

Abstract
The Pro-Ser-Ala-Pro (PSAP) motif in the p2 domain of feline immunodeficiency virus (FIV) Gag is required for efficient virus release, virus replication, and Gag binding to the ubiquitin-E2-variant (UEV) domain of Tsg101. As a result of this direct interaction, expression of an N-terminal fragment of Tsg101 containing the UEV domain (referred to as TSG-5’) inhibits FIV release. In these respects, the FIV p2Gag PSAP motif is analogous to the PTAP motif of HIV-1 p6Gag. To evaluate the feasibility of a late domain-targeted inhibition of virus replication, we created an enriched Crandell-Rees feline kidney (CRFK) cell line (T5’hi) that stably expresses high levels of TSG-5’. Here we show that mutations in either the V3 loop or the second heptad repeat (HR2) domain of the FIV envelope glycoprotein (Env) rescue FIV replication in T5’hi cells without increasing FIV release efficiency. TSG-5’-resistance mutations in Env enhance virion infectivity and the cell-cell spread of FIV when diffusion is limited using a semi-solid growth medium. These findings show that mutations in functional domains of Env confer TSG-5’-resistance, which we propose enhances specific infectivity and the cell-cell transmission of virus to counteract inefficient virus release.
Introduction
The efficient release of retroviruses and many other enveloped viruses relies on a direct interaction with endosomal sorting complexes required for transport (ESCRTs) or ESCRT-associated proteins.1 Specifically, interactions with the ESCRT-I component TSG101, Nedd4 and Nedd4-like HECT ubiquitin ligases associated with ESCRT, or the ESCRT-I/III-interacting protein Alix appear to be the most highly conserved for this function.2-9 The four known ESCRT complexes (ESCRT-0,I,II,III) normally function in the turnover of ubiquitinated membrane-bound protein cargo through a series of interactions with each ESCRT that ultimately targets the cargo for degradation by vesicular budding into late endosomal compartments called multivesicular bodies (MVB) and subsequent delivery to the lysosome. 10-13 ESCRTs also play a role in other membrane scission events that occur during cytokinesis and the budding of enveloped viruses from the plasma membrane.14-18 Interactions with ESCRT occur through binding ubiquitin and short peptide motifs that are unique for each ESCRT.19 Viruses tap into this system by encoding one or sometimes multiple ESCRT-recognizing motifs in their major structural protein, which is often ubiquitinated.20-28 Invariably, these viral motifs consist of one or two critical proline residues (PT/SAP, PPxY, YPXnL, FPIV). Due to the profound defects in events that occur late in the virus replication cycle if these motifs are altered, specifically virus release, these motifs have been termed “late” domains.29 The best characterized mechanism for late domain function is that of HIV-1 Gag p6, which contains a PTAP sequence that directly interacts with the ubiquitin E2-like variant (UEV) domain of TSG101.30 This mimics the function of a PSAP sequence in the heptocyte growth factor-regulated tyrosine kinase substrate (HRS), which is an ESCRT-0 component associated with early endosomes that also binds the UEV domain of TSG101.22,31 The PT/SAP late domain has been widely studied because it is so highly conserved in the Gag protein of retroviruses, including most primate lentiviruses (sometimes in tandem), and most other lentiviruses.21,32 A functional PTAP late domain is also found in the matrix protein of at least two highly pathogenic viruses – the filovirus Ebola VP40 protein and the arenavirus Lassa Z protein.33,34 Often each virus only contains one late domain; however there are several examples of viruses that contain two or even three, which sometimes overlap. In primate lentiviruses, the PTAP motif often plays a more dominant role over Alix-binding motifs. For example, HIV-1 Gag p6 contains a secondary late domain consisting of a YPXnL sequence that binds the V domain of Alix (Alix-V), which enhances virus release and virus replication under specific conditions and cellular environments.35,36 Similar Alix-binding late domains have also recently been identified in SIV.37,38
FIV causes AIDS in domestic cats and is a model for HIV-1-associated pathogenesis, cellular biology, and vaccine development.32,39-44 Despite having very little sequence homology with HIV-1, we and others have shown that the molecular mechanisms and organization of functional domains for FIV virion assembly and release, proteolytic maturation of Gag and Env, Env-mediated receptor/co-receptor binding and membrane fusion, reverse transcription, integration, latency, Rev-dependent export of genomic RNA, and targeting of antiviral restriction factors are all highly conserved with those of HIV-1 and other primate lentiviruses. 32,44-62 For example, the PSAP motif in the C-terminal p2 domain of FIV Gag functions equivalently to the PTAP motif in HIV-1 Gag p6 in its role in FIV release and direct interaction with TSG101.5,46 Unlike HIV-1, however, FIV does not contain a secondary YPXnL-type late domain and is insensitive to agents that would disrupt interactions with the Alix-V domain.46 We previously demonstrated that expression of the N-terminal half of TSG101 (TSG-5’) containing the UEV domain functions in a dominant manner to inhibit HIV-1 and FIV release.15,46,63 We also developed feline cell lines that constitutively express TSG-5’ and described their restriction of FIV replication. In this report, we have now identified two mutations in the FIV Env glycoprotein that rescue FIV replication. One of these involves mutation of the V3 loop in the surface (SU) subunit of FIV Env, which is analogous to the V3 loop of HIV-1 SU in terms of its role in coreceptor binding, heparan sulfate proteoglycan (HSPG) binding, and as an epitope for neutralizing antibodies.64-71 The other mutation is in the second heptad repeat (HR2) of the transmembrane subunit (TM). This region is analogous to the HR2 of HIV-1 TM, which is the site of the fusion inhibitor enfuvirtide (T20) peptide sequence, where mutations that accompany T20 resistance can also develop, and has received interest as a site for neutralizing antibody epitopes.72-74 We propose that these mutations provide resistance to the TSG-5’-mediated inhibition of virus release not by rescuing virus release but rather by enhancing virion infectivity and cell-to-cell transmission.
Materials and Methods
Plasmids and site-directed mutagenesis
pFIV-34TF10 is an infectious molecular clone of the Petaluma isolate75 obtained from J. Elder (Scripps Research Institute, La Jolla, CA) through the NIH AIDS Research and Reference Reagent Program. pFIV-O2R is a derivative of pFIV-34TF10 with a repaired Orf2 gene, which increases infectivity in feline lymphocytes and does not inhibit replication in CRFK cells.76 pFP93 [a gift from E. Poeschla (Mayo Clinic, Rochester, MN)] is an FIV gag-pol expression vector deleted for Env, Vif, and LTRs that uses a CMV promoter to express non-infectious FIV-Petaluma virus-like particles (VLPs) in human cells.77 pHCMV-G, which expresses the G glycoprotein of vesicular stomatitis virus (VSV), was a gift from J. Burns (University of California, San Diego). pFIV-Env is a derivative of pFIV-O2R that expresses the full-length FIV-Petaluma Env and Rev in feline cells using its native promoter, constructed by deletion of an XhoI-KpnI fragment containing the RNA packaging signal, gag, pol, and vif, and an insertion of a compatible SalI-KpnI fragment of pFP93, which encodes the 5’ end of both env and rev. pFIV-Env or pFIV-O2R clones containing TSG-5’ resistance mutations were made by site-directed mutagenesis using the QuikChange II XL mutagenesis kit (Stratagene) and synthetic complementary oligonucleotides (Sigma Genosys). To detect any potential amplification of DNA polymerase errors during mutagenesis, the entire coding region of all mutant clones was fully sequenced prior to use.
