

Abstract
Disorders of lipid metabolism are strongly associated with cardiovascular disease. Recently, there has been significant focus on how tissues process lipid deposits. Impaired cholesterol efflux has been shown to be critical in mediating lipid deposition in atherosclerosis. The inability of macrophages to effectively efflux cholesterol from tissues initiates inflammation, plaque neovascularization and subsequent rupture. Recent studies suggest that inability to effectively efflux cholesterol from tissues may have global implications far beyond atherosclerosis, extending to the pathophysiology of unrelated diseases. We examine the unifying mechanisms by which impaired cholesterol efflux facilitates tissue-specific inflammation and disease progression in age-related macular degeneration, a blinding eye disease and atherosclerosis, a disease associated with significant cardiovascular morbidity.
Keywords: Macrophage, AMD, Atherosclerosis, Cholesterol efflux, Lipids
Age-related macular degeneration (AMD)lipid deposition and innate immunity
AMD (see glossary) is the leading cause of blindness in individuals over 50 years of age in the industrialized world [1]. Accumulation of lipid rich deposits called drusen underneath the retina is a hallmark of AMD and disease progression is often initially characterized by an increase in drusen number and size. Advanced AMD is characterized by photoreceptor loss associated with either atrophic changes in the macula or development of new blood vessels underneath the retina called choroidal neovascularization (CNV) (Figure 1) [1]. Majority of blindness in AMD is secondary to CNV. Although AMD is a multifactorial disease and aging is the major risk factor, inflammation is central to the pathological process [2, 3]. Numerous genetic analyses including several genome wide association studies (GWAS) have strongly linked innate immunity and several complement pathway components to susceptibility to both the development and severity of AMD [4]. There is emerging evidence showing progressive accumulation of macrophages underneath the retina of AMD patients that correlates with the clinical stage of the disease [5, 6], supporting an important role for macrophages in disease pathogenesis in AMD. GWAS studies have also linked lipid metabolism to the pathogenesis of AMD [7, 8]. Indeed, accumulation of intracellular cholesterol in macrophages underneath the retina might be critical for disease pathogenesis, as decreased expression of macrophage cholesterol transporter proteins that result in impaired cholesterol efflux also promote CNV.
Figure 1. Clinical features of AMD.
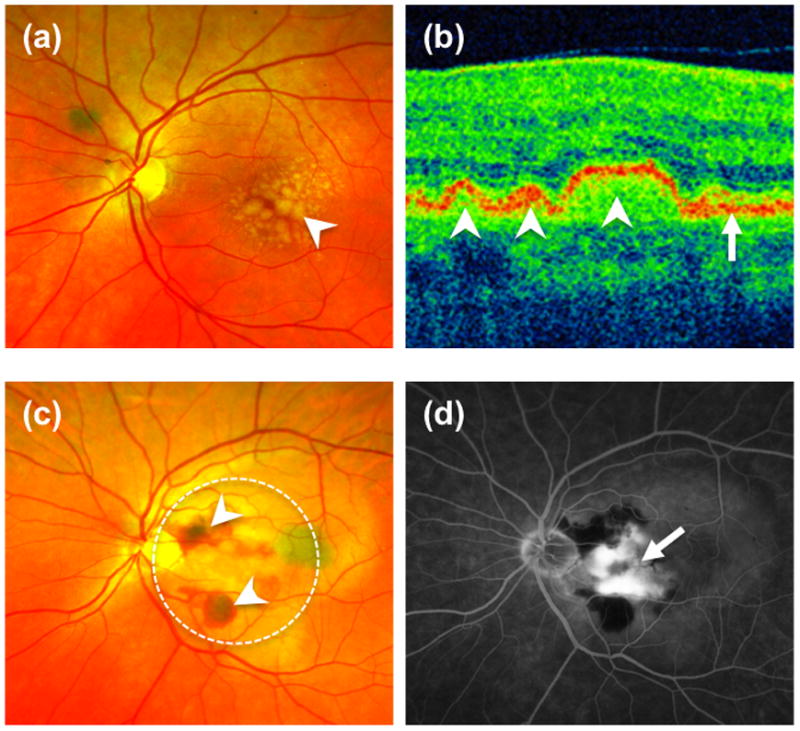
(a) Fundus photograph of the retina of a patient with dry AMD demonstrates large lipid laden drusen (arrowhead) underneath the retina. (b) Corresponding optical coherent tomography (OCT) of the central retina (macula) confirmed the presence of multiple drusen (arrowheads) underneath the retinal pigment epithelium layer (RPE-arrow). (c) Fundus photograph of a patient with the wet form of AMD illustrate subretinal hemorrhage and fluid (arrowhead) secondary to choroidal neovascularization (CNV, dotted circle). A fluorescein angiogram demonstrates leakage of dye from the CNV (arrow).
Here we critically assess new findings that mechanistically connect impaired cholesterol efflux and tissue-specific inflammation, both hallmarks of atherosclerosis, to age-related macular degeneration. We suggest that pharmacotherapeutic, genetic or RNA interference approaches to modify cholesterol metabolic pathways warrant future investigation as potential beneficial therapies for both AMD and atherosclerosis.
