

Abstract
Selective activation of Rho GTPase cascade requires the release of Rho from RhoGDI (GDP‐dissociation inhibitors) complexes. Our previous studies identified RhoGDIα SUMOylation at Lys‐138 and its function in the regulation of cancer cell invasion. In the current study, we demonstrate that RhoGDIα SUMOylation has a crucial role in the suppression of cancer cell anchorage‐independent growth as well as the molecular mechanisms underlying this suppression. We found that ectopic expression of RhoGDIα resulted in marked inhibition of an anchorage‐independent growth with induction of G0/G1 cell cycle arrest, while point mutation of RhoGDIα SUMOylation at residue Lys‐138 (K138R) abrogated this growth suppression and G0/G1 cell cycle arrest in cancer cells. Further studies showed that SUMOylation at Lys‐138 was critical for RhoGDIα down‐regulation of cyclin D1 protein expression and that MEK1/2‐Erk was a specific downstream target of SUMOylated RhoGDIα for its inhibition of C‐Jun/AP‐1 cascade, cyclin d1 transcription, and cell cycle progression. These results strongly demonstrate that SUMOylated RhoGDIα suppressed C‐Jun/AP‐1‐dependent transactivation specifically via targeting MEK1/2‐Erk, subsequently leading to the down‐regulation of cyclin D1 expression and anti‐cancer activity. Our results provide new mechanistic insights into the understanding of essential role of SUMOylation at Lys‐138 in RhoGDIα's biological function.
Keywords: RhoGDIα, SUMOylation, Cyclin D1, Growth arrest, Anchorage‐independent growth
Highlights
SUMOylation at Lys‐138 has a crucial role in RhoGDIα's suppression of cancer cell anchorage‐independent growth.
SUMOylation of RhoGDIα is critical for its down‐regulation of cell cycle transition and cyclin d1 transcription.
MEK1/2‐Erk/AP‐1 is a specific downstream target of SUMOylated RhoGDIα for its inhibition of cyclin d1 transcription.
Abbreviations
- CDK
cyclin-dependent kinase
- Erk
extracellular stimuli mitogen-activated protein kinase
- GDIs
GDP-dissociation inhibitors
- GEFs
guanine nucleotide exchanging factors
- GTP
guanosine triphosphate
- MEK
MAP kinase
- SUMO
small ubiquitin-related modifier
1. Introduction
Cell cycle progression is controlled by the expression of a family of proteins called cyclins (Sherr, 1994). Cyclin D1 is the major D type cyclin in many cell types and is usually the first cyclin to be induced when cells enter the G1 phase from quiescence (G0) (Stacey, 2003). Once expressed, cyclin D1 binds to CDK4 (cyclin‐dependent kinase 4) or CDK6 to form an active holoenzyme that phosphorylates the retinoblastoma 1 (Rb1) protein. Phosphorylation of Rb1 results in its dissociation with E2Fs and then promotes transcription of E2F downstream targets, such as cyclin E and cyclin A, and further resulting in cell cycle progression (Stacey, 2003). Therefore, tight control of cyclin D1 gene expression is a crucial issue in the regulation of G1‐phase progression. In non‐tumor cells, the cyclin D1 gene senses the mitogenic potential of the microenvironment during cell‐cycle entry from quiescence because its induction requires coordinated signaling from the extracellular matrix and soluble growth factors (Assoian and Klein, 2008). These controls can be lost during cellular transformation upon exposure to carcinogens or tumor promoters, resulting in the corresponding overexpression of cyclin D1 in a number of cancers, including those of the breast, liver, lung, and colon (Gillett et al., 1996; Yamamoto et al., 2006; Molenaar et al., 2008; Sanchez‐Mora et al., 2008). Cyclin D1 levels can be regulated in a transcriptional and post‐transcriptional manner (Zhang et al., 2012; Ouyang et al., 2005).
The Rho family contains 20 members (Vega and Ridley, 2008), and over 60 Rho effectors have been identified (Lu et al., 2009). The important role of the Rho family of small GTP (Guanosine Triphosphate) binding proteins in cancer development has been well established (Sahai and Marshall, 2002). The members of the Rho GTPase family are well known for their regulation of actin polymerization and cytoskeletal structures (Vega and Ridley, 2008) and they also contribute to the regulation of many different biological processes, including cell cycle progression with the best‐studied members being RhoA, Rac1, and Cdc42 (Coleman et al., 2004). It has previously been shown that active Rho influences cell cycle progression via the regulation of a number of cell cycle regulatory proteins (Etienne‐Manneville and Hall, 2002). One mechanism is through regulating expression and activity of CDK inhibitors, such as p27 (Hirai et al., 1997; Hu et al., 1999) and p21 (Liberto et al., 2002; Olson et al., 1998). In addition to the regulation of CDK inhibitors, Rho has been reported to influence cyclin levels. Rho and ROCK are necessary for Ras‐GTP loading and lead to increased to increased cyclin D1 transcription following growth factor stimulation (Swant et al., 2005; Welsh et al., 2001). Inhibition of Rho or ROCK function inhibits cyclin A expression and blocks cell proliferation in atrial myofibroblasts (Porter et al., 2004; Croft and Olson, 2006). Moreover, Rho GTPase activity is necessary for cyclin E expression in rat astrocytes (Tanaka et al., 1998).
RhoGDIs have been identified as key regulators of Rho family GTPases as typified by their ability to prevent nucleotide exchange and membrane association (Garcia‐Mata et al., 2011). In resting state, Rho family members are bound to RhoGDIs preventing the conversion from their inactive GDP bound state to the active GTP bound state (Faure and Dagher, 2001; DerMardirossian and Bokoch, 2005). There are three isoforms of RhoGDIs: RhoGDIα, β (also named D4/LyGDI), and γ. Among these RhoGDIs, RhoGDIα is ubiquitously expressed and binds to all of the Rho family proteins thus far examined (Dovas and Couchman, 2005), whereas RhoGDIβ and γ show unique tissue expression patterns, and their substrate specificities have not been precisely determined (Harding and Theodorescu, 2010). RhoGDIs regulate a multitude of cellular phenotypes including cell division, morphology, migration, vesicular trafficking and gene expression (Harding and Theodorescu, 2010). It is likely that they affect these diverse phenotypes principally by controlling the location and activity of members of the Rho family of small GTPases (Dovas and Couchman, 2005). Indeed, there is substantial biochemical and structural evidence showing that Rho GEFs (Guaninenucleotide Exchanging Factors) that cannot act on Rho GTPases form complex with RhoGDI. The GDI‐GTPase complex is thus a major form of RhoGDI regulation of Rho GTPase activity and function (DerMardirossian and Bokoch, 2005).
SUMO (Small Ubiquitin‐related Modifier) modification (SUMOylation) is an important post‐translational protein modification that modulates the biological functions of proteins (Muller et al., 2004; Geiss‐Friedlander and Melchior, 2007). SUMOylation is a highly dynamic process with a three‐step reaction consisting of SUMO activation, transfer, and ligation that are catalyzed by E1 heterodimeric enzyme [SAE1 (SUMO Activating Enzyme E1)/SAE2], E2 enzyme (Ubc9), and E3 SUMO ligases (Melchior, 2000; Kotaja et al., 2002). Unlike ubiquitination, which usually facilitates protein degradation, SUMOylation results in pleiotropic functional consequences that include changes in subcellular localization, protein stability, alterations in DNA binding, and transcriptional activity (Johnson, 2004; Gill, 2005). Transcription factors, co‐activators, and co‐repressors are predominant targets of SUMOylation, which alters their activity and results in changes in gene expression and function (Johnson, 2004; Gill, 2005). Our most recently studies demonstrate that RhoGDIα can be SUMOylated specifically at residue Lys‐138 and that this SUMOylation is crucial for RhoGDIα inhibition of cancer cell motility and invasion (Yu et al., 2012). In the current study, we aimed to investigate the potential biological role of RhoGDIα SUMOylation at Lys‐138 in RhoGDIα regulation of cancer cell anchorage‐independent growth and that molecular mechanisms that lead to this regulation. We found that loss of RhoGDIα SUMOylation by specific K138R point mutation of RhoGDIα at Lys138 impaired its ability to repress cyclin D1 expression and attenuated its induction of G0/G1 growth arrest and inhibition of anchorage‐independent growth. Further studies demonstrated that the suppression of RhoGDIα SUMOylation at Lys‐138 was mediated by its inhibition of MEK1/2/Erk/AP‐1 cascade.