Cell culture
Crandell-Rees feline kidney cells CRFK78 (a gift from S. Le Grice, HIV Drug Resistance Program, National Cancer Institute, Frederick, MD), 293T, and HeLa cells were maintained in either Eagle MEM (E-MEM; ATCC) for CRFK or D-MEM (Gibco) for 293T and HeLa, supplemented with 10% fetal bovine serum (FBS; HyClone), penicillin, streptomycin, and glutamine (Gibco).
Virus release assays
FIV Gag was detected by radioimmunoprecipitation assay (RIPA) as previously described.46,79-81 Briefly, CRFK cells were transfected with vectors expressing FIV proteins or TSG-5’ using Lipofectamine LTX, following the manufacturer’s suggested protocol. Cells were metabolically labeled at 37°C with [35S] Met/Cys (Express Protein-Labeling Mix; Perkin-Elmer) for 4 hr. Released virions or VLPs were collected by filtration of cultured cell supernatant and ultracentrifugation. Cell and virion samples were solubilized in Tris-buffered saline containing 0.5% Triton X-100. Cell lysates were pre-cleared by adsorption with protein G-agarose (Invitrogen). Virion and pre-cleared cell lysates were immunoprecipitated with mouse anti-FIV p24gag (clone PAK3-2C1) bound to protein G-agarose at 4°C. Immunoprecipitated proteins were washed, then resolved by SDS-PAGE, fixed, and dehydrated. Labeled proteins were detected by autoradiography on phosphorimaging plates (Fujifilm) and quantitated using QuantityOne software (BioRad). Virus release efficiency was calculated as the ratio of released Gag over total Gag protein, normalized to the positive control (uninhibited WT Gag).
Fusion assays
Assays to measure Env-mediated cell-cell fusion were based on those described previously.82 Briefly, CRFK cells were transfected in duplicate with Lipofectamine LTX PLUS reagent, and FIV Env expression vectors (pFIV-Env) containing either the WT sequence or mutations that confer resistance to TSG-5’, to produce CRFK-Env using the native FIV promoter. HeLa cells were seeded onto a 96-well plate at a density of 104 cells/well and allowed to adhere overnight at 37°C. HeLa cultures were seeded in replicates with 500-1,000 CRFK-Env cells, respectively, and incubated overnight at 37°C. Hela cells were stained with a crystal violet-Giemsa solution in 80% methanol and destained with water. Syncytia were quantified as giant cells containing five or more nuclei by phase contrast microscopy. Results among replicates were averaged and normalized based on the number of CRFK-Env cells added.
Cell-cell transmission assays
CRFK cells that were chronically infected with each TSG-5’-resistant FIV mutant were mixed with uninfected CRFK or T5’hi cells at a 1:20 ratio, aliquoted into 6-well plates at 0.4 million cells per well, and incubated for 15 hr at 37°C. A semi-solid overlay consisting of normal growth medium and 0.5% high-melting agarose was prepared by boiling 1% agarose and mixing with an equal volume of 2X growth medium (2X Eagle MEM and 20% FBS supplemented with penicillin, streptomycin, and glutamine) at room temperature, which was then cooled to ~50C in a water bath. The cells were washed with PBS, overlain with the freshly prepared semisolid medium, cooled to room temperature, and incubated at 37°C. For each time interval, the overlay was removed with a spatula and adherent cells were resuspended by trypsinization. Cell suspensions were washed in PBS + 5% FBS, then PBS alone, resuspended in 4% formaldehyde, incubated for 15 min, then diluted with 9 volumes of PBS and stored at 4°C. Fixed samples were treated in a series of 1 min incubations with 0.1M glycine, PBS alone, 0.1% Triton X-100, PBS alone, and resuspended in 3% BSA/PBS, then incubated for 30 min with 0.5 μg mouse anti-FIVp24 (PAK3-2C1 clone) conjugated to Alexa Fluor 488 using Zenon labeling technology (Invitrogen). Samples were washed with PBS and resuspended in 5% FBS/PBS for FACS analysis. Labeled cells were detected with a BD FACSCalibur and gated for live cells by forward and side scatter.
Cell-free infectivity and reverse transcriptase assays
FIV was produced by transfection of CRFK-zeo or T5’hi cells with Lipofectamine LTX reagent and infectious pFIV-O2R containing either the WT or TSG-5’-resistant genomes, which establishes a chronic infection. Cell-free virus was harvested from cultured supernatants by 0.4 μm filtration. 12-well culture plates were seeded with 0.3 × 106 uninfected target cells (CRFK-zeo or T5’hi) and serial dilutions of cell-free FIV in 1 ml growth medium containing 20 μg/ml DEAE-Dextran. Cells were incubated at 37°C for ~24 hr and virus inputs were normalized by reverse transcriptase (RT) assay, using methods previously described for HIV-1.83 Cultured supernatant was replaced at 1 day post-infection with fresh growth medium. Cells were fixed at 2 days post-infection in 4% formaldehyde. FIV Gag-positive cells were stained and counted by FACS using the same methods described above for cell-cell transmission assays.
Results
Enrichment for TSG-5’-positive cells enhances inhibition of FIV release
We previously described the stable and constitutive expression of TSG-5’ in a CRFK/TSG-5’(zeo) cell line, hereafter referred to as T5’ cells, which inhibits FIV release and replication relative to control CRFK-zeo cells.46 In T5’ cells infected with ~100 times the minimum infectious dose of wild-type (WT) FIV, virus replication is detectable by RT assay after a one month delay relative to control cells. Virus harvested under these conditions did not replicate more efficiently than WT in T5’ cells in subsequent rounds of infection (Figure 1A), suggesting no acquisition of mutations conferring resistance. Immunofluorescence microscopy (IFM) of T5’ cells probed with an anti-HA antibody revealed that approximately 1/3 of cells are TSG-5’-negative, despite selection for a linked zeocin resistance marker (zeo) (Fig. 1B). To eliminate cells expressing low levels of TSG-5’, which are likely permissive to WT FIV replication, clonal colonies of T5’ cells were screened for TSG-5’ expression by western blot with anti-HA and normalized with anti-tubulin (data not shown). None of the 58 viable clones screened was entirely negative for TSG-5’ expression, however we did observe a wide range of expression levels. There was also no correlation between high TSG-5’ expression and low cell viability, based on cell morphology, colony size, tubulin expression, and cell doubling times (data not shown). IFM of several clones was also entirely consistent with western blotting results (data not shown). Twenty-four TSG-5’-positive clones were pooled to create a TSG-5’-enriched cell line, hereafter referred to as T5’hi to distinguish it from the parental T5’ cell line. TSG-5’ expression levels of T5’ and T5’hi cells, relative to control cells, were quantified by flow cytometry (Figure 2A). T5’hi cells inhibited FIV release more efficiently than did the parental T5’ cell line (Fig. 2B). An increase in the proportion of cellular Gag p50 to Gag p24 in T5’hi cells relative to T5’ cells was also consistently observed, suggesting that the extent of p50 Gag proteolysis also correlates with TSG-5’ levels and virus release efficiency.
Figure 1. Serial passage of FIV in T5’ cells did not yield TSG-5’ resistance.