Macrophage-mediated inflammation: the mechanistic link
Extensive characterization of existing mouse models that exhibit some of the clinical features of AMD has revealed that defective chemotaxis of macrophages in the eye resulted in accelerated accumulation of drusen-like deposits under the retina [9, 10]. Furthermore, in support of the central role of macrophages in the disease process, studies using murine models of injury-induced CNV that accurately demonstrate pathophysiologic characteristics of neovascular AMD seen in human patients, have clearly established the determinant role of macrophages in the progression of pathological angiogenesis [11–13]. However, their precise contribution to the AMD phenotype was initially unclear; in early studies, there was conflicting evidence regarding whether macrophages were involved in promoting or repressing CNV in murine models of AMD. It is now apparent that these results could be attributed to macrophage heterogeneity and the status of their activation and polarization. Indeed, in response to microenvironmental signals, macrophages have been shown to exhibit classic (M1) or alternative (M2) activation characterized by differential cytokine production, receptor expression, and effector function [14, 15]. A variety of specific markers have been identified for the different populations of activated macrophages. Pro-inflammatory M1 macrophages express high levels of TNF-α, IL-12, iNOS, IL-6, IL-1β, PTGS2, CCL2, and MMP9. Conversely, pro-angiogenic M2 macrophages mediate wound healing and are characterized by low M1 signature markers but increased expression of IL-10, CD163, and TGF-β. Previous studies in murine models of AMD demonstrated that the switch of macrophage polarization from M1 to M2, also seen during normal aging, is a key event in CNV progression [11, 16]. In contrast, macrophages recruited under the retina at the initial stage of disease, exhibit a pro-inflammatory M1 phenotype [17]. An analysis of the histopathological features of human AMD with subsequent phenotypic profiling of infiltrated macrophages also revealed that in early stages of AMD, macrophages are polarized to M1 whereas in later stages they are alternatively M2 activated [5, 6] (Figure 2).
Figure 2. The central role of macrophage-mediated immunity in disease pathogenesis of AMD.

Under baseline conditions, non-polarized macrophages (M0) traffic through the choriocapillaris underneath the retina sampling the tissue microenvironment. Aging and complex genetic factors impair macrophage function including reverse cholesterol transport (RCT) and lead to the deposition of lipid rich drusen underneath the retina and RPE in non-neovascular (dry) AMD. In dry AMD, dysfunctional classically activated macrophages (M1) can also induce inflammation driven cell death that leads to advanced stages of dry AMD characterized by loss of RPE cells, called geographic atrophy (GA) and subsequent loss of rod and cone photoreceptor neurons (PR). In the more aggressive form of disease, impaired macrophage RCT in the drusen rich sub-retinal micromilieu polarizes these cells to an alternatively activated (M2) phenotype. Alternatively activated macrophages are disease promoting and pro-angiogenic and facilitate the development of CNV. The sine qua non of wet AMD is the development of CNV, characterized by the growth of new blood vessels from the choroid into the sub-retinal space. These neovascular complexes are leaky and lead to exudation and hemorrhage with disruption of the retinal architecture, interference with the central visual axis, loss of PR and ultimately, irreversible vision loss.
The efficacy of macrophages to efflux and remove intracellular cholesterol engulfed from lipid rich drusen underneath the retina might also be critical for their ability to regulate pathological angiogenesis. Along these lines, an age-associated decrease in the expression of the cholesterol transporter ABCA1 resulted in impaired cholesterol efflux in macrophages and promoted CNV [18]. Moreover, cholesterol buildup in senescent macrophages polarizes them to a pro-angiogenic and disease promoting phenotype [18]. Dysregulation of macrophage cholesterol metabolism play a determinant role in AMD pathogenesis.
Atherosclerosis
Atherosclerosis is the underlying pathological process in ischemic cardiovascular disease, the leading cause of death in many parts of the world [19]. Progressive alteration of the laminar flow and vascular remodeling at specific sites creates a nidus for cellular and lipid accumulation in the sub-endothelial space that initiates atherosclerotic lesions. The development and progression of atherosclerotic plaques can result in complications such as neovascularization and plaque rupture associated with higher risk of occlusive luminal thrombi [20, 21]. It is now well accepted that inflammatory processes are major pathogenic factors underlying the disease [22]. Disturbance of the vessel wall at sites of predilection, infiltration of modified low-density lipoproteins (LDL) and subsequent activation of endothelial cells is the first crucial pathogenic event that modulates the expression of several adhesion molecules and secretion of chemoattractants like CCL2, by altered endothelial cells [20].
