

Abstract
Keratoacanthomas (KAs) and cutaneous squamous cell carcinomas (cuSCCs) develop in 15–30% of patients with BRAFV600E metastatic melanoma treated with BRAF inhibitors (BRAFi). These lesions resemble mouse skin tumors induced by the two‐stage DMBA/TPA skin carcinogenesis protocol; in this protocol BRAFi accelerates tumor induction. Since prior studies demonstrated cyclooxygenase 2 (COX‐2) is necessary for DMBA/TPA tumor induction, we hypothesized that COX‐2 inhibition might prevent BRAFi‐accelerated skin tumors. Celecoxib, a COX‐2 inhibitor, significantly delayed tumor acceleration by the BRAFi inhibitor PLX7420 and decreased tumor number by 90%. Tumor gene expression profiling demonstrated that celecoxib partially reversed the PLX4720‐induced gene signature. In PDV cuSCC cells, vemurafenib (a clinically approved BRAFi) increased ERK phosphorylation and soft agar colony formation; both responses were greatly decreased by celecoxib. In clinical trials trametinib, a MEK inhibitor (MEKi) increases BRAFi therapy efficacy in BRAFV600E melanomas and reduces BRAFi‐induced KA and cuSCC frequency. Trametinib also reduced vemurafenib‐induced PDV soft agar colonies, but less efficiently than celecoxib. The trametinb/celecoxib combination was more effective than either inhibitor alone. In conclusion, celecoxib suppressed both BRAFi‐accelerated skin tumors and soft‐agar colonies, warranting its testing as a chemopreventive agent for non‐melanoma skin lesions in patients treated with BRAFi alone or in combination with MEKi.
Keywords: Squamous cell carcinomas, COX‐2, celecoxib, vemurafenib, trametinib
Highlights
Celecoxib delays squamous cell carcinoma (SCC) tumor acceleration by PLX7420.
It also decreases SCC tumor number by 90%.
It partially reversed the PLX4720‐induced gene signature.
Trametinb/celecoxib combination is more effective than either inhibitor alone.
1. Introduction
Treatment of patients bearing BRAF V600 mutant metastatic melanoma with the BRAF inhibitors vemurafenib (formerly PLX4032) or dabrafenib (formerly GSK2118436) is a highly effective therapy, resulting in unprecedentedly high tumor response rates (Flaherty et al., 2010; Sosman et al., 2012; Hauschild et al., 2012) and improvement in overall survival (Chapman et al., 2011). The most frequent grade 3 or greater side effect of the BRAF inhibitors is the development of cutaneous squamous cell carcinomas (cuSCC), most of which are of the keratoacanthoma (KA) subtype. cuSCCs and KAs develop in approximately one fourth of patients treated with vemurafenib (Sosman et al., 2012). These tumors most frequently appear early in the course of therapy, within weeks, and are associated with a high frequency of HRAS mutations (Su et al., 2012; Oberholzer et al., 2012). Functional studies demonstrated that these tumors are mediated by the paradoxical activation of the mitogen‐activated protein kinase (MAPK) pathway, through the transactivation of CRAF by drug‐inhibited wild type BRAF (Su et al., 2012; Oberholzer et al., 2012). The same mechanism is active in the development of cuSCC/KAs in a lower proportion of patients treated with sorafenib, a pan‐RAF inhibitor (Arnault et al., 2012).
With the approval by health authorities of vemurafenib and dabrafenib for the treatment of BRAF mutant metastatic melanoma, and the approval of sorafenib for the treatment of renal cell carcinoma and hepatocellular carcinoma, there are an increasing number of patients at risk for the development of RAF inhibitor‐induced skin squamoepidermic lesions. The development of skin pre‐malignant and malignant lesions through the activation of the MAPK pathway downstream of RAF can be inhibited by allosteric MEK inhibitors (Su et al., 2012; Arnault et al., 2012) currently in clinical development for cancer treatment both as single agents and in combination with RAF, PI3K or AKT inhibitors (Friday and Adjei, 2008). However, a randomized phase II study using the combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib compared to trametinib alone failed to demonstrate a statistically significant decrease in the development of these secondary skin cancers (Flaherty et al., 2012). These results suggest that, even with the combination of a BRAF and a MEK inhibitor, there is a continued need to prevent the appearance of skin epithelioid malignant lesions.
