scanning electron microscopy (FIB-SEM) to address this issue. We increase the speed, robustness and automation of the process, and achieve consistent z slice thickness of ~3 nm. We introduce “keyframe imaging” to simultaneously image large fields of view and obtain high-resolution 3D images of targeted sub-volumes. We apply these advances to image post-fusion cytoplasmic intermediates of the HIV core. Using fluorescently labeled cell membranes, proteins and HIV cores, we first produce a “target map” of an HIV infected cell by fluorescence microscopy. We then generate a correlated 3D EM volume of the entire cell as well as high-resolution 3D images of individual HIV cores, achieving correlative imaging across a volume scale of 109 in a single automated experimental run.
Current technologies such as X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy are capable of determining the 3D structure of biological specimens ranging in size from small protein complexes to intact viruses at resolutions of ~ 0.2 to 10 nm. Likewise, cells and tissues can be routinely imaged with light microscopy at 3D resolutions as high as ~100 nm. However, there is no satisfactory technology that can efficiently image an entire cell in 3D at both light and electron microscopic resolution, where regions of interest identified by fluorescence microscopy can subsequently be imaged by electron microscopy at resolutions of ~ 10 nm or better. Traditionally, 3D ultrastructure has been obtained using serial section - transmission electron microscopy (TEM), a technique that is labor intensive, prone to artifacts and suffers from low resolution along the axis perpendicular to the cutting plane. Serial block face scanning electron microscopy provides a more automated solution, combining microtomy and imaging using a scanning electron microscope (SEM) (Briggman et al., 2011; Denk and Horstmann, 2004; Shu et al., 2011). Nevertheless, this technology still suffers from the problems of lower resolution along the z-axis axis and artifacts arising from manual sectioning. In focused ion beam scanning electron microscopy (FIB-SEM, also referred to as ion abrasion scanning electron microscopy or IA-SEM) (Drobne et al., 2008; Heymann et al., 2009; Knott et al., 2008), resin-embedded samples are subjected to an iterative process of milling (slicing) with a focused ion beam (typically gallium), followed by imaging by the SEM. This generates a stack of 2D EM images that is computationally converted to a 3D ultrastructural volume of the sample. FIB-SEM has been used recently to describe various structures in biology, including virus-cell interactions, mammalian and non-mammalian cells and tissue architecture at 3D resolutions approaching 10 nm (Drobne, 2013; Heymann et al., 2006; Hildebrand et al., 2009; Murphy et al., 2011).
While FIB-SEM is already a powerful tool in the arsenal for 3D biological imaging using electron microscopy, important limitations remain in the use of this nascent technology. First, the speed of data collection is limited by the time required for sequential SEM image acquisition and ion beam milling: imaging entire cellular volumes at 3D resolutions of 10 nm can take many days for completion. Second, the resolution of imaging is generally anisotropic and while effective pixel sizes as low as 3 nm can be obtained in the xy plane, obtaining the same resolution reproducibly in the z-direction has proven problematic. Finally, a problem that plagues imaging techniques in general is the forced trade-off between resolution and size. Small areas can be viewed at the highest possible resolution afforded by the technique, but larger fields of view must either be imaged at low resolution, or at high resolution by inefficient procedures such as tiling (Schroeder et al., 2011). These limitations in turn result in significant challenges for applications requiring true correlative imaging, where nanoscale objects are located using fluorescence microscopy, and the ultrastructure of the same regions are then determined by 3D electron microscopy.
In order to address these issues, we have used a new “keyframe” imaging strategy that enables point-and-click high resolution 3D ultrastructural imaging of local regions of interest (ROIs), while also obtaining lower resolution ultrastructural information of the entire field of view. The technical advances that enable this are the following: (i) we target high-resolution imaging to only those regions which are of interest within a given field of view, (ii) we speed up the rate of acquisition by milling and imaging simultaneously instead of consecutively, (iii) we correct for drift in 3D, allowing the recording of images that consistently achieve resolutions better than 10 nm in all three planes and (iv) we combine this with light microscopy to identify regions of interest within a given volume. We show that all of these goals can be achieved for imaging both bacterial and mammalian cells using a commercially available microscope and demonstrate that a small nanoscale object such as a 100 nm-sized HIV core can be localized and imaged at 3D resolutions better than 10 nm within the cytoplasm of a 40 μm wide mammalian cell (Supplementary Figure S1).