Macrophage polarization and atherosclerotic lesions
In early stages of atherosclerosis, most of the recruited monocytes at lesion sites differentiate into macrophages. This maturation process is critical for the clearance of atherogenic lipoprotein particles by macrophages and further resolution of lesions [23]. Recent identification of proliferative lesional macrophages provided new insights into the interplay between monocytes and macrophage for atherosclerosis progression and highlighted a novel sequential event that led to local macrophage buildup. This study demonstrated that the accumulation of macrophages observed in atherosclerotic lesions was due to macrophage proliferation rather than a continuous recruitment of circulating monocytes that differentiated into macrophages [24]. In addition, this process is strongly dependent on the local tissue microenvironment as lesional macrophage proliferation is reduced during early atherosclerosis [24]. In the context of massive lipoprotein deposition and cytokine expression, macrophages are activated and polarized to either the pro-inflammatory M1 or atheroprotective M2 phenotype that potentially correlates with their contribution to the evolution of the disease. However, it is still unclear whether the heterogeneity and polarization of macrophages in the context of atherosclerotic lesions is causative or simply reflects disease progression [25, 26]. There is substantial evidence showing the continuous presence of both M1 and M2 macrophages during atherosclerotic plaque development and progression. Recent mouse and human studies revealed that the functional implication of macrophages of different phenotype is directly correlated with their spatial distribution and microenvironment [27, 28]. Although macrophages of both phenotypes accumulate similarly in the fibrous cap region, M1 macrophages are predominantly located in the plaque rupture-prone site whereas M2 macrophages are in vascular adventitia underlying advanced plaques. These findings suggest that excessive production of tissue proteases and inflammatory cytokines by M1 macrophages lead to weakening of plaque shoulders that may become susceptible to rupture. In contrast, profibrotic M2 macrophages located in more stable areas of the plaque promote adventitia neovascularization and possibly increasing monocyte infiltration [28]. These specific distributions within distinct morphological locations in atherosclerotic lesion further implicate M1 and M2 macrophages in atherosclerosis complications [29].
Drusen and atherosclerotic plaques: lipid-laden time bombs
In both AMD and atherosclerosis, inflammation associated with accumulation of lipid-rich deposits is a crucial pathogenic event in the development and progression of these diseases. In multiple studies using different approaches, analysis of the molecular composition of drusen and atherosclerotic plaques revealed similar constituents including lipids, lipoproteins and components of the complement pathway (Figure 3) [30–34]. However, the sources of deposited components such as apoB lipoproteins differ between drusen and atherosclerotic plaques. Although some proteins and lipids originating in the plasma are found in drusen other constituents have an exclusive intraocular origin. Indeed, analysis of retinal pigment epithelium expression profile combined with protein trafficking studies revealed that most of drusen components are synthetized locally and suggest an active role for local cells in drusen biogenesis [31, 35].
Figure 3. Drusen composition and similarities with atherosclerotic plaques.
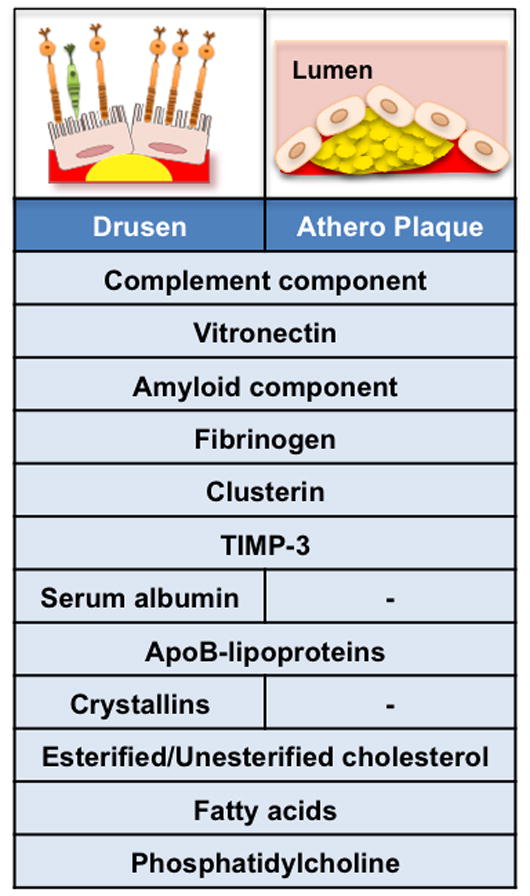
Analysis of the composition of lipid-rich deposits (yellow) in drusen underneath the retina (left upper panel), or within atherosclerotic plaques (right upper panel) demonstrates similar constituents. Many complement proteins have been identified in drusen and atherosclerotic plaque suggesting an activation of the complement pathway within the deposits. Vitronectin, amyloid component, fibrinogen, clusterin and tissue inhibitor of metalloproteinase 3 (TIMP-3) are molecular constituents of both drusen and atherosclerotic plaques. In contrast, serum albumin and crystallins proteins are exclusive drusen components. ApoB lipoproteins are major constituents of drusen and atherosclerotic plaques. In addition to the plasma, the apoB deposited in drusen also have an intraocular source. Lipid profiling of drusen and atherosclerotic plaques revealed that they both contain esterified and unesterified cholesterol, fatty acids and phosphatidylcholine.