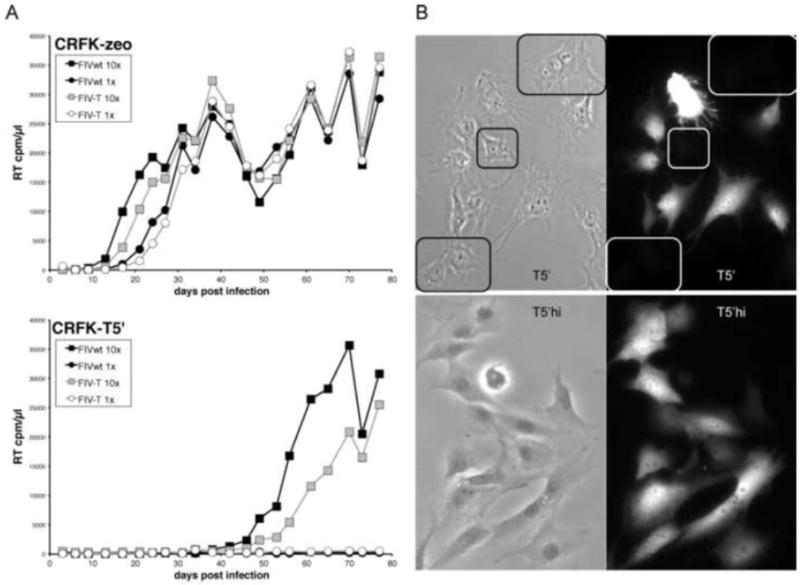
A) CRFK-zeo or T5’ cells in 6-well plates were infected with cell-free preparations of FIV produced from chronically infected CRFK-zeo cells (FIVwt) or T5’ cells (FIV-T) at either 1 × 106 RT cpm) or 10X virus input. B) Expression of TSG-5’ was detected by anti-HA IF microscopy in T5’ and T5’hi cells. Cells expressing undetectable levels of TSG-5’ are highlighted.
Figure 2. Enrichment for TSG-5’-expressing cells improved inhibition of virus release.
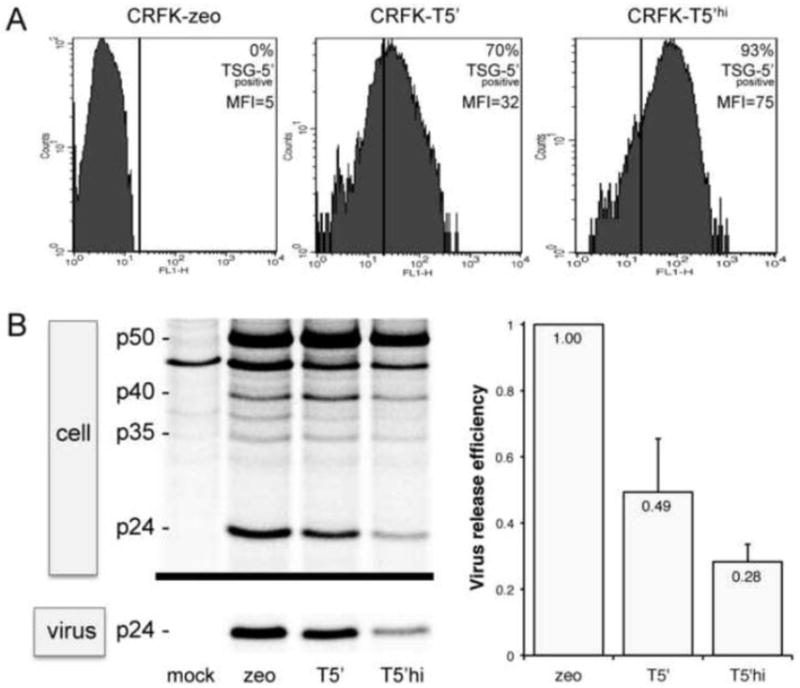
A) Expression of TSG-5’ in CRFK cell lines was detected by anti-HA FACS analysis. Average level of TSG-5’ expression is indicated by the mean fluorescence intensity (MFI), and the proportion of TSG-5’-positive cells is relative to control CRFK-zeo cells. B) Inhibition of FIV release in TSG-5’-expressing CRFK cell lines relative to control CRFK zeo cells is shown as the ratio of virion-associated p24 to total Gag, as detected by metabolic labeling, radio-immunoprecipitation, and phosphorimaging. p50 is the full-length FIV Gag polyprotein and p24 is FIV capsid protein. Average levels of virus release are indicated.
Mutations in FIV Env confer resistance in T5’hi cells
As observed previously with the parental T5’ cells, WT FIV replication in infected T5’hi cultures was significantly delayed, relative to replication in control cells (Fig. 3). Although TSG-5’ expression clearly delays virus release, and TSG-5’ expression was stable throughout each experiment, we consistently observed RT activity in the supernatant of infected T5’hi cells that eventually accumulated to levels equal to those of infected control cells. Cellular DNA was obtained from cultures of infected T5’hi cells that contained replicating FIV, PCR-amplified with FIV-specific primers, and sequenced. Similar samples were prepared from FIV-infected parental CRFK cells to control for genetic variation in the absence of TSG-5’ expression. Mutations in FIV Env were consistently observed in virus obtained from infected T5’hi cells, but not in infected control cells (Fig. 4A; data not shown). One of these mutations (K410N) is in the V3 loop of FIV Env, which has been shown to bind heparan sulfate proteoglycans (HSPGs) and CXCR4 at the surface of CRFK cells to promote membrane attachment and fusion.66 The other mutation found in Env (T762I) is at the membrane-proximal end of the predicted heptad repeat 2 (HR2) region, which is also involved in CXCR4-mediated membrane fusion.82,84 Each of these mutations was identified three times in independent isolates, and no isolate contained both. Mutations in Gag were also identified, including one isolate with a G34E mutation in the nucleocapsid (NC) domain, and another with a conservative R36K mutation in the matrix domain. However, each of these Gag mutations were observed in the context of either the K410N or T762I Env mutations. FIV isolates containing either Env mutation (T762I or K410N) replicated more rapidly than WT FIV in newly infected T5’hi cells (data not shown). Several of the mutations identified through selection in T5’hi cells were introduced into the WT FIV proviral clone by site-directed mutagenesis, including an FIV clone bearing only the G34E mutation in NC, which failed to replicate efficiently in T5’hi cells (data not shown). In contrast, FIV clones containing either the K410N or T762I Env mutation showed peak RT activity in T5’hi cells within 3-4 weeks posttransfection, whereas WT FIV failed to replicate efficiently during the course of each experiment (Fig. 4B, lower panel). This was repeated in three independent experiments in duplicate with the same result, illustrating that either Env mutation is sufficient to rescue FIV replication in T5’hi cells. In control CRFK-zeo cells (not expressing TSG-5’) the WT and K410N and T762I mutants replicated with similar kinetics (Fig. 4B, top panel).
Figure 3. Selection for replicating FIV in T5’hi cells.
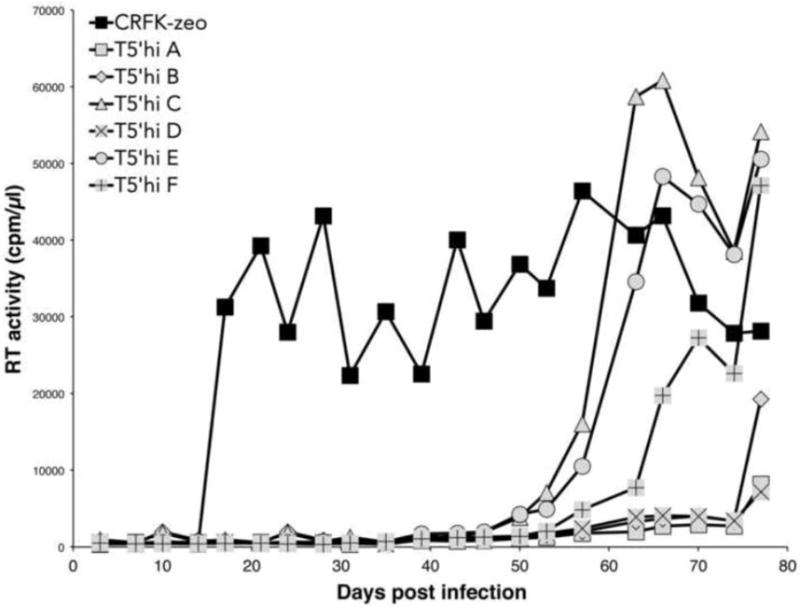
CRFK-zeo or T5’hi cells in 6-well plates were infected with an equal amount of FIV in parallel samples (A-F) and passaged twice per week. RT activity was detected from cultured supernatants harvested prior to each passage.