2. Materials and methods
2.1. Cell culture and plasmids
HCT116 cells and its transfectants, including HCT116 (GFP‐vector), HCT116(RhoGDIα‐WT), HCT116(RhoGDIα‐K105R), and HCT116 (RhoGDIα‐K138R) were established in our previous studies (Yu et al., 2012). The HCT116 cells were cultured in McCoy's 5A medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 2 mM L‐glutamine (Life Technologies, Inc., Rockville, MD) at 37 °C in 5% CO2 incubator. The constructs of ‐963 cyclin D1 promoter‐driven luciferase reporter (‐963 CD1 Luc) and its AP‐1 binding site mutant reporter (‐963 AP‐1mut CD1 Luc) were gifts from Dr. Richard G Pestell, Thomas Jefferson University Jefferson Medical College. The plasmid TAM67, dominant negative C‐Jun mutant, dominant negative Erk1 mutant (Erk1‐K71R), cyclin d1 promoter‐driven luciferase reporter, and AP‐1‐dependent luciferase reporter were described in our previously studies (Ouyang et al., 2005; Zhang et al., 2006; Huang et al., 1999; Wang et al., 2005).
2.2. Antibodies and other reagents
Antibodies against GFP, phospho‐C‐Jun, C‐Jun, cyclin A, phospho‐p65, p65, phospho‐STAT3, STAT3, phospho‐STAT5, STAT5, phospho‐JNKs, JNKs, phospho‐Erks, Erks, and GAPDH were purchased from Cell Signaling Technology Inc. (Beverly, MA); against cyclin D1, CDK4, CDK6, cyclin E, c‐Fos, Jun‐B, Jun‐D, SP1 and ubiquitin were bought from Santa Cruz Biotechnology (Santa Cruz, CA); against p27 and p50 were purchased from Abcam (Cambridge, MA); against RhoGDIα was from Millipore (Billerica, MA). The kinase inhibitor PD98059 was obtained from Sigma Chemical (St. Louis, MO).
2.3. Cell transfection
All of the stable and transient transfections were performed with PolyJet™ DNA in vitro transfection reagent (SignaGen Laboratories, Rockville, MD), according to manufacturer's instructions. For stable transfection, cultures were subjected to either blasticidin or hygromycin B drug selection and cells surviving from the selection were pooled as stable mass cultures (Fang et al., 2012). These stable transfectants were cultured in the selected antibiotic‐free medium for at least two passages before utilization for experiments.
2.4. Anchorage‐independent growth assay
Anchorage‐independent growth ability was determined in soft agar as described in our previous studies (Luo et al., 2008). Briefly, 3 ml of 0.5% agar in basal modified Eagle's medium supplemented with 10% FBS was layered onto each well of 6‐well tissue culture plates. Cell suspensions (1 ml, 1 × 104 cells/well) were mixed with 2 ml of 0.5% agar‐basal modified Eagle's medium supplemented with 10% FBS, and 1 ml of mixture was added into each well over top of the 0.5% agar layer. Plates were incubated at 37 °C in 5% CO2 for 2–3 weeks, and the colonies with more than 32 cells of each were scored and presented as colonies/104 cells.
2.5. Cell cycle assay
After indicated treatment, cells were stained with propidium iodide (PI) solution, as mentioned in the previous report (Song et al., 2007). Cell cycle distribution was determined by flow cytometry, utilizing a Beckman–Coulter EpicsXL flow cytometer. Twenty thousand events were counted for each analysis, and two to four independent experiments were conducted in each group.
2.6. Western blot
Cell extracts were prepared with cell lysis buffer (10 mM Tris–HCl, pH 7.4, 1% SDS, and 1 mM Na3VO4). Protein concentrations were determined by NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE). Proteins (30–60 μg) were subjected to SDS‐PAGE and were subsequently probed with the indicated primary antibodies and AP‐conjugated second antibody (Cell Signaling Technology, Boston, MA), as described in our publications (Zhang et al., 2006; Ouyang et al., 2008). Signals were detected by the enhanced chemifluorescence Western blot system (Model Storm 860, Molecular Dynamics, Kent City, MI) as described in our previous publications (Song et al., 2006, 2007).
2.7. Reverse transcription polymerase chain reaction (RT‐PCR)
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA), and cDNAs were synthesized with the ThermoScript™ RT‐PCR system (Invitrogen). To detect cyclin d1 induction, a pair of oligonucleotides (5′‐GAG GTC TGC GAG GAA CAG AAG TG‐3′ and 5′‐GAG GGC GGA TGG AAA TGA ACT TCA‐3′) were synthesized and used as the specific primers. Human gapdh cDNA was amplified by the primers (5′‐AGA AGG CTG GGG CTC ATT TG‐3′ and 5′‐AGG GGC CAT CCA CAG TCT TC‐3′).
2.8. Luciferase reporter assay
Cells stably transfected with luciferase‐reporter constructs were seeded into 96‐well plates. After the cell density reached 70–80%, cells were treated as indicated in the figure legends, and were then extracted with luciferase assay lysis buffer (Promega, Madison, WI). The luciferase activity was determined with the Dual‐Luciferase Reporter Assay System (Promega), according to the manufacturer's instructions as described (Li et al., 2004; Huang et al., 1997).
2.9. Immunoprecipitation
Cells were lysed in cell lysis buffer (1% Triton X‐100, 150 mM Nacl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, 0.2 mM Na3VO4, 0.5% NP‐40, and complete protein cocktail inhibitors from Roche) on ice. Lysate (0.5 mg) was incubated with Protein A/G plus‐agarose (Santa Cruz Biotechnology, Inc.) and then incubated with specific antibody at 4 °C for 12 h. Protein A/G plus‐agarose (40 μl) were added to the mixture and incubated with agitation for an additional 4 h at 4 °C. The immunoprecipitate was washed three times with cell lysis buffer and subjected to Western Blotting assay (Liu et al., 2012).
2.10. Statistical methods
Student's t‐test was employed to determine the significance of differences between the different groups in each experiment. The differences will be considered significant at p < 0.05.
3. Results
3.1. SUMOylation of RhoGDIα at Lys138 was required for its suppression of anchorage‐independent growth in cancer cells
To delineate the biological significance of RhoGDIα SUMOylation at Lys138 in cancer cell growth, we established stable polyclonal cell lines that overexpress GFP‐vector or GFP fusions with RhoGDIα‐WT, RhoGDIα‐K138R (SUMOylation site point mutation), and RhoGDIα‐K105R (non‐SUMOylation site point mutation as control). Western blot analysis showed equivalent expressions of RhoGDIα‐WT, RhoGDIα‐K105R, and RhoGDIα‐K138R in the various stable transfectants (Figure 1A). Since SUMO often competes with ubiquitin for the same lysine residues (Park et al., 2007), we examined whether RhoGDIα‐K138R mutation affected RhoGDIα ubiquitination. The results showed that there was no difference of RhoGDIα ubiquitination between the transfectants of RhoGDIα‐WT and RhoGDIα‐K138R (Figure 1B).
Figure 1.