HDL biogenesis and cellular cholesterol efflux
HDL genetics and risk modulation
In many complex diseases, GWAS have been used to identify variants that correlate with the disease and potential complications. For instance, several polymorphisms in complement pathway genes could explain about half of the excess risk of AMD to siblings of affected individuals [36]. Association studies can also unravel new pathways essential to the understanding of the etiology of a disease [37].
Recent GWAS revealed new genetic variants that are strongly associated with increased risk for AMD. Analysis of the underlying biological pathways associated with the genes identified involved high-density lipoprotein (HDL) cholesterol metabolism [7, 8]. Polymorphisms in enzymes critical for the synthesis and degradation of HDL, such as hepatic lipase (LIPC) and cholesterylester transfer protein (CETP) significantly contributed to increased risk for advanced AMD. Previous studies have also shown the contribution of these AMD associated polymorphisms at LIPC and CETP loci to plasma HDL-cholesterol concentration and risk of coronary artery disease [38]. Modulation of LIPC activity or expression has a direct effect on LDL-cholesterol and HDL-cholesterol plasma levels [39]. CETP promotes the transfer of cholesteryl ester from HDL to LDL particles. Human and animal studies consistently demonstrated that overexpression of CETP elevates LDL-cholesterol concentration [40]. On the other hand, deficiency in CETP increases plasma HDL-cholesterol, a significant observation because plasma HDL-cholesterol level is inversely related to atherosclerosis [41, 42], owing to its ability to promote reverse cholesterol transport, endothelial cell repair and its anti-inflammatory and antithrombotic properties. Thus, HDL-cholesterol levels are considered highly predictive for cardiovascular events and attempts that increased HDL-cholesterol level in humans and animal models of atherosclerosis resulted in significant reduction of cardiovascular risk [43]. Findings in recent studies suggested that combined changes in LIPC and CETP enzyme activity or expression modulated HDL-cholesterol levels that further influenced the risk of carotid atherosclerosis [44]. Currently, HDL-raising strategies are being explored for their therapeutic potential [41, 42].
Despite numerous genetic studies showing a risk association of AMD with genes involved in lipoproteins biology, the role of HDL in AMD pathogenesis remains unclear. Unlike atherosclerosis, association of these lipid pathway genes with AMD seems independent of plasma HDL-cholesterol levels [7, 8]. Furthermore, conflicting reports exist concerning the impact of elevated HDL-cholesterol in AMD development and progression [45–49]. Some recent studies showed that elevated serum HDL-cholesterol could be associated with lower risk of advanced AMD characterized by geographic atrophy or neovascularization, and therefore be an indicator of AMD complications [47]. Together, these findings demonstrate that although serum lipids are important in AMD pathogenesis, the exact mechanism by which they modulate the disease process is complex. Furthermore, the discrepancy in the beneficial effect of elevated serum HDL-cholesterol level among various studies supports the distinct possibility of an additional layer of local regulation of lipid metabolism that impacts AMD development and progression.
Cellular Cholesterol efflux
The promotion of reverse cholesterol transport by HDL, allows for the transport of cholesterol from extrahepatic cells and tissues to the liver for removal from the body. Macrophages and in particular foam macrophages, play a determinant role in transferring excess cholesterol to serum lipoproteins [50, 51]. Both HDL and its major structural protein component ApoA1 have been shown to stimulate macrophage cholesterol efflux and reduce foam cell formation. In macrophages, cholesterol efflux is mediated by the ATP binding cassette transporters A1 (ABCA1) and G1 (ABCG1). A combined deletion of both transporters resulted in strong reduction of cellular cholesterol efflux from macrophage to ApoA1 and HDL, decreased reverse cholesterol transport and lipid accumulation in tissues [52–55]. This impairment in macrophage cholesterol transport induced chemokine and inflammation gene expression and promoted atherosclerosis. A similar phenotype was also observed in selective ABCA1 deficiency but not with ABCG1 deficiency [52, 54].
Interestingly, several studies have shown that variations at the ABCA1 locus were associated with increased risk of AMD, although they did not achieve genome-wide significance [7, 8]. However, multivariate analysis revealed that ABCA1 alleles were related to intermediate AMD characterized by lipid rich drusen as well as advanced AMD [56]. Recent studies showed that deletion of CYP27A1, a ubiquitous enzyme that catalyzes the hydroxylation of cholesterol and other sterols at position C27, and subsequent dysregulation of cholesterol metabolism, resulted in retinal lesions [57]. Deficiency in CYP27A1 induces cholesterol deposition underneath the retina and is associated with pathological neovascularization, a hallmark of advanced AMD [57]. Aging is the main risk factor in AMD and previous studies have consistently demonstrated that aged macrophages promote pathological neovascularization [16]. Comprehensive analysis of macrophage cholesterol metabolism showed that senescence impaired the ability of macrophages to effectively efflux cholesterol. Macrophages isolated from mice over 18 months of age, exhibited reduced ABCA1 and ABCG1 expression, when compared to macrophages from young, less than 3 months of age, mice [18]. Reduced expression of these transporters in old mice translated into accumulation of cholesterol and oxysterols that in turn promoted CNV. Interestingly, gene deletion of ABCA1, but not ABCG1 in young macrophages accelerated their aging process and transformed them into disease promoting macrophages [18]. These finding suggested that single targeted gene inactivation of either of these transporters has different impact on AMD complications.