Figure 4. Mutations in FIV Env confer resistance to TSG-5’.
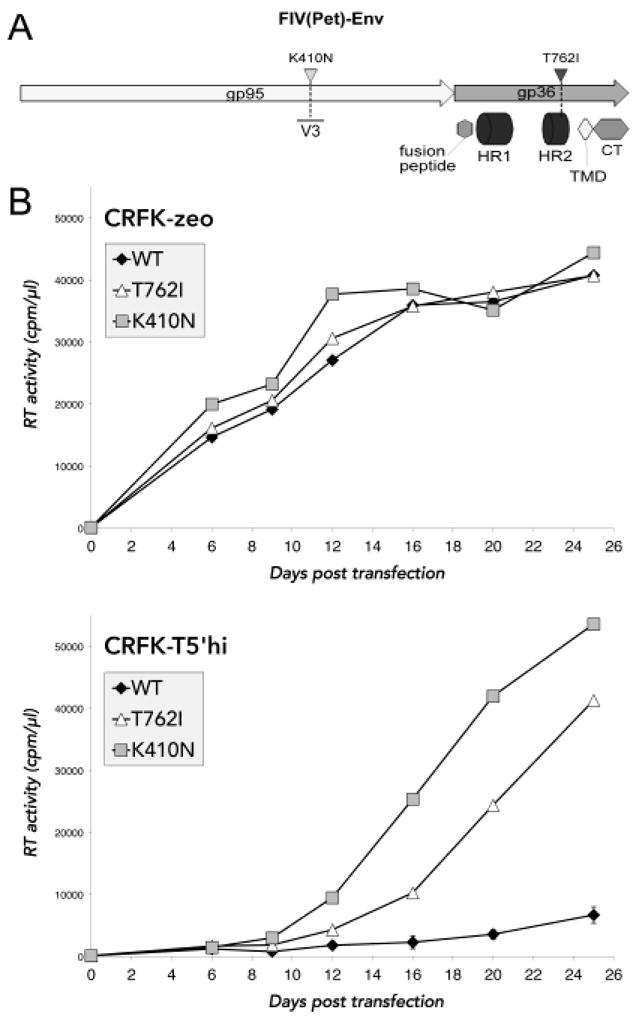
A) The two mutations in FIV Env that were detected by sequencing cDNA from infected T5’hi cells are shown to scale with functional domains labeled. gp95 is the surface subunit, analogous to HIV-1 gp120. gp36 is the transmembrane subunit analogous to HIV-1 gp41, which contains the two heptad repeats (HR) involved in membrane fusion, the transmembrane domain (TMD), and the cytoplasmic tail (CT). B) Mutations in FIV Env that putatively conferred resistance to TSG-5’ were introduced into the WT molecular FIV clone by site-directed mutagenesis. Mutant clones were then tested against WT FIV for replication capacity in transfected CRFK-zeo and T5’hi cells.
TSG-5’-resistance mutations do not rescue virus release inhibition in T5’hi cells and do not increase Env fusogenicity
To begin to determine the mechanism of potential TSG-5’ resistance conferred by each mutation, we first examined whether the resistant mutant clones were insensitive to TSG-5’-mediated restriction of virus release. T5’hi cells were transfected with equal amounts of each mutant proviral clone and compared with the WT in a virus release assay (Fig. 5A). The TSG-5’-resistant FIV clones failed to release virion-associated p24 more efficiently than the WT, demonstrating that these mutations do not overcome the inhibition of virus release imposed by TSG-5’. To determine whether mutations in FIV Env that confer resistance to TSG-5’ expression might alter FIV infectivity by modulating Env fusogenicity with target cells, we assayed FIV Env membrane fusion activity in a syncytial assay that has been previously described (ref). Briefly, CRFK cells transfected with a plasmid vector expressing either WT or mutant FIV Env were cocultured with HeLa cell monolayers overnight, then fixed and stained to visualize cell membranes and nuclei. HeLa cells express sufficient CXCR4 co-receptor to fuse with plasma membrane-associated FIV Env and form syncytia in a CD134-independent manner.47,82,85 Numbers of syncytia from multiple replicates of each sample were averaged. Syncytia were defined as giant cells containing at least 5 nuclei. Cells expressing the FIV Env K410N mutant consistently fused less efficiently with uninfected HeLa cells than cells expressing either the WT FIV Env or the T762I Env mutant (Fig. 5B). Similar results were obtained with CRFK cells chronically infected with WT or Env-mutant FIV cocultured with HeLa cells (data not shown). Thus, mutations in Env that confer resistance to TSG-5’ do not simply upregulate Env fusogenic activity.
Figure 5. TSG-5’ resistance mutations do not rescue virus release or enhance membrane fusion.
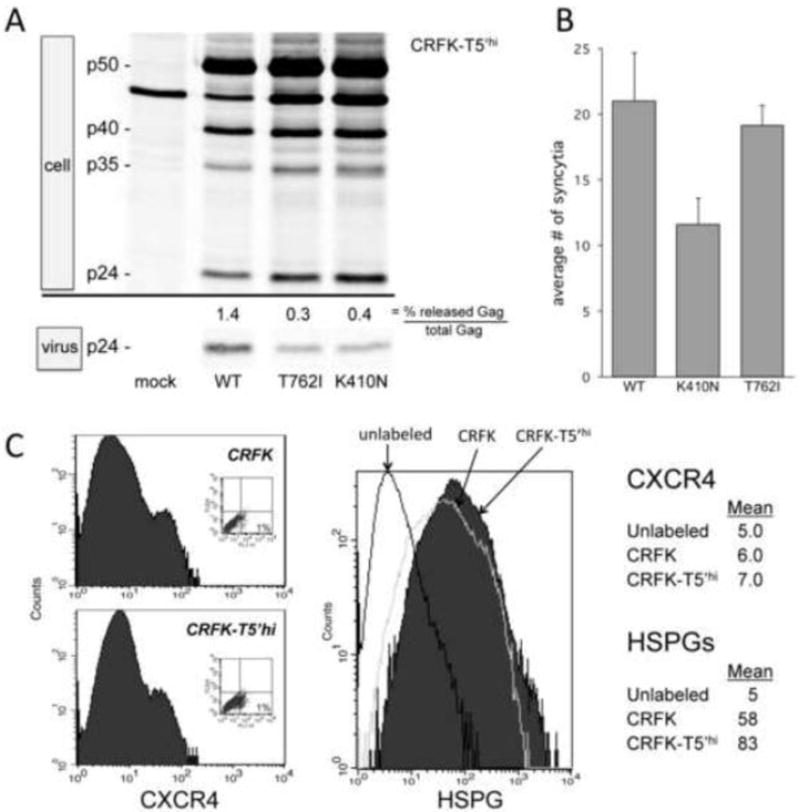
A) T5’hi cells were transfected with mutant FIV clones and compared with WT FIV for virus release efficiency, as detected by metabolic labeling, radio-immunoprecipitation, and phosphorimaging. B) Results from coculture of FIV Env-expressing CRFK cells and HeLa cells, fixed and stained with crystal violet, are shown as the average number of syncytia counted per well in a 96-well plate. C) Flow cytometry of CXCR4 and HSPG expression in CRFK and T5’hi cells. Primary antibodies were labeled with AlexaFluor 488-conjugated anti mouse IgG (Invitrogen). CXCR4 was detected with mouse anti-human CXCR4 (clone 44717) obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. HSPGs were detected with mouse anti-heparan sulfate clone F58-10E4 (Seikagaku America), as described previously.47
CrFK T5’hi cells do not express a higher level of HSPGs or CXCR4
It has been shown that heparan sulfate proteoglycans (HSPG) and CXCR4 serve as the primary and coreceptor for FIV infection in CRFK cells, which lack expression of the true primary receptor CD134 that is utilized by primary isolates of FIV.47,52,53,85-88 Since mutations that confer resistance to TSG-5’ expression mapped to the Env glycoprotein of FIV, we considered whether this was possibly a compensation for any specific decrease in HSPG or CXCR4 expression in T5’hi cells relative to controls. To evaluate this we measured CXCR4 and HSPG expression in T5’hi and control CRFK cells using methods described previously.47 Our results show no decrease in the expression of these cell surface markers as a function of TSG-5’ (Fig. 5C). The low expression of CXCR4 in CRFK cells is consistent with a previous report.47 The significance of the very slight increase in HSPG expression in T5’hi cells is not clear, as this would tend to render these cells even more permissive to infection, which we also did not observe.