The two‐stage mouse skin carcinogenesis model has been very useful in understanding the process of cuSCC development. Exposure to a single sub‐carcinogenic topical treatment with the carcinogen 7,12‐dimethylbenz[a]anthracene (DMBA) results in rare HRAS Q61 mutations in the mouse skin, but does not induce tumors (Quintanilla et al., 1986; Balmain et al., 1984). Subsequent topical treatment with the tumor promoter tetradecanoyl phorbol acetate (TPA) leads to the initial development of papillomas, many of which progress to tumors that histologically resemble human KAs and invasive cuSCCs (Abel et al., 2009). Administration of a BRAF inhibitor (e.g. the vemurafenib analogue PLX4720) concurrently with TPA results in a marked acceleration in the appearance of papillomas, an increase in their frequency, and enhanced progression to KAs and cuSCCs that resemble the KAs and cuSCCs induced clinically by BRAF inhibitors (Su et al., 2012). Prior research in the DMBA/TPA two‐stage skin carcinogenesis model demonstrated the critical role of COX‐2 in the development of these tumors from DMBA initiated epithelial cells (Tiano et al., 2002). In particular, topical application of the COX‐2 inhibitor celecoxib inhibited the development of papillomas and cuSCCs (Muller‐Decker et al., 1998; Chun et al., 2006).
In the current work on the two‐stage skin carcinogenesis model, we tested the use of systemic celecoxib treatment as a chemopreventive approach to reduce the burden of BRAF inhibitor‐induced epidermal squamoepithelial tumors. Our results demonstrate that the administration of celecoxib prevents, nearly completely, the appearance of DMBA/TPA‐induced skin tumors accelerated by the BRAF inhibitor.
2. Materials and methods
2.1. Mice and reagents
Female FVB/N mice were from Charles River Laboratory (Wilmington, MA). Tumor induction procedures were carried out in accordance with the UCLA animal care policy and with the Animal Research Committee approval. Two‐stage carcinogenesis was performed as described (Abel et al., 2009, 2010, 2010).}; 10 mice per group. DMBA and TPA were from Sigma (St. Louis, MO). Celecoxib chow was from Newco Lab (Rancho Cucamonga, CA). PLX4720, obtained under a material transfer agreement with Plexxikon (Berkeley, CA) and Roche (Nutley, NJ), was dissolved in dimethylsulfoxide (DMSO; Fisher Scientific, Morristown, NJ) and phosphate buffered saline (PBS; 1:1) and injected intraperitoneally (20 mg/kg, twice/week). PLX4720 was given intraperitoneally due to low oral bioavailability of the vemurafenib formulation (Flaherty et al., 2010; Su et al., 2012). PDV cells were provided by Dr. Miguel Quintanilla (Madrid, Spain). Vemurafenib, obtained under an MTA with Plexxikon, was dissolved in DMSO (10 mM) and used at a final concentration of 1 μM. Trametinib (GSK1120212, MEK1‐2 inhibitor), purchased from Selleck Chemicals (Houston, TX), was dissolved in DMSO to a 100 mM stock. Celecoxib (LKT laboratories, Inc; St. Paul, MN) was dissolved in DMSO to a stock concentration of 100 mM and used in culture at 32 μM. TPA (20 ng/ml) and PGE2 (2 μM) (both from Cayman Chemical, Ann Arbor, MI), were used for culture studies.
2.2. Cell proliferation assays
24‐well plates were covered with 300 μl of serum‐free RPMI 1640 (Fisher Scientific, Hampton, NH) with 0.6% Noble agar (BD Biosciences, San Jose, CA) and incubated at 37 °C overnight, until solid. 300 μl of a suspension of PDV cells (15,000 cells/ml) in a 1:1 mixture of growth medium and growth factor reduced matrigel (BD Biosciences San Jose, CA) was added to each well. After one week, automated colony quantification was performed using a BioSpot Series 5 UV analyzer (Cellular Technology Limited, Cleveland, OH).
2.3. Cell viability assays
Melanoma cell lines were treated in triplicate with vemurafenib and, trametinib, either with or without celecoxib, at the concentrations shown. Cell viability was measured after five days, using The CellTiter‐Glo® Luminescent Cell Viability Assay (Promega, Madison, WI) according to the manufacturers' instructions.
2.4. PGE2 measurement
PDV cells were treated in duplicate as described in figure legends. Twenty‐four hours later, medium was changed to serum‐free medium containing 10 μM arachidonic acid. The medium, harvested 1 h later, was assayed for PGE2 using the Prostaglandin E2 Express EIA Kit (Cayman Chemical).