The dissimilar functional outcomes of ABCA1 deficiency or ABCG1 deficiency have also been reported in mouse models of atherosclerosis. LDL receptor (LDLr) deficient mice transplanted with bone marrow of Abca1−/− mice exhibited an increase of atherosclerotic lesions whereas moderate or no changes were found in recipients of Abcg1−/− marrow [52, 58]. In contrast, other studies reported atheroprotection with ABCG1 deficiency [59, 60]. The unexpected protective effect of ABCG1 deletion could be due to the concomitant overexpression of ABCA1 and increased apoptosis of macrophages lacking ABCG1. Furthermore, bone marrow lacking both ABCA1 and ABCG1 induced a strong increase of atherosclerotic lesions when transplanted to LDLr deficient mice [61]. Interestingly, analysis of ABCA1/G1 expression in peripheral blood mononuclear cells (PBMC) of patients diagnosed with AMD revealed that only ABCA1 gene expression and protein levels were decreased, suggesting a predominant role of ABCA1 in AMD pathogenesis [18].
Regulation of cholesterol efflux mediated by ABCA1/G1
LXR pathway
The ability of ABCA1 and ABCG1 to efflux cholesterol from macrophages to ApoA1 and functional HDL particles combined with subsequent anti-inflammatory properties emphasizes the therapeutic potential of cellular cholesterol modulation by targeting these transporters. In presence of excess cholesterol loading, macrophages can coordinately regulate genes involved in cholesterol metabolism by activating the Liver X Receptor (LXR) pathway [62]. The nuclear hormone receptor LXR is a crucial regulator of lipid and cholesterol metabolism, and LXR activity has been linked to the pathogenesis of atherosclerosis. The two LXR isoforms LXRα and LXRβ, are activated by oxysterols to induce the expression of several genes including Abca1 and Abcg1 [63–65]. A variety of synthetic ligands have been identified for these nuclear receptors and their ability to increase cellular cholesterol efflux and reverse cholesterol transport has been analyzed in multiple studies. It is now well established that treatment with LXR agonists significantly decreases atherosclerotic lesion development and progression in different mouse models of atherosclerosis [66]. Interestingly, treatment of old mice with synthetic LXR agonists also resulted in a strong reduction of injury-induced CNV suggesting that increasing cellular cholesterol efflux and subsequent reverse cholesterol transport could be a valuable strategy in preventing AMD complications [18]. Although targeting LXR signaling pathways using synthetic ligands is attractive for the potential resolution of atherosclerosis and AMD complications, systemic treatment with LXR agonists is also associated with many adverse effects including induction of lipogenesis, hypertriglyceridemia and hepatic steatosis [67, 68]. The overall effect on lipid metabolism is driven by the induction of the LXR target genes sterol regulatory element binding protein 1 (SREBP-1), and carbohydrate response-element binding protein (ChREBP) [67, 69]. However, atheroprotective LXR agonists with a novel profile have now been identified, with negligible effects on inducing hepatic and plasma triglyceride levels [70, 71]. In a mouse model of neovascular AMD, local administration was sufficient to circumvent the undesirable effects of systemic treatment with an LXR agonist. Indeed, treatment of old mice with a synthetic LXR agonist in eyedrop formulation restored their tissue specific cellular cholesterol efflux capacities and strongly reduced CNV [18].
Silencing miR-33
The above reports highlight the need to develop approaches to selectively target cellular pathways without affecting overall lipid metabolism. Therefore, specifically targeting ABCA1/G1 or cellular cholesterol efflux pathways may represent a beneficial strategy for the prevention or treatment of atherosclerosis and AMD. It is now well established that microRNAs (miRs) are key players in the regulation of a variety of signaling pathways by modulating the levels of their target mRNAs. Recent studies have identified miR-33a/b, located in intronic regions of SREBP-2 and 1 respectively. Both miRs regulate the expression of genes involved in cellular cholesterol metabolism including ABCA1 and ABCG1 [72–76]. MiR-33a is highly conserved among animal species while miR-33b is absent in rodents, and both miRs are co-transcribed with their host genes [72–76]. Several studies have demonstrated that treatment with anti-miR-33 and inhibition of miR-33 resulted in increased expression of Abca1, increased plasma HDL-cholesterol levels and promoted reverse cholesterol transport, in mice [72, 73, 76]. Moreover, similar results were noted in miR-33 knockout mice [75]. These preliminary results confirmed the therapeutic potential of miR-33 silencing for enhancing cellular cholesterol efflux, reverse cholesterol transport and HDL biogenesis. Using different mouse models of atherosclerosis, recent studies showed that either genetic miR-33 deficiency or in vivo antagonism of miR-33 induced a significant decrease in the development and severity of atherosclerotic lesions [77–79]. By contrast, in another study, long-term silencing of miR-33 had no effect on the development and progression of atherosclerosis suggesting that prolonged inhibition of miR-33 can impact the efficacy of this therapeutic approach [80]. As mentioned above, the expression of ABCA1/G1 in macrophages isolated from old mice is strongly reduced. Further analysis of miR-33 biology showed that aging significantly increased the expression of miR-33, possibly explaining the repression of ABCA1. In vivo antagonism of miR-33 in old mice restored the expression level of ABCA1 in macrophages and several tissues including the eye [18]. As expected, silencing of miR-33 improved the ability of macrophages to inhibit pathological vessel growth as anti-miR-33 treated mice developed less CNV when compared to control treated mice [18]. These studies demonstrated that an miR-33 silencing approach could be effective and therapeutically relevant for the treatment of atherosclerosis and AMD. Future investigations will determine the impact of anti miR-33 treatment duration in its efficacy/safety and provide more insights in the effect of long-term silencing on the overall lipid metabolism.