TSG-5’-resistant FIV mutants have greater cell-free infectivity
Considering that resistance to TSG-5’ mapped to FIV Env, it is likely that virion infectivity may have increased in order to enhance the spread of infection. To address this possibility, we first examined whether TSG-5’-resistant FIV mutants have a higher specific infectivity. Cell-free FIV was isolated from chronically infected CRFK cells and used to infect an equal number of control CRFK or T5’hi cells over a range of virus inputs. In parallel, RT assays were performed on each virus isolate. The number of Gag-positive cells was then enumerated at two days postinfection, and results were normalized to the RT activity of each input inoculum, expressed as RT cpm per cell (Figure 6). To compare infectivity directly with WT without genetic selection, we only examined viruses produced in the absence of TSG-5’. Our results show that TSG-5’-expressing cells are ~2-fold more resistant to cell-free FIV infection than control cells, but mutations that confer resistance to TSG-5’ did not overcome this effect. Infections of T5’hi cells also began to saturate at a lower virus input than control cells. Importantly, there was a significant increase in the infectivity of each TSG-5’-resistant mutant relative to that of the WT, especially for the Env T762I mutant, which was entirely consistent with other independent experiments.
Figure 6. TSG-5’ resistance mutations enhance FIV specific infectivity.
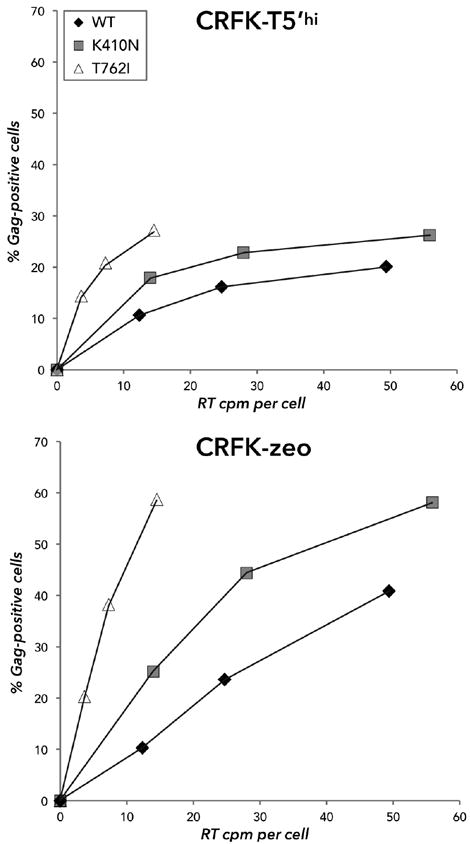
TSG-5’-resistant FIV mutant clones and WT FIV were harvested from the supernatant of chronically infected CRFK-zeo cells and added to uninfected CRFK-zeo or T5’hi cells in 2-fold serial dilutions and mixed with DEAE-Dextran. At two days postinfection, cells were resuspended and fixed in 4% formaldehyde, then stained with mouse anti-p24 conjugated to Alexa Fluor 488 and quantified by flow cytometry.
TSG-5’-resistant FIV mutants show more efficient cell-cell transmission
Based on the TSG-5’-mediated inhibition of virus release that we have demonstrated, we speculated that FIV-infected T5’hi cells contain a high proportion of cell-associated virions, and mutations that enhance cell-cell transmission would be preferentially selected. To address this possibility, we developed an assay that indirectly quantifies the rate of FIV cell-cell transmission by restricting the diffusion of cell-free virions with a semi-solid medium. This assay is based on methods previously described for poxviral plaque assays.89 With this approach, the cells continue to grow and divide normally to form cell-cell contacts that allow for a spreading infection. FIV-infected and uninfected CRFK cells divide at the same rate (data not shown) and we have found that FIV infection does not kill CRFK cells, so the proportion of FIV Gag-positive cells increases only as a result of infectious virus replication and a spreading infection. In T5’hi cells, the spread of WT FIV from infected to uninfected cells remained restricted to only ~8% Gag-positive cells within the 7 days of coculture, whereas TSG-5’-resistant Env mutants K410N and T762I expanded 4- to 8-fold to attain 20-40% Gag-positive cells (Fig. 7A). Even in the absence of TSG-5’ expression, TSG-5’-resistant mutants consistently spread more rapidly under semi-solid overlays to infect ~35-45% of cells within 5 days, whereas the spread of WT FIV was relatively restricted at 15% within the same time frame (Fig. 7B). This contrasts with the results shown in Fig. 4B, perhaps because the spread of infection is limited by agarose and the amount of cell-free virus is no longer in excess, similar to conditions where virus release is inhibited. The detection of Gag-positive cells as a measure of a spreading infection with this method is also more accurate and precise than detecting RT activity released into the supernatant. These results suggest that TSG-5’-resistant mutants are more efficient in cell-cell transmission, and that this property is associated with Env function.
Figure 7. TSG-5’ resistance mutations enhance cell-cell FIV transmission.
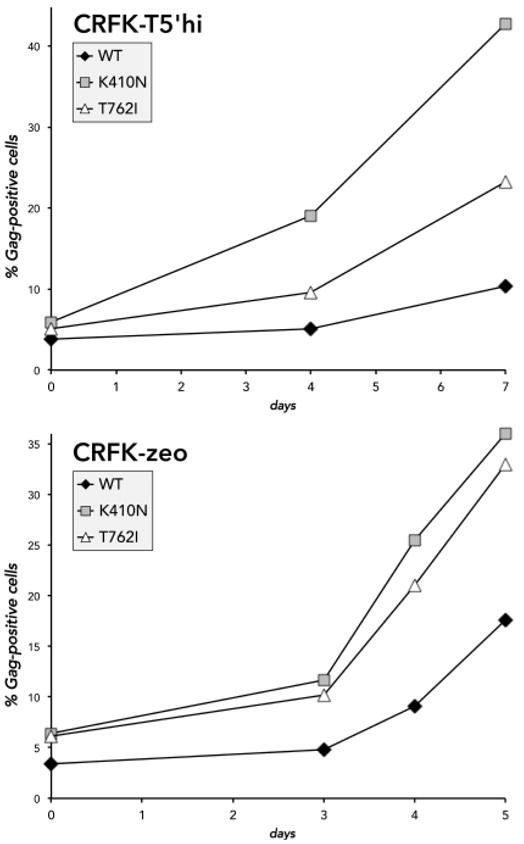
CRFK-zeo cells chronically infected with either WT FIV or one of the TSG-5’-resistant mutants were counted and mixed with uninfected CRFK-zeo or T5’hi cells in suspension at a final ratio of 1:20 (5% Gag-positive cells). Cell mixtures were aliquoted in cell culture plates and incubated overnight. Adherent cells were washed to remove any released cell-free virions and covered with melted growth medium containing 0.5% high-melting agarose, then incubated at 37°C. For each time interval, overlays were removed and the cell monolayer was resuspended, washed, and fixed. FIV p24 was later detected in all fixed samples by immunofluorescence and flow cytometry. Cells from the initial coculture were also fixed and analyzed to verify the baseline ratio of infected cells.