In Figure 1a, we show a schematic of the keyframe method of imaging. In this example, the field of view encompasses several adherent and non-adherent cells, and the high resolution ROI is the cell-cell contact zone. As we progress with imaging, we obtain two sets of images. One set, named the “keyframe”, includes the entire field of view imaged rapidly at a resolution of ~ 20 nm in the imaging (xy) plane, and ~100 nm in the z direction. Between successive keyframe images, we obtain images of the ROI at much higher pixel sampling, typically ranging from 3 – 10 nm in the x, y, and z directions. This is achieved without changing magnification, thus the optics are kept stable throughout the run. The ROIs can be altered in real time: in an experiment examining cell-cell contacts between dendritic cells and adherent carcinoma cells, a circular ROI was set up based on an initial keyframe (Figure 1b, d); however, subsequent keyframes revealed a new area of interest (Figure 1c). The shape of the high resolution ROI being imaged was then altered during the course of the run to now include the new area, alowing it to be imaged at high resolution in 3D (Figure 1e). To further increase the speed of data collection on the FIB-SEM, we addressed the use of the two beams. In conventional FIB-SEM imaging, resin milling by the FIB and image acquisition by the SEM are performed sequentially. By synchronously imaging a face and milling it, with both beams constantly turned on, we can achieve significant gains in speed, reducing the time required for data acquisition by ~ 2-fold, with little or no loss in image quality (Supplementary Figure S2).
Figure 1. Targeted 3D FIB-SEM by keyframe imaging.
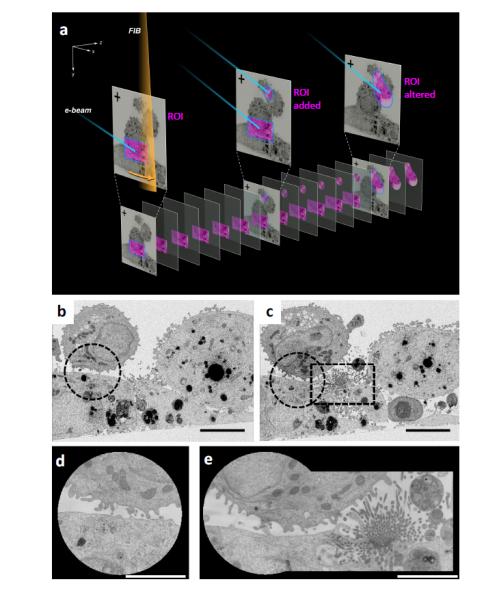
(a) FIB milling (orange beam) and SEM imaging (blue beam) of resin embedded cells generates two image classes. Lower resolution keyframes of the entire field of view (top) are collected at sparse z intervals, and higher resolution images of ROIs (pink) are collected more frequently. The shape and number of ROIs can be altered during a run. (b,c) Keyframes of mammalian cell-cell contacts reveal a new area of interest midway through the experiment, ROI sub-areas indicated with dotted lines. (d) High resolution ROI image of initial synapse, (e) ROI image altered to include new area of interest.