Concluding remarks and future perspectives
Upon cursory examination AMD and atherosclerosis seem to be complex, unrelated diseases. AMD affects the eye and causes blindness in people over 50 years of age while atherosclerosis affects large vessels and is the leading cause of cardiovascular morbidity and mortality. A more careful evaluation of disease pathogenesis in these conditions provides some very interesting and valuable insights (Figure 3). Both disorders are characterized by extracellular lipid deposition, impaired cholesterol efflux and reverse cholesterol transport, macrophages unable to effectively handle intracellular lipid transport and efflux, ultimately leading to tissue inflammation and pathologic neovascularization. Although there are some subtle differences in macrophage polarization and activation depending on tissue context, the phenotypic and genotypic similarities are quite uncanny. In vitro and in vivo data clearly suggest that lipid pathway modification by pharmacotherapeutic, genetic or RNA interference approaches may influence pathogenic inflammation and angiogenesis in both AMD and atherosclerosis, and merit further translational investigation.
There is a great deal that can also be learned from the minefields of lipid modification therapies designed for atherosclerosis and cardiovascular disease. Many agents such as CETP inhibitors have not lived up to the initial promise mostly due to toxicity secondary to off-target effects associated with systemic delivery, and on occasion, an incomplete understanding of disease pathogenesis. As the important role of lipids within drusen in AMD pathogenesis is just beginning to be elucidated, similarities in how dysfunction in lipid metabolism plays a key role in AMD progression and atherosclerosis start to emerge. Thus, existing agents that have been shown to improve reverse cholesterol transport and cholesterol efflux may potentially be re-formulated for local use in the treatment of AMD. Since the eye is suitable for local drug delivery, some of the undesirable adverse effects of systemic delivery of these therapeutic agents can be circumvented while maintaining therapeutic efficacy. The eye may also provide a novel opportunity to investigate additional compounds that affect lipid deposition, cholesterol efflux and inflammation in diseases such as AMD, potentially opening novel therapeutic vistas in atherosclerosis therapy.
Highlights.
Drusen and atherosclerotic plaques share similar components
Altered macrophage cholesterol efflux is a determinant pathogenic event in AMD and atherosclerosis
Targeting macrophage mediated inflammation is a relevant therapeutic approach
Emerging Questions and Trends.
Questions
What is the molecular basis for altered cholesterol sensing capacities in aged macrophages?
Is the age-associated accumulation of macrophages in the subretinal space driven by defective chemotaxis or local macrophage proliferation?
What are the precise implications of M1/M2 macrophages in AMD development and progression?
Does systemic dysregulation of cholesterol metabolism result in drusen biogenesis?
What is the effect of cholesterol lowering drugs on drusen development?
What are the molecular mechanisms by which aging regulates lipid metabolism in the eye
Trends
Regulation of ocular cholesterol metabolism
Modulation of age-associated cholesterol buildup in macrophage and subsequent inflammation
Determine the contribution of plasma HDL/LDL cholesterol in AMD pathogenesis
Acknowledgments
This work was supported by NIH grant K08EY016139; NIH grant R01EY019287; NIH Vision Core Grant P30EY02687, U.S. Civilian Research and Development Foundation; the Carl Marshall Reeves and Mildred Almen Reeves Foundation Inc. Award; the Research to Prevent Blindness Inc. Career Development Award; the International Retina Research Foundation; the American Health Assistance Foundation; the Lacy Foundation Research Award and the Thome Foundation. We thank Nicole Zapata for helping with illustrations and Dr. Clay Semenkovich for his insightful comments.