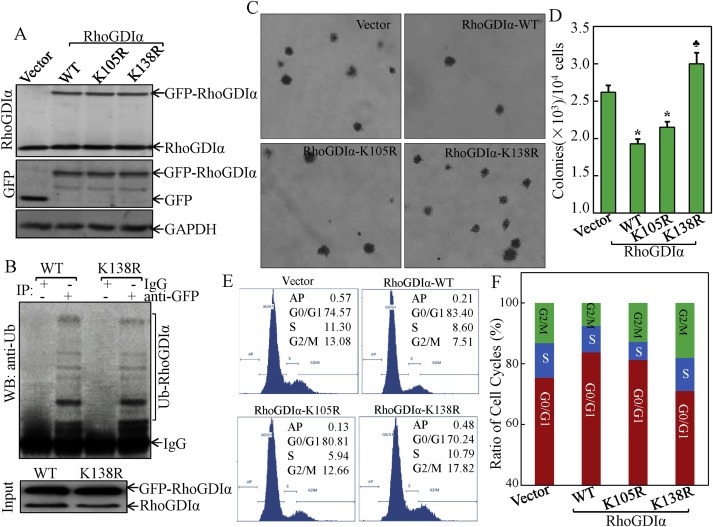
RhoGDIα SUMOylation at Lys‐138 was essential for RhoGDIα inhibition of cancer cell anchorage‐independent growth and cell cycle progression. (A) stable HCT116 cell transfectants of GFP‐RhoGDIα‐WT, GFP‐RhoGDIα‐K138R, GFP‐RhoGDIα‐K105R, and the empty control vector, were identified by Western blot. (B) HCT116 cells were transfected with various constructs as indicated for detection of RhoGDIα ubiquitination. (C & D) anchorage‐independent cell growth of above stable transfectants was determined by soft agar assay. The colony formation was observed under an inverted microscope and photographed (C) and numbers of colonies were scored, and presented as colonies per 10,000 seeded cells (D). The symbol (*) indicates a significant repression in comparison to that of the control vector transfectant (p < 0.05); and (♣) indicates a significant increase compared with control vector transfectant (p < 0.05). Each bar indicates the mean and standard derivation of three independent experiments. (E & F) Cell cycle profile was determined by PI staining and FACS analysis in the transfectants of the control vector, RhoGDIα‐WT, RhoGDIα‐K105R, and RhoGDIα‐K138R. Representative histograms of cell cycle profiles were presented from three independent experiments (E). Percentage of cells in the G0/G1, S, and G2/M phases were presented from three independent experiments (F).
We performed anchorage‐independent cell growth assay with these stable transfectants. The results indicated that overexpression of RhoGDIα‐WT resulted in significant reduction in colony numbers as compared with those in control vector‐transfected cells (Figure 1C). Interestingly, in contrast to RhoGDIα‐WT, cells stably transfected with RhoGDIα‐K138R showed an increase in colony formation, while those transfected with RhoGDIα‐K105R showed a similar inhibition on colony formation (Figure 1D), suggesting that SUMOylation of RhoGDIα at Lys138 was essential for its inhibition of anchorage‐independent cancer cell growth. To examine the underlying mechanisms, we then examined the cell cycle profile of HCT116 cells stable transfectants of RhoGDIα‐WT, RhoGDIα‐K105R, and RhoGDIα‐K138R by flow cytometry (Figure 1E). Ectopic expression of RhoGDIα‐WT exhibited delayed progression in the G1/S phase of the cell cycle, resulting in G0/G1 arrest (83.40% ± 1.99 vs 74.57% ± 1.23) in comparison to vector control transfectant, whereas the cell cycle profile of RhoGDIα‐K138R transfectant did not show this delayed progression in the G0/G1 phase (70.24% ± 1.94 vs 74.57% ± 1.23) (Figure 1E and F). These results suggested that SUMOylation at K138 was essential for RhoGDIα inhibition of anchorage‐independent cancer cell growth and that this RhoGDIα inhibition might be associated with its effect on G0/G1 arrest.
3.2. RhoGDIα SUMOylation at Lys138 mediated its inhibition of cyclin d1 transcription
To examine the molecular basis underlying RhoGDIα SUMOylation‐regulated cancer cell growth, we next examined the potential effects of RhoGDIα SUMOylation‐regulated the expression levels of key cell cycle regulatory proteins. The results showed that ectopic expression of RhoGDIα‐WT or RhoGDIα‐K105R significantly inhibited cyclin D1 expression in HCT116 cells, while RhoGDIα‐K138R did not show any observable inhibition of cyclin D1 protein expression (Figure 2A). Moreover, none of the RhoGDIα‐WT or its mutants showed an observable effect on the expression of other cell cycle regulators, including p21, p27, CDK4, CDK6, cyclin E, and cyclin A. These results suggested that SUMOylated RhoGDIα inhibited cancer cell growth and induced G0/G1 growth arrest specific associated with the reduction of cyclin D1 expression.
Figure 2.
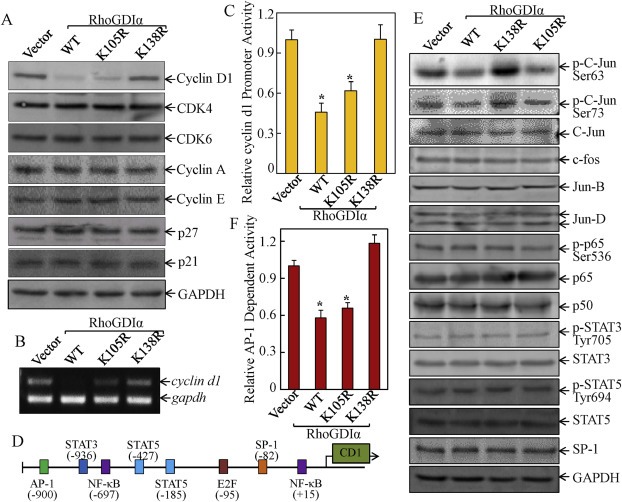
Point mutation of the RhoGDIα at SUMOylation site Lys‐138 lost its suppression of cyclin d1 transcription. (A & B) The cells were synchronized by incubation of cells with 0.1% FBS medium for 24 h. The cells were then cultured in 2% FBS medium for 24 h. Cells were then extracted for determination of protein expression by Western blot (A) or cyclin d1 mRNA expression by RT‐PCR (B). (C) cells (1 × 104) stably transfected with cyclin d1 promoter‐driven luciferase reporter were seeded into each well of a 96‐well plate. After synchronization, the cells were extracted for determination of the luciferase activity, as described in our previous studies (Ouyang et al., 2008). The symbol (*) indicates a significant repression in comparison to that of the control vector transfectant (p < 0.05); (D) schematic representation of transcription factor binding sites in human cyclin d1 promoter region (Klein and Assoian, 2008). (E) Western blot was performed to determine the transcription factor expression and activation in the indicated transfectants. (F) the indicated transfectants that were stably transfected with AP‐1 luciferase reporter (1 × 104) were seeded into a 96‐well plate and subjected to luciferase activity assay, as described in Materials and Methods. The results were presented as luciferase activity relative to medium control (relative AP‐1 activity). The symbol (*) indicates a significant repression in comparison to the control vector transfectant (p < 0.05).
Since cyclin D1 levels can be regulated transcriptionally and post‐transcriptionally (Zhang et al., 2012; Ouyang et al., 2005; Musgrove, 2006), we evaluated cyclin d1 mRNA levels among various transfectants. Consistent with cyclin D1 protein expression, cyclin d1 mRNA expression was profoundly downregulated in cells transfected with RhoGDIα‐WT and RhoGDIα‐K105R, while RhoGDIα‐K138R did not show an inhibitory effect on cyclin d1 mRNA (Figure 2B). Furthermore, cyclin d1 promoter‐driven luciferase reporter (Ouyang et al., 2005) was co‐transfected with RhoGDIα‐WT, RhoGDIα‐K105R, RhoGDIα‐K138R, or control vector into HCT116 cells, respectively, and the transfectants was used to test whether RhoGDIα SUMOylation regulated cyclin d1 mRNA at the transcriptional level. Consistent with repression of cyclin d1 mRNA levels, RhoGDIα‐WT and RhoGDIα‐K105R repressed the cyclin d1 promoter activity, as compared with control vector transfectant, and RhoGDIα‐K138R did not show this suppression (Figure 2C), indicating that RhoGDIα SUMOylation downregulated cyclin d1 transcription.