Discussion
We have demonstrated that FIV can develop partial resistance to a constitutive inhibition of virus release, which in this study was mediated by expression of a dominant interfering N-terminal fragment of cellular TSG101 (TSG-5’). Our results suggest that enhancements of virion infectivity and possibly cell-to-cell transmission by mutations in either the V3 loop or the HR2 domain of the Env glycoprotein provide mechanisms for this resistance. We had initially speculated the acquisition of TSG-5’-resistance by mutation of FIV Env (K410N) in the V3 loop would possibly affect virion binding affinity to CXCR4 and HSPGs, either positively or negatively, based on the reported correlations between overall net charge in this region and coreceptor affinity.65,90 It has been shown that the net charge in the V3 loop differs between lab-adapted isolates grown in cell culture and primary isolates obtained from infected cats, which is actively maintained by selective pressures in both environments.68-70,91 Since our study focused on FIV replication mediated by CD134-independent entry, the selection for resistance may have favored mutations in the HSPG or CXCR4-binding regions of FIV Env that are more highly utilized by the 34TF10 molecular clone. To avoid some of these limitations, we had engineered the FIV molecular clone used in this study to encode a full-length orf-2/orf-A, which expands its host range to include CD134-positive feline T cell lines and potentially PBMCs.76 However, our efforts to stably express high levels of TSG-5’ in the MCH5-4 feline T cell line have not yet been successful. We did not observe any significant change in HSPG or CXCR4 levels in CRFK cells expressing TSG-5’ relative to parental cells that would explain the advantage of acquiring a mutation in this domain, however any specific changes in affinity remain to be evaluated. During the course of this study the CXCR4 and HSPG-binding sites of FIV Env were more clearly indentified by Hu et al., which includes the lysine residue at position 410 of the same molecular clone of FIV Petaluma (34TF10) used in our study.65,66 We had also speculated that the acquisition of TSG-5’ resistance by mutation of the HR2 domain of FIV Env (T762I) would possibly affect Env-mediated membrane fusion, since the coiled-coil interaction between HR2 and HR1 is critical for fusion.92 Notably, the HR2 domain of HIV-1 Env is the source of the antiviral peptide enfuvirtide (T20, fuzeon), which functions as a competitive inhibitor of HR1 binding.93,94 We were surprised to find that fusion activity of the T762I mutant is paradoxically diminished, given that our HR2 mutant is more infectious and spreads more efficiently from cell to cell. In parallel studies, we have also observed mutations in HIV-1 Env that are selected in the presence of Alix binding site mutants, which again appear to enhance cell-to-cell transmission (unpublished results).
Consistent with our previous report,46 the inhibition of virus release through stable expression of TSG-5’ had no deleterious effect on cell viability. We have also developed a Jurkat cell line stably expressing a significant level of TSG-5’, which has no effect on cell viability and moderately inhibits the replication of the HIV-1 clone NL4-3 (unpublished results). Given that our FIV molecular clone did not acquire any mutations that fully restored virus replication kinetics in the presence of this late domain inhibitor, and that mutations that did provide resistance did not restore virus release efficiency, this study provides proof of principle that interference of late domain function is a difficult block for the virus to circumvent through any single nucleotide change. Serial passages of resistant FIV isolates also did not lead to any secondary mutations that would possibly have improved viral fitness (data not shown). This stresses the importance of the TSG101-Gag interaction to efficient virus release, and that there is little redundancy in this pathway. This is encouraging if development of small molecule inhibitors of this interface is to be pursued further as an antiviral therapy. Furthermore, our results suggest that interference of late domain function should be combined with inhibitors of cell-to-cell transmission to be even more effective.
Highlights.
Overexpression of dominant-negative Tsg101 fragment (TSG-5’) inhibits FIV budding
Stable expression of TSG-5’ in the feline cell line CRFK impairs FIV replication
Passage of FIV in TSG-5’-expressing cells leads to TSG-5’-resistant FIV mutants
TSG-5’ resistance mutations all map to the FIV envelope glycoprotein (Env)
Env mutations conferring TSG-5’ resistance enhance cell-cell transfer and infectivity
Acknowledgments
We thank members of the Freed laboratory for helpful discussion, and John Elder and Eric Poeschla for providing valuable reagents. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH, and by the Intramural AIDS Targeted Antiviral Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martin-Serrano J, Neil SJD. Host factors involved in retroviral budding and release. Nat Rev Microbiol. 2011;9:519–31. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 3.Garrus JE, von Schwedler UK, Pornillos O, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Côté M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 4.VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag) Proc Natl Acad Sci USA. 2001;98:7724–9. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci USA. 2002;99:955–60. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–99. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 7.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol. 2005;168:89–101. doi: 10.1083/jcb.200408155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usami Y, Popov S, Gottlinger HG. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J Virol. 2007;81:6614–22. doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RD, Chung H-Y, Zhai Q, Robinson H, Sundquist WI, Hill CP. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell. 2007;128:841–52. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt O, Teis D. The ESCRT machinery. Curr Biol. 2012;22:R116–20. doi: 10.1016/j.cub.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Ren X, Hurley JH. Proline-rich regions and motifs in trafficking: from ESCRT interaction to viral exploitation. Traffic. 2011;12:1282–90. doi: 10.1111/j.1600-0854.2011.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad Sci USA. 2008;105:7687–91. doi: 10.1073/pnas.0801567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–12. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 15.Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol. 2007;5:912–6. doi: 10.1038/nrmicro1790. [DOI] [PubMed] [Google Scholar]
- 17.Caballe A, Martin-Serrano J. ESCRT machinery and cytokinesis: the road to daughter cell separation. Traffic. 2011;12:1318–26. doi: 10.1111/j.1600-0854.2011.01244.x. [DOI] [PubMed] [Google Scholar]