We next extended the technology to achieve isotropic resolution in all three planes. Pixel sizes of ~3 nm are possible in the xy plane, but to achieve this level of resolution in the z-direction, it is necessary to measure and maintain ion beam stability over the entire imaging session. Although the beam can mill in increments as small as 0.1 nm, the stability of ion beam milling can be thwarted by thermal and mechanical fluctuations that cause slow drifts as well as unpredictable movements as large as +/− 200 nm or more in all 3 directions. To measure and correct for this drift, we deposited an approximately 1 μm thick, patterned platinum-carbon double layer on the top surface of the volume. Because the cross sections of the patterns (Supplementary Figure S3) are visible in each successive image, they can be used to dynamically correct for drift in all three axes, and serve as markers for automated focus and stigmation correction. Using this approach we were able to obtain images at a consistent z-spacing of ~3 nm, correcting for changes in drift, focus and stigmation during data collection runs spanning several days. To test this system, we collected a stack of 2D images of the sporulating bacteria B. subtilis. These bacterial forespores are surrounded by a proteinaceous coat, comprising at least 20 different crosslinked polypeptides and organized into several distinct layers, ranging from <20 nm to several hundred nanometers. Shown in Figure 2a is a plot of the slice thickness for an image acquisition run of a large volume of resin-embedded bacteria, where the user-defined pixel sampling was 3×3×3 nm. The average measured thickness of the z slices for this run lasting nearly 24 hours (excluding the earliest period during system stabilization) was 3.25 nm, with a standard deviation of 0.54 nm, demonstrating an exquisite control of the FIB over long periods of time, even with soft biological material. The data are presented in a 3D volume resampled with 5 nm-sized voxels, establishing that the quality of the 3D image is similar in all three viewing planes (Figure 2b). At least 5 coat layers surrounding the spore core are resolved in each plane (Figure 2c-e), and features as small as ~20 nm proximal to the central core (arrowheads) are resolved. Image slices through representative bacteria from the original xy, xz, and yz planes, without resampling and aligning, are shown in Supplementary Figure S4. The fine features of the spore coat layers are visible in all 3 axes. Importantly, the ability to image a given volume in 3D with isotropic pixel sampling means that the image resolution of any given sub-region (in this case, individual bacteria within the pellet) is independent of its orientation within the larger volume.
Figure 2. High resolution in 3D by controlled FIB milling.
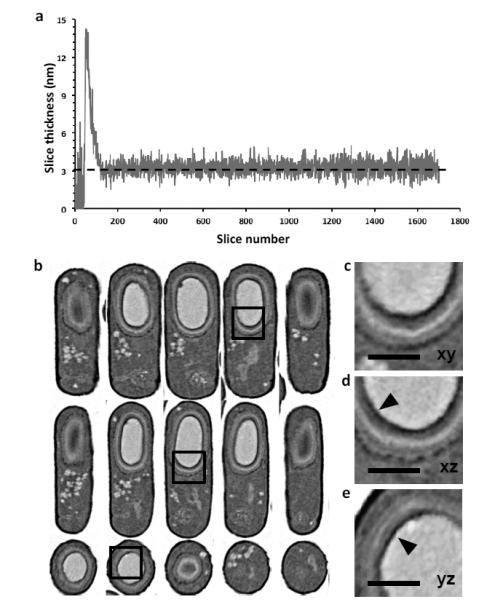
(a) Plot of slice thickness over time during an overnight run of resin-embedded B.subtilis bacteria; the user defined thickness was 3 nm (dotted line). The calculated slice thickness measurements during autofocus and stigmation procedures were excluded. (b) Resampled slices through a representative B.subtilis bacterium, with orthogonal planes oriented to the principal axes of the cylindrical cell (xy: top; xz: middle and yz: bottom). Quality of data is comparable across all planes (c-e) Zoomed in images of the same region of the cell shown as slices in xy, xz and yz planes corresponding to the boxed region of the images in (b), revealing at least five clear concentric spore coat layers around the dark spore core. Small 20-30 nm electron dense features coating the core (arrowheads) can be resolved. Scale bar 400 nm.