3.3. Transcription factor C‐Jun/AP‐1 was downstream mediator of RhoGDIα SUMOylation for repressing cyclin d1 transcription
To identify the transcription factor(s) responsible for RhoGDIα SUMOylation‐regulated cyclin d1 transcription, TFANSFAC® Transcription Factor Binding Sites Software (Biological Database, Wolfenbüttel, Germany) was used for bioinformatics analysis of the cyclin d1 promoter region. The results revealed that the promoter region of the human cyclin d1 gene contains the multiple putative DNA‐binding sites of transcription factors, including NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells), STAT3/5 (signal transducers and activators of transcription), AP‐1 (activator protein‐1), E2F1, and SP‐1 (specificity protein‐1) (Figure 2D). We next examined changes in the expression of related transcription factors or their activated form among the transfectants of RhoGDIα variants. The results are consistent with cyclin d1 transcription, showing that inhibition of C‐Jun phosphorylation and AP‐1‐dependent transactivation was clearly observed in the transfectants of RhoGDIα‐WT and RhoGDIα‐K105R, whereas point mutation of RhoGDIα at Lys138 (RhoGDIα‐K138R) abolished this inhibitory effect (Figure 2E and F). Significantly, there were no observed effects on other putative transcription factors, such as c‐Fos, C‐Jun, NF‐κB (phosphorylated p65, p65 and p50), STAT3, STAT5, and SP‐1 by ectopic expression of either RhoGDIα‐WT, RhoGDIα‐K105R, or RhoGDIα‐K138R. Our results revealed that C‐Jun/AP‐1 might be a SUMOylated RhoGDIα downstream target for mediating inhibition of cyclin d1 transcription.
To determine the role of C‐Jun/AP‐1 in cancer cell growth repression by SUMOylated RhoGDIα, we overexpressed the dominant negative mutant form of C‐Jun (TAM67) (Zhang et al., 2006) in HCT116 stable RhoGDIα‐K138R transfectant to abolish C‐Jun activation (Figure 3A). Inhibition of C‐Jun activation by TAM67 attenuated cyclin D1 protein expression (Figure 3A). TAM67 overexpression also impaired cyclin d1 promoter activity in RhoGDIα‐K138R transfectant (Figure 3B). To further validate the important role of C‐Jun/AP‐1 in mediating cyclin d1 transcription activity, we transfected cyclin d1 promoter luciferase reporter (‐963 CD1‐Luc) and ‐963 CD1‐Luc reporter with AP‐1 binding site mutation (‐963 AP‐1mut CD1‐Luc) into RhoGDIα‐K138R transfectant. As shown in Figure 3C, cyclin d1 promoter transcription activity was dramatically reduced in the transfectant of ‐963 AP‐1mut CD1‐Luc in comparison to that in ‐963 CD1‐Luc transfectant in RhoGDIα‐K138R transfectant (Figure 3C), indicating that AP‐1 transactivation is crucial for cyclin d1 transcription in cancer cells. Consistent with the inhibition of cyclin D1 expression, ectopic expression of TAM67 also blocked G1/S phase transition in RhoGDIα‐K138R transfectant (Figure 3D). These results demonstrated that C‐Jun/AP‐1 was crucial for RhoGDIα SUMOylation‐dependent repression of cyclin d1 transcription.
Figure 3.
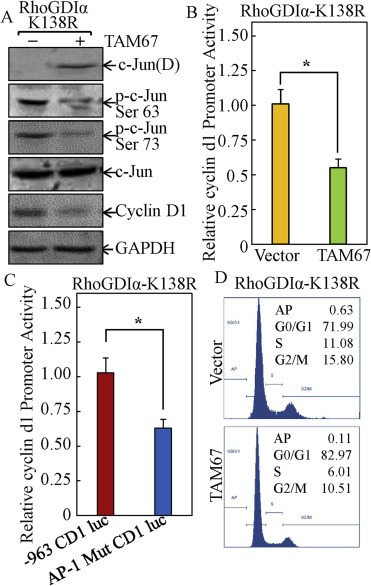
The C‐Jun/AP‐1 pathway mediated SUMOylated RhoGDIα‐repressed cyclin d1 transcription and expression, as well as cell cycle regulation. (A) the RhoGDIα‐K138R cells stably transfected with either TAM67 construct or the control empty vector were cultured in 2% FBS medium for 24 h and the cell extracts were subjected to Western blot. (B) the RhoGDIα‐K138R stable transfectants were established by co‐transfection of cyclin d1 promoter luciferase reporter with TAM67 and blasticidin selection. After synchronization, cells were cultured in 2% FBS medium for 24 h and the cells were then extracted for determination of luciferase activity. The symbol (*) indicates a significant repression in comparison to that of the control vector transfectant (p < 0.05); (C) the RhoGDIα‐K138R cells' stable transfectants were established by transfection of ‐963 CD1 Luc and ‐963 AP‐1mut CD1 Luc and blasticidin selection. After synchronization, cells were cultured in 2% FBS medium for 24 h, and the cells were then extracted for determination of luciferase activity. The symbol (*) indicates a significant repression as compared with that of ‐963 CD1 Luc reporter transfectant (p < 0.05); (D) the cell cycle profile in the indicated cell transfectants was determined by flow cytometry analysis.
3.4. SUMOylated RhoGDIα suppression of AP‐1/cyclin D1 transcription and growth arrest was specific mediated by targeting Erks, but not JNKs
To investigate the possible upstream kinase cascade involved in the SUMOylated RhoGDIα inhibition of AP‐1 activation and cyclin D1 expression, we determined Erks and JNK (C‐Jun N‐terminal kinases), two well‐known MAPKs responsible for AP‐1 activation. The results showed that phosphorylation of Erks and not JNKs, was markedly decreased in either RhoGDIα‐WT or RhoGDIα‐K105R transfectants. However, the level of phosphorylated Erk in RhoGDIα‐K138R transfectant was similar to that observed in control vector transfectant (Figure 4A). Moreover, our results also indicated that SUMOylated RhoGDIα inhibited phosphorylation of MEK1/2 (Figure 4A), an upstream kinase mediating Erks phosphorylation and activation. Our results indicated that SUMOylation at Lys‐138 was essential for RhoGDIα repression of MEK1/2/Erk cascade.
Figure 4.
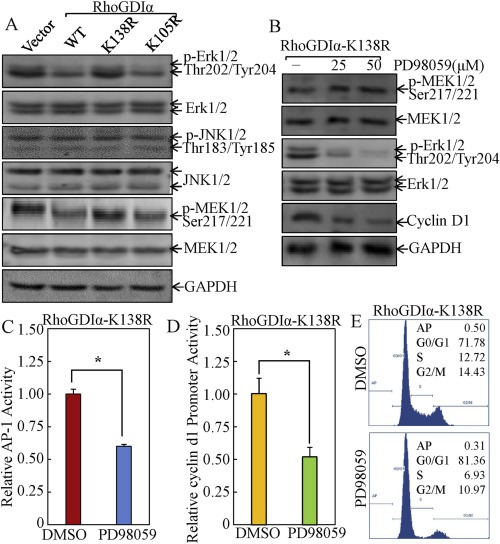
MEK1/2‐Erk cascade was specifically targeted by SUMOylated RhoGDIα for repression of C‐Jun/AP‐1 activation, cyclin D1 expression, and cancer cell cycle progression. (A) after synchronization, the indicated cells were cultured in 2% FBS medium for 12 h and the cell extracts were subjected to Western blot assay. (B) the RhoGDIα‐K138R cells were treated with 25 μM or 50 μM of PD98059 or control medium containing 0.1% DMSO for 12 h and the cell extracts were subjected to Western blot analysis. (C) the indicated AP‐1‐luciferase reporter stable transfectants were treated with PD98059 (50 μM) or control medium containing 0.1% DMSO and the cells were then subjected to luciferase activity assay. The results were presented as luciferase activity relative to medium control (relative AP‐1 activity). The symbol (*) indicates a significant repression in comparison to that in cells without PD98059 treatment (p < 0.05). (D) the indicated stable transfectants of cyclin d1 promoter‐driven luciferase reporter were treated with PD98059 (50 μM) or control medium containing 0.1% DMSO and the cells were then subjected to luciferase activity assay. The results were presented as luciferase activity relative to medium control (relative cyclin d1 promoter activity). The symbol (*) indicates a significant repression as compared with that in cells without PD98059 treatment (p < 0.05). (E), with PD98059 (50 μM) or control medium containing 0.1% DMSO and the cells were then subjected to flow cytometry analysis of cell cycle progression.