GLOSSARY
- Age-related macular degeneration (AMD)
a medical condition and major cause of blindness and visual impairment in older adults. It results in a loss of vision due to damage to the retina. AMD may occur in “dry” and “wet” forms. Dry AMD is characterized by accumulation of drusen between the retina and the choroid. The more sever wet form, is characterized by abnormal blood vessel growth (choroidal neovascularization)
- ATP binding cassette transporters
are members of a superfamily of transmembrane proteins that utilize ATP hydrolysis to transport a wide variety of substrates such as lipids, sterols, and drugs, across membranes. ABCA1 and ABCG1 are involved in macrophage cholesterol and phospholipids transport, and may regulate cellular lipid homeostasis in other cell types as well
- Cholesterylester transfer protein (CETP)
is a plasma protein that helps transport of cholesteryl esters and triglycerides between lipoprotein molecules
- Choroidal neovascularization (CNV)
refers to the creation of new blood vessels in the choroid layer of the eye often seen in AMD
- Drusen
are tiny yellow or white deposits made up of lipids in a layer of the retina called Bruchs membrane. They are considered an early sign of age-related macular degeneration
- Hepatic lipase (LIPC)
LIPC is a lipolytic enzyme that promotes the hydrolysis of triglycerides and phospholipids in lipoproteins. This enzyme has a dual function since its binding to lipoproteins also facilitates their uptake and metabolism through cell surface receptors
- Liver X Receptor (LXR)
is a member of the nuclear receptor superfamily of transcription factor that regulates cholesterol, fatty acid, and glucose homeostasis. LXR exists in two isoforms, LXRα and LXRβ, both of which to and are activated by endogenous oxysterol ligands
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedman DS, et al. Prevalence of age-related macular degeneration in the United States. Archives of ophthalmology. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 2.Hageman GS, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Progress in retinal and eye research. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 3.Whitcup SM, et al. The role of the immune response in age-related macular degeneration. International journal of inflammation. 2013;2013:348092. doi: 10.1155/2013/348092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zipfel PF, et al. The role of complement in AMD. Advances in experimental medicine and biology. 2010;703:9–24. doi: 10.1007/978-1-4419-5635-4_2. [DOI] [PubMed] [Google Scholar]
- 5.Cherepanoff S, et al. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. The British journal of ophthalmology. 2010;94:918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- 6.Cao X, et al. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathology international. 2011;61:528–535. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen W, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7401–7406. doi: 10.1073/pnas.0912702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neale BM, et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC) Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7395–7400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambati J, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nature medicine. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 10.Luhmann UF, et al. The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Investigative ophthalmology & visual science. 2009;50:5934–5943. doi: 10.1167/iovs.09-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apte RS, et al. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS medicine. 2006;3:e310. doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espinosa-Heidmann DG, et al. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Investigative ophthalmology & visual science. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- 13.Sakurai E, et al. Macrophage depletion inhibits experimental choroidal neovascularization. Investigative ophthalmology & visual science. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- 14.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends in immunology. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Mosser DM. The many faces of macrophage activation. Journal of leukocyte biology. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 16.Kelly J, et al. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. The Journal of clinical investigation. 2007;117:3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruz-Guilloty F, et al. Infiltration of proinflammatory m1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. International journal of inflammation. 2013;2013:503725. doi: 10.1155/2013/503725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sene A, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell metabolism. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahlof B. Cardiovascular disease risk factors: epidemiology and risk assessment. The American journal of cardiology. 2010;105:3A–9A. doi: 10.1016/j.amjcard.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 21.Asakura T, Karino T. Flow patterns and spatial distribution of atherosclerotic lesions in human coronary arteries. Circulation research. 1990;66:1045–1066. doi: 10.1161/01.res.66.4.1045. [DOI] [PubMed] [Google Scholar]
- 22.Mayerl C, et al. Atherosclerosis research from past to present--on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Archiv: an international journal of pathology. 2006;449:96–103. doi: 10.1007/s00428-006-0176-7. [DOI] [PubMed] [Google Scholar]
- 23.Qiao JH, et al. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. The American journal of pathology. 1997;150:1687–1699. [PMC free article] [PubMed] [Google Scholar]
- 24.Robbins CS, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nature medicine. 2013 doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, et al. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- 26.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khallou-Laschet J, et al. Macrophage plasticity in experimental atherosclerosis. PloS one. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoger JL, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson LV, et al. A potential role for immune complex pathogenesis in drusen formation. Experimental eye research. 2000;70:441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- 31.Mullins RF, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2000;14:835–846. [PubMed] [Google Scholar]
- 32.Crabb JW, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curcio CA, et al. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Progress in retinal and eye research. 2009;28:393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kramsch DM, et al. The protein and lipid composition of arterial elastin and its relationship to lipid accumulation in the atherosclerotic plaque. The Journal of clinical investigation. 1971;50:1666–1677. doi: 10.1172/JCI106656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curcio CA, et al. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. Journal of lipid research. 2010;51:451–467. doi: 10.1194/jlr.R002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maller J, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nature genetics. 2006;38:1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 37.Jakobsdottir J, et al. Interpretation of genetic association studies: markers with replicated highly significant odds ratios may be poor classifiers. PLoS genetics. 2009;5:e1000337. doi: 10.1371/journal.pgen.1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature genetics. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santamarina-Fojo S, et al. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1750–1754. doi: 10.1161/01.ATV.0000140818.00570.2d. [DOI] [PubMed] [Google Scholar]