Because specific, intracellular protein labeling is difficult to achieve for FIB-SEM, it is useful to correlate fluorescence microscopy with this technique. In previous correlative FIB-SEM work, we reported methods for image registration that enabled the detection of a single 100 – 200 nm-sized spherically shaped fluorescently labeled virus particle in contact with a T cell, where the entire region was imaged at resolutions of ~ 20 nm (Murphy et al., 2011). Given the improvements in imaging described above, we applied our methods for keyframe imaging and isotropic resolution to correlative imaging of the HIV core. The HIV core is a conically shaped entity filled with the packaged genome of the HIV virus, which enters cells during the process of infection. To test whether we could localize and image HIV cores following their entry into a target cell, we infected cells expressing YFP-labeled TRIM5α with mCherry labeled fluorescent HIV. TRIM5α is a critical host-specific retroviral restriction factor (Perron et al., 2004; Stremlau et al., 2004) known to associate with the HIV capsid (Ganser-Pornillos et al., 2011). When proteasome function is inhibited, TRIM5α forms “TRIM bodies” that capture apparently intact HIV cores in the cytoplasm of infected cells (Anderson et al., 2006; Campbell et al., 2007). Locating and visualizing, in a single imaging session, the mCherry labeled 100 nm-sized HIV core, the YFP-labeled TRIM5α, as well as the entire cell, which is many orders of magnitude larger, is a stringent test of the keyframe imaging methodology. Starting from a fluorescence microscopic image that allowed clear visualization of both HIV and TRIM5α (Figure 3a), we carried out 3D imaging of the entire cell, collecting keyframe images at lower resolution (xyz pixels of 12, 12 and 120 nm respectively, Supplementary Movie M1), combined with targeted imaging of the regions surrounding each of the TRIM bodies at higher resolution (xyz pixels of 4, 4 and 12 nm respectively; a generously drawn ROI that captured a TRIM body as well as the leading edge of the cell is shown in Supplementary Movie M2). The resulting data, collected in a single overnight session, contains information on the 3D structure of the entire cell, including visualization of various subcellular organelles (Figure 3b, Supplementary Figure S5), the TRIM bodies identified by fluorescence microscopy (Figure 3c), as well as correlated maps at higher resolution of individual HIV cores associated with TRIM bodies (Figure 3d-g). The resolution of the imaging is high enough in 3D to visualize different shapes of the cores, some which are curved (Figure 3d, Supplementary Figure S6), and some which are conical (Figure 3g), the curvature likely the result of interactions of HIV capsid (CA) multimers with TRIM5α on the TRIM body. The information obtained in this experiment thus includes images of HIV cores (approximately 0.0001 μm3), TRIM bodies (~ 1 μm3) as well as the entire infected cell itself (~ 10,000 μm3), spanning a volume range of 109 in a single dataset.
Figure 3. Identification of cytoplasmic HIV cores.
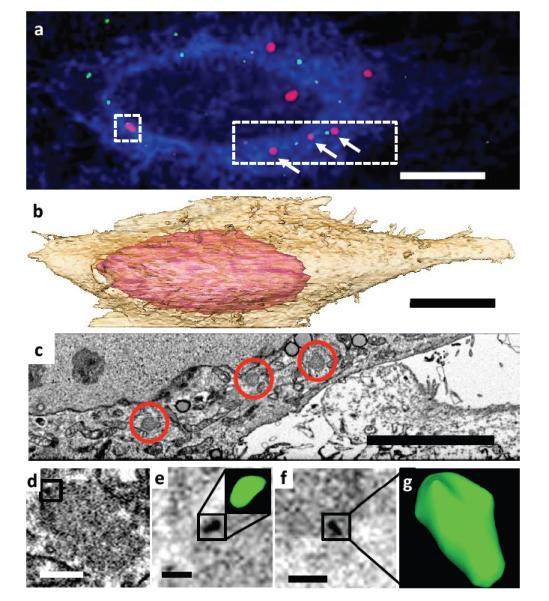
(a) Fluorescence image of HeLa-TRIM5αYFP cells (membrane, blue; TRIM5α, red) infected with HIV-Vpr-mCherry (green). Scale bar 10 μm. (b) Segmented representation of the 3D volume of the entire cell shown in (a), reconstructed from keyframe images. Scale bar 10 μm. (c) Single xy plane from 3D EM volume (collected at 4 × 4 × 12 nm) corresponding to dotted area in (a). Individual TRIM bodies are circled in red, corresponding to TRIM bodies indicated by white arrows in (a). Scale bar 5 μm. (d) Correlated image slice from high resolution “region of interest” image stack containing TRIM5α-associated HIV corresponding to square boxed region in (a), with putative core boxed. (e) Scale bar 400 nm., (e-g) Oblique image planes indicating detection and visualization of individual HIV cores with bent (e, corresponding to boxed area in d) and canonical (g) shapes. Scale bar 200 nm.
In summary, the advances we present here provide a general and broadly applicable set of tools for correlative 3D imaging in cell biology, enabling efficient imaging of selected subcellular regions at resolutions of better than 10 nm in addition to obtaining contextual information from lower resolution imaging of the entire cell using light and electron microscopy. The methods we have developed are implemented in commercially available instruments using procedures that we believe are general enough to be adapted for use with a range of optical and FIB-SEM microscopes supplied by different manufacturers.