To examine whether the inhibition of Erks by SUMOylated RhoGDIα was a key factor for RhoGDIα‐mediated repression of AP‐1 activation, cyclin D1 expression, and cancer cell growth, RhoGDIα‐K138R transfectant were treated with PD98059 (25–50 μM), an inhibitor specific for inhibiting MEK1/2 kinase enzyme activity. This was followed by determination of AP‐1 activation, cyclin d1 transcription and protein expression, as well as cell cycle alteration. The results indicated that inhibition of Erk activation by PD98059 impaired AP‐1 activation, downregulation of cyclin d1 promoter activity, and protein expression in RhoGDIα‐K138R transfectant (Figure 4B–D). Consistently, the PD98059 treatment also increased G0/G1‐phase cells from 71.8% ± 1.4 to 81.4% ± 2.0 in RhoGDIα‐K138R transfectant (Figure 4E). To further evaluate the role of Erks in RhoGDIα‐regulated biological effects, a dominant negative mutant of Erk1 (Huang et al., 1999) was employed. As shown in Figure 5, ectopic expression of dominant negative mutant of Erk1 (DN‐Erk1) consistently reversed AP‐1 transcription activation, cyclin d1 promoter activity and protein expression, as well as G0/G1 cell cycle progression in RhoGDIα‐K138R transfectant, implicating that SUMOylation‐dependent repression role of RhoGDIα in AP‐1 activation, cyclin D1 expression, cell cycle progression in colon cancer HCT116 cells was specific via Erk pathway.
Figure 5.
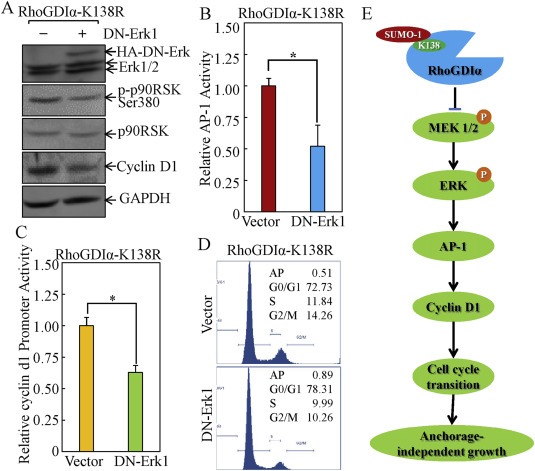
Ectopic expression with DN‐Erk1 reversed the AP‐1 transactivation, cyclin D1 transcription and expression, and cancer cell cycle progression in RhoGDIα‐K138R stable transfectant. (A) indicated stable transfectants were cultured in 2% FBS medium for 12 h and the cell extracts were subjected to Western blot. (B) the indicated cells were stably co‐transfected with AP‐1‐luciferase reporter and DN‐Erk1, then subjected to luciferase activity assay. The results were presented as luciferase activity relative to medium control (relative AP‐1 activity). The symbol (*) indicates a significant repression in comparison to that in the cells without DN‐Erk1 transfection (p < 0.05). (C) indicated cells were stably co‐transfected with cyclin d1 promoter‐luciferase reporter and DN‐Erk1, then subjected to luciferase activity assay. The results were presented as luciferase activity relative to medium control (relative cyclin d1 promoter activity). The symbol (*) indicates a significant repression in comparison to that in cells without DN‐Erk1 transfection (p < 0.05). (D) the Indicated cells were stably co‐transfected with cyclin d1 promoter‐luciferase reporter and DN‐Erk1, then subjected to flow cytometry analysis. (E) A model for RhoGDIα SUMOylation at Lys‐138 regulated modulation of cancer cell anchorage‐independent growth.
4. Discussion
The Rho GTPase family is well known for its regulation of actin polymerization and cytoskeletal structures (Vega and Ridley, 2008), and its role in regulation of G1 cell cycle progression has also been reported (Van Aelst and D'Souza‐Schorey, 1997). Rho GDP dissociation inhibitors (RhoGDIs), endogenous inhibitors of Rho GTPases, play an important role in regulating the biological activities of Rho GTPases (Sasaki and Takai, 1998). Since our most recently studies demonstrate that RhoGDIα can be SUMOylated at Lys138 and that this SUMOylation is crucial for RhoGDIα inhibition of cancer cell invasion (Yu et al., 2012), here we demonstrated that RhoGDIα SUMOylation at Lys138 was required for its inhibition of cyclin D1 expression and cell cycle G1 progression, as well as anchorage‐independent growth in human cancer cells. Our study also identified that the MEK1/2/Erk/AP‐1 cascade was a downstream target of SUMOylated RhoGDIα for mediation of this inhibition in cancer cells.
Protein SUMOylation is an important mechanism for modulation of cellular function (Muller et al., 2001). As SUMO modification is a reversible and highly dynamic process, the SUMOylation status of some target proteins changes markedly in response to various stimuli (Dadke et al., 2007). Recent studies have revealed that the presence of SUMOylated proteins occurs not only in the nucleus, but also in other cellular compartments, including the cytoplasm, mitochondria, endoplasmic reticulum and the plasma membrane (Geiss‐Friedlander and Melchior, 2007). RhoGDIα have been proven to negatively regulate the activities of small G proteins of the Rho family by shutting off their GDP/GTP cycling and cytosol/membrane translocation (Snyder et al., 2002; Hoffman et al., 2000). The Rho family of proteins cycle between active GTP‐bound form to function as molecular switch to regulate the downstream signal transduction process (Tzima, 2006). The essential role of RhoGDIs is to form the Rho/RhoGDIs complex, thus inhibiting guanine nucleotide exchange factors that stimulate GDP/GTP exchange (Snyder et al., 2002). RhoGDIs also shut down the activity of Rho proteins by keeping them in the cytosol, where these proteins are attached by an isoprenoid moiety located at their C terminus (Hoffman et al., 2000). It has been reported that Rho family proteins participate in the regulation of polarity, proliferation, adhesion, spreading, migration, and cytoskeleton organization (Etienne‐Manneville and Hall, 2002). In this study, we established stable transfectants that overexpressed GFP or GFP fusions with RhoGDIα‐WT or its point mutants. Western blot showed comparable expressions of RhoGDIα‐WT, RhoGDIα‐K105R, and RhoGDIα‐K138R in the various stable transfectants (Figure 1A) and the exogenous protein was present at similar levels to endogenous RhoGDIα. RhoGDIα overexpression did not decrease Rho GTPases activity in the studies from other groups (Moissoglu et al., 2009; Wang and Thurmond, 2010) and in our group (Yu et al., 2012). Moreover, RhoGDIα‐K138R mutation did not result in changed its ubiquitination status (Figure 1B). Our studies identified that site‐directed mutagenesis of RhoGDIα SUMOylation site at Lys138 not only lost its inhibition of the MEK/Erk/C‐Jun/AP‐1/cyclin D1 cascade, subsequently reducing G0/G1 cell cycle arrest and promotion of cancer cell anchorage‐independent growth, it also exhibited a slight dominant negative effect on the signaling cascade and biological effect. These findings strongly indicate that SUMOylation as a key modification of RhoGDIα that impacts RhoGDIα‐mediated cyclin D1 repression with an overt impact on its function in cancer cell cycle progression and cancer properties.