- 18.Agromayor M, Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends in Cell Biology. 2013 doi: 10.1016/j.tcb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Shields SB, Piper RC. How ubiquitin functions with ESCRTs. Traffic. 2011;12:1306–17. doi: 10.1111/j.1600-0854.2011.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strack B, Calistri A, Accola MA, Palu G, Gottlinger HG. A role for ubiquitin ligase recruitment in retrovirus release. Proc Natl Acad Sci USA. 2000;97:13063–8. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freed EO. Viral late domains. J Virol. 2002;76:4679–87. doi: 10.1128/JVI.76.10.4679-4687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pornillos O, Higginson DS, Stray KM, Fisher RD, Garrus JE, Payne M, He G-P, Wang HE, Morham SG, Sundquist WI. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J Cell Biol. 2003;162:425–34. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA. 2003;100:12414–9. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pincetic A, Leis J. The Mechanism of Budding of Retroviruses from Cell Membranes. Advances in Virology. 2009 doi: 10.1155/2009/623969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jäger S, Gottwein E, Kräusslich H-G. Ubiquitination of human immunodeficiency virus type 1 Gag is highly dependent on Gag membrane association. J Virol. 2007;81:9193–201. doi: 10.1128/JVI.00044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci USA. 2008;105:3974–9. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calistri A, Del Vecchio C, Salata C, Celestino M, Celegato M, Gottlinger HG, Palù G, Parolin C. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: involvement of ubiquitin and Nedd4-2s ligase in viral egress. J Cell Physiol. 2009;218:175–82. doi: 10.1002/jcp.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhadina M, Bieniasz PD. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathogens. 2010;6:e1001153. doi: 10.1371/journal.ppat.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, Wills JW. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol. 1995;69:5455–60. doi: 10.1128/jvi.69.9.5455-5460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002;9:812–7. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- 31.Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–42. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luttge BG, Freed EO. FIV Gag: virus assembly and host-cell interactions. Vet Immunol Immunopathol. 2010;134:3–13. doi: 10.1016/j.vetimm.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strecker T, Eichler R, Meulen Jt, Weissenhorn W, Dieter Klenk H, Garten W, Lenz O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected] J Virol. 2003;77:10700–5. doi: 10.1128/JVI.77.19.10700-10705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, Harty RN. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J Virol. 2003;77:1812–9. doi: 10.1128/JVI.77.3.1812-1819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Schwedler UK, Stuchell M, Müller B, Ward DM, Chung H-Y, Morita E, Wang HE, Davis T, He G-P, Cimbora DM, Scott A, Kräusslich H-G, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114:701–13. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH. Structural basis for viral late-domain binding to Alix. Nat Struct Mol Biol. 2007;14:194–9. doi: 10.1038/nsmb1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhai Q, Landesman MB, Robinson H, Sundquist WI, Hill CP. Identification and structural characterization of the ALIX-binding late domains of simian immunodeficiency virus SIVmac239 and SIVagmTan-1. J Virol. 2011;85:632–7. doi: 10.1128/JVI.01683-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bello NF, Wu F, Sette P, Dussupt V, Hirsch VM, Bouamr F. Distal leucines are key functional determinants of Alix-binding simian immunodeficiency virus SIV(smE543) and SIV(mac239) type 3 L domains. J Virol. 2011;85:11532–7. doi: 10.1128/JVI.05284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willett BJ, Flynn JN, Hosie MJ. FIV infection of the domestic cat: an animal model for AIDS. Immunol Today. 1997;18:182–9. doi: 10.1016/s0167-5699(97)84665-8. [DOI] [PubMed] [Google Scholar]
- 40.Hohdatsu T, Okada S, Motokawa K, Aizawa C, Yamamoto JK, Koyama H. Effect of dual-subtype vaccine against feline immunodeficiency virus infection. Vet Microbiol. 1997;58:155–65. doi: 10.1016/S0378-1135(97)00164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burkhard MJ, Dean GA. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr HIV Res. 2003;1:15–29. doi: 10.2174/1570162033352101. [DOI] [PubMed] [Google Scholar]
- 42.Coats KS. The feline immunodeficiency virus-infected cat: a model for lentivirus-induced placental immunopathology and reproductive failure (mini-review) Am J Reprod Immunol. 2005;54:169–85. doi: 10.1111/j.1600-0897.2005.00296.x. [DOI] [PubMed] [Google Scholar]
- 43.Münk C, Hechler T, Chareza S, Löchelt M. Restriction of feline retroviruses: lessons from cat APOBEC3 cytidine deaminases and TRIM5alpha proteins. Vet Immunol Immunopathol. 2010;134:14–24. doi: 10.1016/j.vetimm.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Elder JH, Sundstrom M, de Rozieres S, de Parseval A, Grant CK, Lin Y-C. Molecular mechanisms of FIV infection. Vet Immunol Immunopathol. 2008;123:3–13. doi: 10.1016/j.vetimm.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammadi H, Bienzle D. Pharmacological Inhibition of Feline Immunodeficiency Virus (FIV) Viruses. 2012;4:708–724. doi: 10.3390/v4050708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luttge BG, Shehu-Xhilaga M, Demirov DG, Adamson CS, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. Molecular characterization of feline immunodeficiency virus budding. J Virol. 2008;82:2106–19. doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Parseval A, Elder JH. Binding of recombinant feline immunodeficiency virus surface glycoprotein to feline cells: role of CXCR4, cell-surface heparans, and an unidentified non-CXCR4 receptor. J Virol. 2001;75:4528–39. doi: 10.1128/JVI.75.10.4528-4539.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elder JH, Schnölzer M, Hasselkus-Light CS, Henson M, Lerner DA, Phillips TR, Wagaman PC, Kent SB. Identification of proteolytic processing sites within the Gag and Pol polyproteins of feline immunodeficiency virus. J Virol. 1993;67:1869–76. doi: 10.1128/jvi.67.4.1869-1876.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips TR, Lamont C, Konings DA, Shacklett BL, Hamson CA, Luciw PA, Elder JH. Identification of the Rev transactivation and Rev-responsive elements of feline immunodeficiency virus. J Virol. 1992;66:5464–71. doi: 10.1128/jvi.66.9.5464-5471.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J Virol. 2004;78:9524–37. doi: 10.1128/JVI.78.17.9524-9537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006;314:461–4. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 52.de Parseval A, Grant CK, Sastry KJ, Elder JH. Sequential CD134-CXCR4 interactions in feline immunodeficiency virus (FIV): soluble CD134 activates FIV Env for CXCR4-dependent entry and reveals a cryptic neutralization epitope. J Virol. 2006;80:3088–91. doi: 10.1128/JVI.80.6.3088-3091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willett BJ, Picard L, Hosie MJ, Turner JD, Adema K, Clapham PR. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J Virol. 1997;71:6407–15. doi: 10.1128/jvi.71.9.6407-6415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dunham SP. Lessons from the cat: development of vaccines against lentiviruses. Vet Immunol Immunopathol. 2006;112:67–77. doi: 10.1016/j.vetimm.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Savarino A, Pistello M, D’Ostilio D, Zabogli E, Taglia F, Mancini F, Ferro S, Matteucci D, De Luca L, Barreca ML, Ciervo A, Chimirri A, Ciccozzi M, Bendinelli M. Human immunodeficiency virus integrase inhibitors efficiently suppress feline immunodeficiency virus replication in vitro and provide a rationale to redesign antiretroviral treatment for feline AIDS. Retrovirology. 2007;4:79. doi: 10.1186/1742-4690-4-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.North TW, Cronn RC, Remington KM, Tandberg RT, Judd RC. Characterization of reverse transcriptase from feline immunodeficiency virus. J Biol Chem. 1990;265:5121–8. [PubMed] [Google Scholar]
- 57.Auwerx J, Esnouf R, De Clercq E, Balzarini J. Susceptibility of feline immunodeficiency virus/human immunodeficiency virus type 1 reverse transcriptase chimeras to non-nucleoside RT inhibitors. Mol Pharmacol. 2004;65:244–51. doi: 10.1124/mol.65.1.244. [DOI] [PubMed] [Google Scholar]