Materials and Methods
Virus and cells
Fluorescently labeled VSV-G pseudotyped HIV-1 was produced by polyethylenimine (PEI, Polysciences) transfection of 293T cells with an HIV-1 proviral plasmid (R9Δenv), the vesicular stomatitis virus envelope (VSV-G), and mCherry-Vpr. Virus was purified by ultracentrifugation through a sucrose cushion. Preparations of VSV-G pseudotyped HIV-1 viruses used for these experiments contained 1857.91 ng p24/mL as measured by ELISA. HeLa cells stably expressing YFP-tagged rhTRIM5α(Campbell et al., 2007) and 293T cells were cultured in DMEM or High Modified medium without phenol red (HyClone) containing 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 292 μg/mL -glutamine (Gibco). Co-cultures of plasmacytoid dendritic cells and Huh-7 cells were fixed in situ and processed directly for electron microscopy according to standard protocols. B.subtilis cells were centrifuged at 14,000 r.p.m on a tabletop centrifuge into a pellet, washed, and processed for electron microscopy.
Chamber preparation and infection for correlative microscopy
8-well chamber slides (Lab-Tek) were manually separated into their chambers and slide portions. Gridded chambers were prepared by affixing a 23x23 mm gridded coverslip (Electron Microscopy Sciences) to the top of the chambers using Super Silicone Sealant (3M) and curing at room temperature for three days. Gridded chambers were treated with fibronectin (Sigma-Aldrich) for 20 minutes before seeding 40,000 HeLa-YFP-TRIM5αcells per well. The following day, cells were infected with mCherry-Vpr labeled virus in the presence of 10 μg/mL DEAE-dextran (Sigma-Aldrich) and the proteasome inhibitor MG132 (Sigma-Aldrich), at a final concentration of 1 μg/mL. Infection was carried out at 37°C for 4 hours, with the equivalent of 87ng p24 added per well. After removing virus, membranes were labeled with 0.05 ug/uL CellMask Deep Red (Invitrogen) for 5 minutes at 37°C. Cells were rinsed thrice with PBS and then fixed in 4% EM grade paraformaldehyde (Electron Microscopy Sciences) in PIPES buffer (100 mM PIPES, 2 mM MgCl2, 1 mM EGTA) for 15 minutes at room temperature. After fixation cells were rinsed thrice in PBS and then immediately imaged.
Fluorescence Microscopy
Fluorescent image stacks containing 20 sections in the z plane, 0.2 μm apart, were acquired and deconvolved using softWoRx software (Applied Precision) on a DeltaVision microscope. After deconvolution, images were shifted in the Z-plane to correct for chromatic aberration, according to values obtained after imaging virus on glass on the same microscope with the same parameters. After fluorescent imaging, cells were rinsed in 0.1M sodium cacodylate buffer (Electron Microscopy Sciences) before fixing with 2.5% EM grade glutaraldehyde (Electron Microscopy Sciences) in 0.1M sodium cacodylate buffer for one hour. Cells were rinsed thrice in 0.1M sodium cacodylate buffer, and then dishes were returned to the microscope. The same cells were located and imaged with transmitted light such that both the cells and the alphanumeric grid location were imaged.
Electron Microscopy
Sample preparation
A series of resin and staining combinations were used before the optimal conditions for FIB-SEM were ascertained. Cells imaged by fluorescence were processed for imaging by FIB-SEM in the chambers themselves, as described previously (Murphy et al., 2011), or in the absence of correlation, were processed in Beem capsules. The samples were post-fixed in 1% OsO4 in 0.1M sodium cacodylate for 1 hour at room temperature, washed twice in cacodylate buffer and once with cold 0.1N acetate buffer, then en-bloc stained with 0.5% UA in cold acetate buffer for 1 hour, and washed thrice in acetate buffer. The samples were dehydrated through graded ethyl alcohol, with 2 changes and 10 minute incubations each of 35%, 50%, 70%, 95% and 100%. Pure epoxy resin (Polysciences) was added to the sample, transferred to an oven at 55°C, and replaced with fresh resin after 10 minutes. This cycle was executed thrice, and then samples were allowed to incubate in resin overnight at room temperature. A final exchange with fresh degassed resin was performed the next day and allowed to polymerize for 48 hours at 55°C. The resin was trimmed using a Dremel tool (Bosch), separated from the glass coverslip (for correlative experiments), affixed to a stub and sputter coated with a thin layer of gold.