Cyclin d1 gene induction is a key event in G1 phase progression. The levels of cyclin d1 mRNA and protein are low in quiescent cells and increase during progression through G1 phase (Welsh et al., 2001; Bohmer et al., 1996). The increase in cyclin d1 mRNA requires cooperative signaling by growth factor receptors and adhesion receptors (Assoian and Schwartz, 2001). Erk activity has also been linked to cyclin d1 gene expression in several cell types (Ramakrishnan et al., 1998; Talarmin et al., 1999; Watanabe et al., 1996). The Erk‐MAPK pathway mediates mitogenic signaling and is essential for the control of cell fate, differentiation and proliferation (Fang and Richardson, 2005). Erk signaling is initiated by activation of cell‐surface‐receptor tyrosine kinases that, in turn induce the small G protein Ras to exchange guanosine diphosphate (GDP) for guanosine triphosphate (GTP). The Raf family of MAPKKK is recruited to the plasma membrane and is activated by GTP‐bound Ras through a multistage process including phosphorylation and dimerization (Rajakulendran et al., 2009; Heidorn et al., 2010; Hatzivassiliou et al., 2010). Activated Raf phosphorylates and activates MEK1/2, which then activates Erk. Activated Erk translocates to the nucleus where it activates transcription factors, such as AP‐1, to induce the expression of growth‐promoting genes such as that encoding cyclin d1 (Balmanno and Cook, 1999; Cook et al., 1999). Genetic alterations resulting in constitutive Erk activation are frequently observed in cancer patients. For example, Ras proteins are activated by mutations in approximately 30% of all human cancers (Malumbres and Barbacid, 2003; Schubbert et al., 2007; Young et al., 2009).
Although most SUMO targets are nuclear proteins, a number of non‐nuclear substrates have recently been reported, many of which are signaling molecules. For instance, SUMOylation regulates the subcellular localization of Dictyostelium DdMEK1 as well as activities of the human type I TGF (transforming growth factor)‐receptor and protein tyrosine phosphatase 1B (Dadke et al., 2007; Desterro et al., 1998; Sobko et al., 2002). Thus, altered SUMOylation of intracellular signaling modules may be associated with pathological conditions that include carcinogenesis. Indeed, it has been shown that oncogenic Ras inhibits MEK SUMOylation to enhance cell transformation (Kubota et al., 2011). Unlike other oncogenes in the Erk pathway, hyperactive Ras alone can both initiate and sensitize the Erk cascade, potentially explaining the striking etiological feature of Ras oncogenes in human cancers. Given the well‐defined role of the Erk pathway in tumor growth, therapeutic targeting of Erk signaling components is an area of intense investigation.
The Erk1/2‐MAPK is evolutionarily conserved signaling module by which cells transduce extracellular signals into intracellular responses, and is essential for the control of cell death, differentiation, and proliferation (Park et al., 2007). The Raf has been extensively studied as the upstream kinase linking Ras activation to the MEK phosphorylation. Raf has also been implicated as a downstream target of the small GTPase Rac (Leng et al., 1999). Rac regulates Raf‐1 activation via its downstream target PAK, which phosphorylates Raf‐1 at serine 338 (King et al., 1998; Li et al., 2001). A role of Rho in Raf activation has also been described, but the effectors are unknown (Li et al., 2001). Furthermore, organization of the actin cytoskeleton is implicated in MEK autophosphorylation through Raf‐independent mechanism (Park et al., 2007). There are three major families of MAPK cascade, i.e., Ras‐Raf1‐MEK1/2‐Erk1/2, Rac‐MEKK1‐MEK4/7‐JNK1/2, and MEK3/6‐p38 MAPK, have been defined in mammalian cells (Krens et al., 2006), and MAPK cascades are activated via upstream kinases‐mediated sequential phosphorylations of cascades. The results obtained from current studies indicate that SUMOylation of RhoGDIα is specifically required for activation of the MEK1/2‐Erk cascade, but not the MEK4/7‐JNK cascade. As a result, RhoGDIα SUMOylation mediated cyclin D1 repression and cell growth arrest in cancer cells. Although the mechanisms underlying the specific regulation of MEK1/2‐Erk cascade by RhoGDIα SUMOylation has not been elucidated yet, the potential involvement of Ras, Raf, Rac, as well as specific kinase phosphatases, will be investigated in our future studies.
In summary, the results from the current study demonstrate that SUMOylated RhoGDIα is the active form for its inhibition of cancer cell anchorage‐independent growth, and that this inhibition was mediated by suppression of the MEK1/2‐Erk‐AP‐1 cascade, in turn leading to a reduction of cyclin D1 transcription and protein expression, as well as cancer cell growth arrest. These new findings provide significant novel insight into our understanding of the nature of RhoGDIα SUMOylation at Lys‐138 in its regulation of cancer cell property of anchorage‐independent growth, in addition to its regulation of cancer invasion and metastasis, as demonstrated in our previous studies (Yu et al., 2012; Liu et al., 2011). Moreover, modulation of RhoGDIα through SUMOylation might be used as a potential approach for cancer prevention and therapy.
Conflict of interest
The authors have no conflict of interest to disclose.
Author contribution
Zipeng Cao, acquisition of data and drafting of the manuscript; Xueyong Li, Jingxia Li and Peibei Kang, acquisition of data; Jingyuan Chen, Wenjing Luo, critical discussion and revision of the manuscript; Chuanshu Huang, study concept and design, critical revision of the manuscript for important intellectual content and study supervision.
Acknowledgments
We thank Dr. Richard G Pestell from Thomas Jefferson University Jefferson Medical College for generous gift of cyclin D1 luciferase reporters. This work was supported in part by grants from NIH/NCI RO1 CA112557 and CA177665, NIH/NIEHS ES000260; NSFC81229002, NSFC9102970 and NBRPC2012CB525004.
Cao Zipeng, Li Xueyong, Li Jingxia, Kang Beipei, Chen Jingyuan, Luo Wenjing and Huang Chuanshu, (2014), SUMOylation of RhoGDIα is required for its repression of cyclin D1 expression and anchorage‐independent growth of cancer cells, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.11.006.
References
- Assoian, R.K. , Klein, E.A. , 2008. Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol.. 18, 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian, R.K. , Schwartz, M.A. , 2001. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev.. 11, 48–53. [DOI] [PubMed] [Google Scholar]
- Balmanno, K. , Cook, S.J. , 1999. Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene. 18, 3085–3097. [DOI] [PubMed] [Google Scholar]
- Bohmer, R.M. , Scharf, E. , Assoian, R.K. , 1996. Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage-dependent expression of cyclin D1. Mol. Biol. Cell. 7, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, M.L. , Marshall, C.J. , Olson, M.F. , 2004. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol.. 5, 355–366. [DOI] [PubMed] [Google Scholar]
- Cook, S.J. , Aziz, N. , McMahon, M. , 1999. The repertoire of fos and jun proteins expressed during the G1 phase of the cell cycle is determined by the duration of mitogen-activated protein kinase activation. Mol. Cell. Biol.. 19, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft, D.R. , Olson, M.F. , 2006. The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol. Cell. Biol.. 26, 4612–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadke, S. , Cotteret, S. , Yip, S.C. , Jaffer, Z.M. , Haj, F. , Ivanov, A. , Rauscher, F. , Shuai, K. , Ng, T. , Neel, B.G. , Chernoff, J. , 2007. Regulation of protein tyrosine phosphatase 1B by sumoylation. Nat. Cell Biol.. 9, 80–85. [DOI] [PubMed] [Google Scholar]