- 40.Barter PJ, et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 41.Gordon DJ, Rifkind BM. High-density lipoprotein--the clinical implications of recent studies. The New England journal of medicine. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 42.Barter P, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. The New England journal of medicine. 2007;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- 43.Barter P. HDL-C: role as a risk modifier. Atherosclerosis Supplements. 2011;12:267–270. doi: 10.1016/S1567-5688(11)70885-6. [DOI] [PubMed] [Google Scholar]
- 44.Soyal SM, et al. Cholesteryl ester transfer protein and hepatic lipase gene polymorphisms: effects on hepatic mRNA levels, plasma lipids and carotid atherosclerosis. Atherosclerosis. 2011;216:374–380. doi: 10.1016/j.atherosclerosis.2011.01.052. [DOI] [PubMed] [Google Scholar]
- 45.Fauser S, et al. Evaluation of serum lipid concentrations and genetic variants at high-density lipoprotein metabolism loci and TIMP3 in age-related macular degeneration. Investigative ophthalmology & visual science. 2011;52:5525–5528. doi: 10.1167/iovs.10-6827. [DOI] [PubMed] [Google Scholar]
- 46.Nowak M, et al. Changes in lipid metabolism in women with age-related macular degeneration. Clinical and experimental medicine. 2005;4:183–187. doi: 10.1007/s10238-004-0054-z. [DOI] [PubMed] [Google Scholar]
- 47.Reynolds R, et al. Serum lipid biomarkers and hepatic lipase gene associations with age-related macular degeneration. Ophthalmology. 2010;117:1989–1995. doi: 10.1016/j.ophtha.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abalain JH, et al. Is age-related macular degeneration associated with serum lipoprotein and lipoparticle levels? Clinica chimica acta; international journal of clinical chemistry. 2002;326:97–104. doi: 10.1016/s0009-8981(02)00288-7. [DOI] [PubMed] [Google Scholar]
- 49.van Leeuwen RKC, Vingerling JR, Hofman A, van Duijn CM, Stricker BH, de Jong PT. Cholesterol and age-related macular degeneration: is there a link? American Journal of Ophthalmology. 2004;137:750–752. doi: 10.1016/j.ajo.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 50.Tall AR, et al. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell metabolism. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nature medicine. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 52.Yvan-Charvet L, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. The Journal of clinical investigation. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Out R, et al. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:258–264. doi: 10.1161/ATVBAHA.107.156935. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. The Journal of clinical investigation. 2007;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adorni MP, et al. The roles of different pathways in the release of cholesterol from macrophages. Journal of lipid research. 2007;48:2453–2462. doi: 10.1194/jlr.M700274-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Yu Y, et al. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Investigative ophthalmology & visual science. 2011;52:4663–4670. doi: 10.1167/iovs.10-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Omarova S, et al. Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. The Journal of clinical investigation. 2012;122:3012–3023. doi: 10.1172/JCI63816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Out R, et al. Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2295–2300. doi: 10.1161/01.ATV.0000237629.29842.4c. [DOI] [PubMed] [Google Scholar]
- 59.Baldan A, et al. Impaired development of atherosclerosis in hyperlipidemic Ldlr−/− and ApoE−/− mice transplanted with Abcg1−/− bone marrow. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2301–2307. doi: 10.1161/01.ATV.0000240051.22944.dc. [DOI] [PubMed] [Google Scholar]
- 60.Ranalletta M, et al. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2308–2315. doi: 10.1161/01.ATV.0000242275.92915.43. [DOI] [PubMed] [Google Scholar]
- 61.Westerterp M, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circulation research. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janowski BA, et al. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 63.Costet P, et al. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. The Journal of biological chemistry. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 64.Repa JJ, et al. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 65.Venkateswaran A, et al. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. A transcriptional role for specific oxysterols. The Journal of biological chemistry. 2000;275:14700–14707. doi: 10.1074/jbc.275.19.14700. [DOI] [PubMed] [Google Scholar]
- 66.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schultz JR, et al. Role of LXRs in control of lipogenesis. Genes & development. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grefhorst A, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. The Journal of biological chemistry. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 69.Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. The Journal of biological chemistry. 2007;282:743–751. doi: 10.1074/jbc.M605023200. [DOI] [PubMed] [Google Scholar]
- 70.Quinet EM, et al. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. Journal of lipid research. 2009;50:2358–2370. doi: 10.1194/jlr.M900037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kratzer A, et al. Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. Journal of lipid research. 2009;50:312–326. doi: 10.1194/jlr.M800376-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Najafi-Shoushtari SH, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marquart TJ, et al. miR-33 links SREBP-2 induction to repression of sterol transporters. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gerin I, et al. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. The Journal of biological chemistry. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horie T, et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rayner KJ, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horie T, et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. Journal of the American Heart Association. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rayner KJ, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. The Journal of clinical investigation. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rotllan N, et al. Therapeutic Silencing of MicroRNA-33 Inhibits the Progression of Atherosclerosis in Ldlr−/− Mice--Brief Report. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1973–1977. doi: 10.1161/ATVBAHA.113.301732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marquart TJ, et al. Anti-miR-33 therapy does not alter the progression of atherosclerosis in low-density lipoprotein receptor-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:455–458. doi: 10.1161/ATVBAHA.112.300639. [DOI] [PMC free article] [PubMed] [Google Scholar]