Imaging parameters
FIB-SEM imaging was performed using a Zeiss NVision 40 microscope, with the SEM operated at 1.5keV landing energy, and backscattered electrons were recorded at an energy-selective back-scattered electron (EsB) detector with a grid voltage of 1200V. The user interface employed was ATLAS 3D from Carl Zeiss, consisting of a dual 16 bit scan generator assembly to simultaneously control both the FIB and SEM beams and dual signal acquisition inputs, as well as the necessary software and firmware to control the system. The imaging parameters were set to optimize for best signal to noise ratios and resolution in images. The EsB detector filtered out all lower energy secondary electrons such as those generated from ion milling; filtering out secondary electrons and recording low energy back-scattered electrons greatly improved image quality. Typically, higher resolution ROI images were collected at 1 μs dwell time, line average 4, and pixel sampling 3 to 6 nm. Efficiency was maximized when the time required to image an area was similar to the time taken to mill the corresponding slice (z slices were usually 5 to 15 nm). Keyframes were collected at lower pixel sampling (usually 12 nm) and at larger intervals (120 nm).
FIB protocols and 3D tracking parameters
To protect the resin volume to be imaged and to correct for stage and/or specimen drift, we executed the following protocol: we deposited a 0.5 μm thick protective platinum layer (700 pA current on the FIB) on top surface, i.e. perpendicular to the image face, of the volume to be milled. Three parallel notches and two notches in the shape of a chevron were then FIB milled into this surface with a finely tuned 80pA beam. These notches were filled in with a layer of carbon (also 0.5 μm thick, deposited at 700 pA), creating a bilayer of differing contrasts when imaged in cross section. FIB milling of this patterned protective pad during an imaging run resulted in the appearance of a specific pattern in cross section (supplementary figure S3); the parallel notches should appear in a constant position in every image, and are used to dynamically correct for any drift in x and y during a run by applying appropriate SEM beam shifts. The notches from the chevrons should appear closer to the center with every subsequent image, displaced from the previous location by a small but constant distance. The difference between their actual and theoretical location (computed from the angle of the chevron and the thickness of the z slice) is used to measure and dynamically correct for drift in z by modifying the FIB mill rate appropriately. The tips of the notches are also used as markers for automated focus and stigmation correction protocols at user defined intervals. The FIB was operated at 700pA during the collection of the dataset.
Correlative imaging
After fluorescence microscopy and fixation, the cells were post-fixed, stained and resin embedded in situ. The alphanumeric pattern of the grid was transferred to the resin and was located by low magnification scanning of the resin face at 3 keV and detection with the secondary (SE2) detector. The outlines of the cells themselves were imaged using higher electron beam energies, typically 15 keV, and cross-referenced with the light microscopic images. Once located, the resin-embedded cells were protected with the patterned layer, as above. An area in front of the cells was milled to form a deep trench using the FIB at high current (usually 13 or 27 nA) and then polished using a lower current in a stepwise manner (usually 1.5 or 3 nA followed by 300 or 700 pA beam current), until the plasma membrane of the cell was reached. The FIB was operated either at 300 or 700 pA for the duration of the imaging run, and the acquisition of the dataset was performed with the FIB and the SEM operated simultaneously, with ROIs drawn or edited by the user with the fluorescence image used as a “target map”. The direction of image acquisition for FIB-SEM datasets is perpendicular to that for fluorescence microscopy.