- 58.Kenyon JC, Lever AML. The molecular biology of feline immunodeficiency virus (FIV) Viruses. 2011;3:2192–213. doi: 10.3390/v3112192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dietrich I, Hosie MJ, Willett BJ. The role of BST2/tetherin in feline retrovirus infection. Vet Immunol Immunopathol. 2011;143:255–64. doi: 10.1016/j.vetimm.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zielonka J, Münk C. Cellular restriction factors of feline immunodeficiency virus. Viruses. 2011;3:1986–2005. doi: 10.3390/v3101986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zielonka J, Marino D, Hofmann H, Yuhki N, Löchelt M, Münk C. Vif of feline immunodeficiency virus from domestic cats protects against APOBEC3 restriction factors from many felids. J Virol. 2010;84:7312–24. doi: 10.1128/JVI.00209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murphy B, Vapniarsky N, Hillman C, Castillo D, McDonnel S, Moore P, Luciw PA, Sparger EE. FIV establishes a latent infection in feline peripheral blood CD4+ T lymphocytes in vivo during the asymptomatic phase of infection. Retrovirology. 2012;9:12. doi: 10.1186/1742-4690-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77:6507–19. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lombardi S, Massi C, Tozzini F, Zaccaro L, Bazzichi A, Bandecchi P, La Rosa C, Bendinelli M, Garzelli C. Epitope mapping of the V3 domain of feline immunodeficiency virus envelope glycoprotein by monoclonal antibodies. J Gen Virol. 1995;76(Pt 8):1893–9. doi: 10.1099/0022-1317-76-8-1893. [DOI] [PubMed] [Google Scholar]
- 65.Hu Q-Y, Fink E, Hong Y, Wang C, Grant CK, Elder JH. Fine definition of the CXCR4-binding region on the V3 loop of feline immunodeficiency virus surface glycoprotein. PLoS ONE. 2010;5:e10689. doi: 10.1371/journal.pone.0010689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu Q-Y, Fink E, Happer M, Elder JH. Identification of amino acid residues important for heparan sulfate proteoglycan interaction within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J Virol. 2011;85:7108–17. doi: 10.1128/JVI.00573-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laakso MM, Lee F-H, Haggarty B, Agrawal C, Nolan KM, Biscone M, Romano J, Jordan APO, Leslie GJ, Meissner EG, Su L, Hoxie JA, Doms RW. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 2007;3:e117. doi: 10.1371/journal.ppat.0030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Jong JJ, de Ronde A, Keulen W, Tersmette M, Goudsmit J. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J Virol. 1992;66:6777–80. doi: 10.1128/jvi.66.11.6777-6780.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siebelink KH, Bosch ML, Rimmelzwaan GF, Meloen RH, Osterhaus AD. Two different mutations in the envelope protein of feline immunodeficiency virus allow the virus to escape from neutralization by feline serum antibodies. Vet Immunol Immunopathol. 1995;46:51–9. doi: 10.1016/0165-2427(94)07005-r. [DOI] [PubMed] [Google Scholar]
- 70.Verschoor EJ, Boven LA, Blaak H, van Vliet AL, Horzinek MC, de Ronde A. A single mutation within the V3 envelope neutralization domain of feline immunodeficiency virus determines its tropism for CRFK cells. J Virol. 1995;69:4752–7. doi: 10.1128/jvi.69.8.4752-4757.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Parseval A, Bobardt MD, Chatterji A, Chatterji U, Elder JH, David G, Zolla-Pazner S, Farzan M, Lee T-H, Gallay PA. A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J Biol Chem. 2005;280:39493–504. doi: 10.1074/jbc.M504233200. [DOI] [PubMed] [Google Scholar]
- 72.Shi W, Bohon J, Han DP, Habte H, Qin Y, Cho MW, Chance MR. Structural characterization of HIV gp41 with the membrane-proximal external region. J Biol Chem. 2010;285:24290–8. doi: 10.1074/jbc.M110.111351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cunyat F, Marfil S, García E, Svicher V, Pérez-Alvárez N, Curriu M, Perno CF, Clotet B, Blanco J, Cabrera C. The HR2 polymorphism N140I in the HIV-1 gp41 combined with the HR1 V38A mutation is associated with a less cytopathic phenotype. Retrovirology. 2012;9:15. doi: 10.1186/1742-4690-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ray N, Blackburn LA, Doms RW. HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J Virol. 2009;83:2989–95. doi: 10.1128/JVI.02496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phillips TR, Talbott RL, Lamont C, Muir S, Lovelace K, Elder JH. Comparison of two host cell range variants of feline immunodeficiency virus. J Virol. 1990;64:4605–13. doi: 10.1128/jvi.64.10.4605-4613.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Waters AK, de Parseval AP, Lerner DL, Neil JC, Thompson FJ, Elder JH. Influence of ORF2 on host cell tropism of feline immunodeficiency virus. Virology. 1996;215:10–6. doi: 10.1006/viro.1996.0002. [DOI] [PubMed] [Google Scholar]
- 77.Saenz DT, Poeschla EM. FIV: from lentivirus to lentivector. J Gene Med. 2004;6(Suppl 1):S95–104. doi: 10.1002/jgm.500. [DOI] [PubMed] [Google Scholar]
- 78.Crandell RA, Fabricant CG, Nelson-Rees WA. Development, characterization, and viral susceptibility of a feline (Felis catus) renal cell line (CRFK) In Vitro. 1973;9:176–85. doi: 10.1007/BF02618435. [DOI] [PubMed] [Google Scholar]
- 79.Samelson LE, Harford JB, Klausner RD. Identification of the components of the murine T cell antigen receptor complex. Cell. 1985;43:223–31. doi: 10.1016/0092-8674(85)90027-3. [DOI] [PubMed] [Google Scholar]
- 80.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol. 1994;68:5311–20. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Willey RL, Bonifacino JS, Potts BJ, Martin MA, Klausner RD. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc Natl Acad Sci U S A. 1988;85:9580–4. doi: 10.1073/pnas.85.24.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garg H, Fuller FJ, Tompkins WAF. Mechanism of feline immunodeficiency virus envelope glycoprotein-mediated fusion. Virology. 2004;321:274–86. doi: 10.1016/j.virol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 83.Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Capon DJ, Martin MA. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol. 1988;62:139–47. doi: 10.1128/jvi.62.1.139-147.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Medinas RJ, Lambert DM, Tompkins WAF. C-Terminal gp40 peptide analogs inhibit feline immunodeficiency virus: cell fusion and virus spread. J Virol. 2002;76:9079–86. doi: 10.1128/JVI.76.18.9079-9086.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poeschla EM, Looney DJ. CXCR4 is required by a nonprimate lentivirus: heterologous expression of feline immunodeficiency virus in human, rodent, and feline cells. J Virol. 1998;72:6858–66. doi: 10.1128/jvi.72.8.6858-6866.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Parseval A, Chatterji U, Sun P, Elder JH. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc Natl Acad Sci USA. 2004;101:13044–9. doi: 10.1073/pnas.0404006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Parseval A, Ngo S, Sun P, Elder JH. Factors that increase the effective concentration of CXCR4 dictate feline immunodeficiency virus tropism and kinetics of replication. J Virol. 2004;78:9132–43. doi: 10.1128/JVI.78.17.9132-9143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shimojima M, Miyazawa T, Ikeda Y, McMonagle EL, Haining H, Akashi H, Takeuchi Y, Hosie MJ, Willett BJ. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science. 2004;303:1192–5. doi: 10.1126/science.1092124. [DOI] [PubMed] [Google Scholar]
- 89.Luttge BG, Moyer RW. Suppressors of a host range mutation in the rabbitpox virus serpin SPI-1 map to proteins essential for viral DNA replication. J Virol. 2005;79:9168–79. doi: 10.1128/JVI.79.14.9168-9179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chandramouli B, Chillemi G, Abbate I, Capobianchi MR, Rozera G, Desideri A. Importance of V3 loop flexibility and net charge in the context of co-receptor recognition. A molecular dynamics study on HIV gp120. J Biomol Struct Dyn. 2012;29:879–91. doi: 10.1080/07391102.2012.10507416. [DOI] [PubMed] [Google Scholar]
- 91.Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, Chesebro B, Goldsmith MA. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J Virol. 1997;71:7136–9. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mizukoshi F, Baba K, Goto Y, Setoguchi A, Fujino Y, Ohno K, Oishi S, Kodera Y, Fujii N, Tsujimoto H. Antiviral activity of membrane fusion inhibitors that target gp40 of the feline immunodeficiency virus envelope protein. Vet Microbiol. 2008 doi: 10.1016/j.vetmic.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 93.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91:9770–4. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kilgore NR, Salzwedel K, Reddick M, Allaway GP, Wild CT. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J Virol. 2003;77:7669–72. doi: 10.1128/JVI.77.13.7669-7672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]