- DerMardirossian, C. , Bokoch, G.M. , 2005. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol.. 15, 356–363. [DOI] [PubMed] [Google Scholar]
- Desterro, J.M. , Rodriguez, M.S. , Hay, R.T. , 1998. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell. 2, 233–239. [DOI] [PubMed] [Google Scholar]
- Dovas, A. , Couchman, J.R. , 2005. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem. J.. 390, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S. , Hall, A. , 2002. Rho GTPases in cell biology. Nature. 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Fang, J.Y. , Richardson, B.C. , 2005. The MAPK signalling pathways and colorectal cancer. Lancet Oncol.. 6, 322–327. [DOI] [PubMed] [Google Scholar]
- Fang, Y. , Yu, Y. , Hou, Q. , Zheng, X. , Zhang, M. , Zhang, D. , Li, J. , Wu, X.R. , Huang, C. , 2012. The Chinese herb isolate isorhapontigenin induces apoptosis in human cancer cells by down-regulating overexpression of antiapoptotic protein XIAP. J. Biol. Chem.. 287, 35234–35243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure, J. , Dagher, M.C. , 2001. Interactions between Rho GTPases and Rho GDP dissociation inhibitor (Rho-GDI). Biochimie. 83, 409–414. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata, R. , Boulter, E. , Burridge, K. , 2011. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol.. 12, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss-Friedlander, R. , Melchior, F. , 2007. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol.. 8, 947–956. [DOI] [PubMed] [Google Scholar]
- Gill, G. , 2005. Something about SUMO inhibits transcription. Curr. Opin. Genet. Dev.. 15, 536–541. [DOI] [PubMed] [Google Scholar]
- Gillett, C. , Smith, P. , Gregory, W. , Richards, M. , Millis, R. , Peters, G. , Barnes, D. , 1996. Cyclin D1 and prognosis in human breast cancer. Int. J. Cancer. 69, 92–99. [DOI] [PubMed] [Google Scholar]
- Harding, M.A. , Theodorescu, D. , 2010. RhoGDI signaling provides targets for cancer therapy. Eur. J. Cancer. 46, 1252–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou, G. , Song, K. , Yen, I. , Brandhuber, B.J. , Anderson, D.J. , Alvarado, R. , Ludlam, M.J. , Stokoe, D. , Gloor, S.L. , Vigers, G. , 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Heidorn, S.J. , Milagre, C. , Whittaker, S. , Nourry, A. , Niculescu-Duvas, I. , Dhomen, N. , Hussain, J. , Reis-Filho, J.S. , Springer, C.J. , Pritchard, C. , Marais, R. , 2010. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 140, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, A. , Nakamura, S. , Noguchi, Y. , Yasuda, T. , Kitagawa, M. , Tatsuno, I. , Oeda, T. , Tahara, K. , Terano, T. , Narumiya, S. , 1997. Geranylgeranylated rho small GTPase(s) are essential for the degradation of p27Kip1 and facilitate the progression from G1 to S phase in growth-stimulated rat FRTL-5 cells. J. Biol. Chem.. 272, 13–16. [PubMed] [Google Scholar]
- Hoffman, G.R. , Nassar, N. , Cerione, R.A. , 2000. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 100, 345–356. [DOI] [PubMed] [Google Scholar]
- Hu, W. , Bellone, C.J. , Baldassare, J.J. , 1999. RhoA stimulates p27(Kip) degradation through its regulation of cyclin E/CDK2 activity. J. Biol. Chem.. 274, 3396–3401. [DOI] [PubMed] [Google Scholar]
- Huang, C. , Ma, W.Y. , Dawson, M.I. , Rincon, M. , Flavell, R.A. , Dong, Z. , 1997. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. U S A. 94, 5826–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Ma, W.Y. , Dong, Z. , 1999. The extracellular-signal-regulated protein kinases (Erks) are required for UV-induced AP-1 activation in JB6 cells. Oncogene. 18, 2828–2835. [DOI] [PubMed] [Google Scholar]
- Johnson, E.S. , 2004. Protein modification by SUMO. Annu. Rev. Biochem.. 73, 355–382. [DOI] [PubMed] [Google Scholar]
- King, A.J. , Sun, H. , Diaz, B. , Barnard, D. , Miao, W. , Bagrodia, S. , Marshall, M.S. , 1998. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 396, 180–183. [DOI] [PubMed] [Google Scholar]
- Klein, E.A. , Assoian, R.K. , 2008. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci.. 121, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaja, N. , Karvonen, U. , Janne, O.A. , Palvimo, J.J. , 2002. PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol.. 22, 5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krens, S.F. , Spaink, H.P. , Snaar-Jagalska, B.E. , 2006. Functions of the MAPK family in vertebrate-development. FEBS Lett.. 580, 4984–4990. [DOI] [PubMed] [Google Scholar]
- Kubota, Y. , O'Grady, P. , Saito, H. , Takekawa, M. , 2011. Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat. Cell Biol.. 13, 282–291. [DOI] [PubMed] [Google Scholar]
- Leng, J. , Klemke, R.L. , Reddy, A.C. , Cheresh, D.A. , 1999. Potentiation of cell migration by adhesion-dependent cooperative signals from the GTPase Rac and Raf kinase. J. Biol. Chem.. 274, 37855–37861. [DOI] [PubMed] [Google Scholar]
- Li, W. , Chong, H. , Guan, K.L. , 2001. Function of the Rho family GTPases in Ras-stimulated Raf activation. J. Biol. Chem.. 276, 34728–34737. [DOI] [PubMed] [Google Scholar]
- Li, J. , Chen, H. , Tang, M.S. , Shi, X. , Amin, S. , Desai, D. , Costa, M. , Huang, C. , 2004. PI-3K and Akt are mediators of AP-1 induction by 5-MCDE in mouse epidermal Cl41 cells. J. Cell Biol.. 165, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberto, M. , Cobrinik, D. , Minden, A. , 2002. Rho regulates p21(CIP1), cyclin D1, and checkpoint control in mammary epithelial cells. Oncogene. 21, 1590–1599. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Zhang, D. , Luo, W. , Yu, Y. , Yu, J. , Li, J. , Zhang, X. , Zhang, B. , Chen, J. , Wu, X.R. , 2011. X-linked inhibitor of apoptosis protein (XIAP) mediates cancer cell motility via Rho GDP dissociation inhibitor (RhoGDI)-dependent regulation of the cytoskeleton. J. Biol. Chem.. 286, 15630–15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Zhang, D. , Luo, W. , Yu, J. , Li, J. , Yu, Y. , Zhang, X. , Chen, J. , Wu, X.R. , Huang, C. , 2012. E3 ligase activity of XIAP RING domain is required for XIAP-mediated cancer cell migration, but not for its RhoGDI binding activity. PloS One. 7, e35682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Q. , Longo, F.M. , Zhou, H. , Massa, S.M. , Chen, Y.H. , 2009. Signaling through Rho GTPase pathway as viable drug target. Curr. Med. Chem.. 16, 1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, W. , Liu, J. , Li, J. , Zhang, D. , Liu, M. , Addo, J.K. , Patil, S. , Zhang, L. , Yu, J. , Buolamwini, J.K. , 2008. Anti-cancer effects of JKA97 are associated with its induction of cell apoptosis via a Bax-dependent and p53-independent pathway. J. Biol. Chem.. 283, 8624–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres, M. , Barbacid, M. , 2003. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 3, 459–465. [DOI] [PubMed] [Google Scholar]
- Melchior, F. , 2000. SUMO – nonclassical ubiquitin. Annu. Rev. Cell Dev. Biol.. 16, 591–626. [DOI] [PubMed] [Google Scholar]
- Moissoglu, K. , McRoberts, K.S. , Meier, J.A. , Theodorescu, D. , Schwartz, M.A. , 2009. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res.. 69, 2838–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar, J.J. , Ebus, M.E. , Koster, J. , van Sluis, P. , van Noesel, C.J. , Versteeg, R. , Caron, H.N. , 2008. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res.. 68, 2599–2609. [DOI] [PubMed] [Google Scholar]
- Muller, S. , Hoege, C. , Pyrowolakis, G. , Jentsch, S. , 2001. SUMO, ubiquitin's mysterious cousin. Nat. Rev. Mol. Cell Biol.. 2, 202–210. [DOI] [PubMed] [Google Scholar]
- Muller, S. , Ledl, A. , Schmidt, D. , 2004. SUMO: a regulator of gene expression and genome integrity. Oncogene. 23, 1998–2008. [DOI] [PubMed] [Google Scholar]