Image Processing
The stack of images generated during an image acquisition run could be separated into two sets of images – keyframes (lower resolution, sparser sampling, but scanning the entire field of view) and ROI files (higher in-plane resolutions, finer slices, and one or more specific areas of interest within the field of view). These two sets were processed separately, but in a similar manner. The images were first binned in x and y to yield isotropic voxels, inverted, tilted and trimmed as required using the IMOD package (Kremer et al., 1996), and finally aligned by cross-correlation (Murphy et al., 2011). The resulting 3D images were segmented manually using Avizo (Visualization Sciences Group) or Slicer 3D (Fedorov et al., 2012), (www.slicer.org) to highlight the desired volumes of interest. In the case of the bacterial cell dataset, the sub-volumes of individual bacteria excised from the original dataset were resampled with a windowed sinc function onto a bounding box oriented to the principle moments of the segmentation of the cells themselves, at 5nm3 resolution. An unsharp mask was then applied and sparse cross-sections were displayed as required.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Kunio Nagashima for assistance with specimen preparation and Prashant Rao for assistance with image processing. Resin embedded B.subtilis samples, and resin-embedded co-cultures of plasmacytoid dendritic cells and Huh-7 cells were kind gifts from Dr Kumaran Ramamurthi and Dr Francis V Chisari, respectively. This work was supported by funds from the Center for Cancer Research, NCI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson JL, Campbell EM, Wu X, Vandegraaff N, Engelman A, Hope TJ. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J Virol. 2006;80:9754–9760. doi: 10.1128/JVI.01052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471:183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- Campbell EM, Dodding MP, Yap MW, Wu X, Gallois-Montbrun S, Malim MH, Stoye JP, Hope TJ. TRIM5 alpha cytoplasmic bodies are highly dynamic structures. Mol Biol Cell. 2007;18:2102–2111. doi: 10.1091/mbc.E06-12-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Horstmann H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS biology. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobne D. 3D imaging of cells and tissues by focused ion beam/scanning electron microscopy (FIB/SEM) Methods in molecular biology. 2013;950:275–292. doi: 10.1007/978-1-62703-137-0_16. [DOI] [PubMed] [Google Scholar]
- Drobne D, Milani M, Leser V, Tatti F, Zrimec A, Znidarsic N, Kostanjsek R, Strus J. Imaging of intracellular spherical lamellar structures and tissue gross morphology by a focused ion beam/scanning electron microscope (FIB/SEM) Ultramicroscopy. 2008;108:663–670. doi: 10.1016/j.ultramic.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Fedorov A, Beichel R, Kalpathy-Cramer J, Finet J, Fillion-Robin JC, Pujol S, Bauer C, Jennings D, Fennessy F, Sonka M, Buatti J, Aylward S, Miller JV, Pieper S, Kikinis R. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magnetic resonance imaging. 2012;30:1323–1341. doi: 10.1016/j.mri.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci U S A. 2011;108:534–539. doi: 10.1073/pnas.1013426108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JA, Hayles M, Gestmann I, Giannuzzi LA, Lich B, Subramaniam S. Site-specific 3D imaging of cells and tissues with a dual beam microscope. J Struct Biol. 2006;155:63–73. doi: 10.1016/j.jsb.2006.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JA, Shi D, Kim S, Bliss D, Milne JL, Subramaniam S. 3D imaging of mammalian cells with ion-abrasion scanning electron microscopy. J Struct Biol. 2009;166:1–7. doi: 10.1016/j.jsb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand M, Kim S, Shi D, Scott K, Subramaniam S. 3D imaging of diatoms with ion-abrasion scanning electron microscopy. J Struct Biol. 2009;166:316–328. doi: 10.1016/j.jsb.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G, Marchman H, Wall D, Lich B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:2959–2964. doi: 10.1523/JNEUROSCI.3189-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Murphy GE, Narayan K, Lowekamp BC, Hartnell LM, Heymann JA, Fu J, Subramaniam S. Correlative 3D imaging of whole mammalian cells with light and electron microscopy. J Struct Biol. 2011;176:268–278. doi: 10.1016/j.jsb.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron MJ, Stremlau M, Song B, Ulm W, Mulligan RC, Sodroski J. TRIM5alpha mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc Natl Acad Sci U S A. 2004;101:11827–11832. doi: 10.1073/pnas.0403364101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JL, Bakalar M, Pohida TJ, Balaban RS. Rapid overlapping-volume acquisition and reconstruction (ROVAR): automated 3D tiling for high-resolution, large field-of-view optical microscopy. J Microsc. 2011;243:103–110. doi: 10.1111/j.1365-2818.2011.03490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS biology. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.