- Musgrove, E.A. , 2006. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors. 24, 13–19. [DOI] [PubMed] [Google Scholar]
- Olson, M.F. , Paterson, H.F. , Marshall, C.J. , 1998. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 394, 295–299. [DOI] [PubMed] [Google Scholar]
- Ouyang, W. , Ma, Q. , Li, J. , Zhang, D. , Liu, Z.G. , Rustgi, A.K. , Huang, C. , 2005. Cyclin D1 induction through IkappaB kinase beta/nuclear factor-kappaB pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res.. 65, 9287–9293. [DOI] [PubMed] [Google Scholar]
- Ouyang, W. , Luo, W. , Zhang, D. , Jian, J. , Ma, Q. , Li, J. , Shi, X. , Chen, J. , Gao, J. , Huang, C. , 2008. PI-3K/Akt pathway-dependent cyclin D1 expression is responsible for arsenite-induced human keratinocyte transformation. Environ. Health Perspect.. 116, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, E.R. , Eblen, S.T. , Catling, A.D. , 2007. MEK1 activation by PAK: a novel mechanism. Cell. Signal.. 19, 1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Kim, K. , Lee, E.J. , Seo, Y.J. , Lim, S.N. , Park, K. , Rho, S.B. , Lee, S.H. , Lee, J.H. , 2007. Elevated level of SUMOylated IRF-1 in tumor cells interferes with IRF-1-mediated apoptosis. Proc. Natl. Acad. Sci. U S A. 104, 17028–17033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, K.E. , Turner, N.A. , O'Regan, D.J. , Balmforth, A.J. , Ball, S.G. , 2004. Simvastatin reduces human atrial myofibroblast proliferation independently of cholesterol lowering via inhibition of RhoA. Cardiovasc. Res.. 61, 745–755. [DOI] [PubMed] [Google Scholar]
- Rajakulendran, T. , Sahmi, M. , Lefrancois, M. , Sicheri, F. , Therrien, M. , 2009. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 461, 542–545. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, M. , Musa, N.L. , Li, J. , Liu, P.T. , Pestell, R.G. , Hershenson, M.B. , 1998. Catalytic activation of extracellular signal-regulated kinases induces cyclin D1 expression in primary tracheal myocytes. Am. J. Respir. Cell Mol. Biol.. 18, 736–740. [DOI] [PubMed] [Google Scholar]
- Sahai, E. , Marshall, C.J. , 2002. RHO-GTPases and cancer. Nat. Rev. Cancer. 2, 133–142. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mora, N. , Presmanes, M.C. , Monroy, V. , Moreno, N. , Lara-Martinez, J.M. , Aladro, M.H. , Alvarez-Fernandez, E. , 2008. Micropapillary lung adenocarcinoma: a distinctive histologic subtype with prognostic significance: case series. Hum. Pathol.. 39, 324–330. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. , Takai, Y. , 1998. The Rho small G protein family-Rho GDI system as a temporal and spatial determinant for cytoskeletal control. Biochem. Biophys. Res. Commun.. 245, 641–645. [DOI] [PubMed] [Google Scholar]
- Schubbert, S. , Shannon, K. , Bollag, G. , 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 7, 295–308. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. , 1994. G1 phase progression: cycling on cue. Cell. 79, 551–555. [DOI] [PubMed] [Google Scholar]
- Snyder, J.T. , Worthylake, D.K. , Rossman, K.L. , Betts, L. , Pruitt, W.M. , Siderovski, D.P. , Der, C.J. , Sondek, J. , 2002. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Biol.. 9, 468–475. [DOI] [PubMed] [Google Scholar]
- Sobko, A. , Ma, H. , Firtel, R.A. , 2002. Regulated SUMOylation and ubiquitination of DdMEK1 is required for proper chemotaxis. Dev. Cell. 2, 745–756. [DOI] [PubMed] [Google Scholar]
- Song, L. , Li, J. , Zhang, D. , Liu, Z.G. , Ye, J. , Zhan, Q. , Shen, H.M. , Whiteman, M. , Huang, C. , 2006. IKKbeta programs to turn on the GADD45alpha-MKK4-JNK apoptotic cascade specifically via p50 NF-kappaB in arsenite response. J. Cell Biol.. 175, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L. , Li, J. , Ye, J. , Yu, G. , Ding, J. , Zhang, D. , Ouyang, W. , Dong, Z. , Kim, S.O. , Huang, C. , 2007. p85alpha acts as a novel signal transducer for mediation of cellular apoptotic response to UV radiation. Mol. Cell. Biol.. 27, 2713–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey, D.W. , 2003. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol.. 15, 158–163. [DOI] [PubMed] [Google Scholar]
- Swant, J.D. , Rendon, B.E. , Symons, M. , Mitchell, R.A. , 2005. Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J. Biol. Chem.. 280, 23066–23072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talarmin, H. , Rescan, C. , Cariou, S. , Glaise, D. , Zanninelli, G. , Bilodeau, M. , Loyer, P. , Guguen-Guillouzo, C. , Baffet, G. , 1999. The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase cascade activation is a key signalling pathway involved in the regulation of G(1) phase progression in proliferating hepatocytes. Mol. Cell. Biol.. 19, 6003–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, T. , Tatsuno, I. , Noguchi, Y. , Uchida, D. , Oeda, T. , Narumiya, S. , Yasuda, T. , Higashi, H. , Kitagawa, M. , Nakayama, K. , 1998. Activation of cyclin-dependent kinase 2 (Cdk2) in growth-stimulated rat astrocytes. Geranylgeranylated Rho small GTPase(s) are essential for the induction of cyclin E gene expression. J. Biol. Chem.. 273, 26772–26778. [DOI] [PubMed] [Google Scholar]
- Tzima, E. , 2006. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ. Res.. 98, 176–185. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L. , D'Souza-Schorey, C. , 1997. Rho GTPases and signaling networks. Genes Dev.. 11, 2295–2322. [DOI] [PubMed] [Google Scholar]
- Vega, F.M. , Ridley, A.J. , 2008. Rho GTPases in cancer cell biology. FEBS Lett.. 582, 2093–2101. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Thurmond, D.C. , 2010. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J. Biol. Chem.. 285, 6186–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Ouyang, W. , Li, J. , Wei, L. , Ma, Q. , Zhang, Z. , Tong, Q. , He, J. , Huang, C. , 2005. Loss of tumor suppressor p53 decreases PTEN expression and enhances signaling pathways leading to activation of activator protein 1 and nuclear factor kappaB induced by UV radiation. Cancer Res.. 65, 6601–6611. [DOI] [PubMed] [Google Scholar]
- Watanabe, G. , Lee, R.J. , Albanese, C. , Rainey, W.E. , Batlle, D. , Pestell, R.G. , 1996. Angiotensin II activation of cyclin D1-dependent kinase activity. J. Biol. Chem.. 271, 22570–22577. [DOI] [PubMed] [Google Scholar]
- Welsh, C.F. , Roovers, K. , Villanueva, J. , Liu, Y. , Schwartz, M.A. , Assoian, R.K. , 2001. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat. Cell Biol.. 3, 950–957. [DOI] [PubMed] [Google Scholar]
- Yamamoto, M. , Tamakawa, S. , Yoshie, M. , Yaginuma, Y. , Ogawa, K. , 2006. Neoplastic hepatocyte growth associated with cyclin D1 redistribution from the cytoplasm to the nucleus in mouse hepatocarcinogenesis. Mol. Carcinog.. 45, 901–913. [DOI] [PubMed] [Google Scholar]
- Young, A. , Lyons, J. , Miller, A.L. , Phan, V.T. , Alarcon, I.R. , McCormick, F. , 2009. Ras signaling and therapies. Adv. Cancer Res.. 102, 1–17. [DOI] [PubMed] [Google Scholar]
- Yu, J. , Zhang, D. , Liu, J. , Li, J. , Yu, Y. , Wu, X.R. , Huang, C. , 2012. RhoGDI SUMOylation at Lys-138 increases its binding activity to Rho GTPase and its inhibiting cancer cell motility. J. Biol. Chem.. 287, 13752–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Song, L. , Li, J. , Wu, K. , Huang, C. , 2006. Coordination of JNK1 and JNK2 is critical for GADD45alpha induction and its mediated cell apoptosis in arsenite responses. J. Biol. Chem.. 281, 34113–34123. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Ouyang, W. , Li, J. , Zhang, D. , Yu, Y. , Wang, Y. , Li, X. , Huang, C. , 2012. Suberoylanilide hydroxamic acid (SAHA) inhibits EGF-induced cell transformation via reduction of cyclin D1 mRNA stability. Toxicol. Appl. Pharmacol.. 263, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]