2.5. Western blotting
PDV cells were treated in duplicate as described in figure legends. Western blotting was performed as described previously (Atefi et al., 2011). Skin tumors excised from FVB/N mice 16 weeks after DMBA administration were also analyzed by Western blotting. Primary antibodies included pBRAF Ser445, p‐ERK Thr204/205, ERK, pMEK Ser217/221, MEK, p‐AKT Ser473 and Thr308, AKT, beta‐actin (all from Cell Signaling Technology, Danvers, MA), COX‐2 and EP4 (both from Cayman Chemical). Immuno‐reactivity was revealed with an ECL‐Plus kit, using a Typhoon scanner (both from Amersham Biosciences Co, Piscataway, NJ).
2.6. Histological analysis
Mouse skin tumors excised at week 16 were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections were cut at 4 μm, deparaffinized with xylene and descendant ethanol, and then incubated in 3% H2O2 for 10 min. Retrievagen A solution (BD Pharmingen, San Jose, CA) was used for antigen retrieval. Sections were stained with COX‐2 antibody (RM‐9121‐R7, Thermo Scientific, Rockford, IL) (Huang et al., 2011), and the anti‐rabbit IgG with ImmPress™reagent kit (MP‐740, Vector Laboratories Burlingame CA) was applied. Then slides were developed with the ImmPACT™ DAB kit (SK‐4105, Vector Laboratories, Burlingame, CA) and counterstained with Hematoxylin, dehydrated, cleared, air dried, and mounted with mounting medium (Richard‐Allan Scientific). Human cuSCC/KA samples, obtained under UCLA IRB approval #11‐003254, were stained for COX‐2 as described for mouse samples. Tumors were obtained from discarded biopsy tissues and surgical resections of cuSCC/KA from either patients who had this diagnosis irrespective of having or not a concomitant melanoma, as well as from patients with metastatic melanoma who were on treatment with vemurafenib and developed cuSCC/KAs as a side effect.
2.7. Mutational analysis
DNA was extracted from mouse cuSCC/KA specimens and sequenced for HRAS (exons 1 and 2) using polymerase chain reaction (PCR) amplification followed by DNA Sanger sequencing (Sanger et al., 1977) as previously described (Su et al., 2012).
2.8. Gene expression microarray analysis
RNA from a mixture of 3 skin tumors in each treatment group was extracted (RNeasy Mini Kit, Qiagen, Valencia, CA) 16 weeks from the study start. Gene expression was measured using Agilent G4852A mouse SurePrint 8 × 60 k gene expression chips (Agilent, Santa Clara, CA). Probes were mapped to UniGene (mouse build number 190) using the first accession number in the Agilent chip annotation, yielding 34,311 Unigene annotated probes. Raw data was normalized among samples by median intensity. Principal component analysis (PCA) was used to study the variation across samples. The data was first mean centered; we then performed PCA by eigenvalue decomposition of the covariance matrix (Alter et al., 2000; Holter et al., 2000). For subsequent analysis, microarray probes with average expression greater than 0.3 across our three samples were retained, yielding 26,374 probes. For the BRAF‐inhibitor mediated changes in gene expression in DMBA/TPA induced skin tumors (DTP versus DT), probes were ranked by their log fold change (log2(DTP/DT)). In the fold change analysis, probes with intensity near background (less than 0.15) were set equal to this threshold so that fold change differences were not over‐represented. For clustering analysis, probes with a coefficient of variation greater than 0.3 across our three samples were retained, yielding 10,332 probes. Probes were clustered by Pearson correlation distance measure and pairwise average‐linkage using Cluster 3.0 (Eisen et al., 1998). Gene expression data has been deposited at the Gene Expression Omnibus (GEO).
2.9. Statistical analysis
Data were analyzed with GraphPad Prism (version 5) software (GraphPad Software, La Jolla, CA). Significance was determined using Mann Whitney t‐test or student t‐test with two‐tailed p‐values and one‐way Anova for comparison of more than two values.
3. Results
3.1. COX‐2 inhibition blocks acceleration of DMBA/TPA‐induced skin tumor development by a BRAF inhibitor
In three replicate experiments, with similar results, we tested the ability of celecoxib to inhibit PLX4720‐accelerated development of DMBA/TPA‐induced skin epithelioid tumors. Four groups of mice receiving DMBA and TPA to induce skin carcinogenesis were examined (Figure 1a). As described previously (Su et al., 2012), PLX4720 accelerated the appearance of DMBA/TPA‐induced skin tumors and increased the number of tumors per mouse. Compared to the DMBA/TPA (DT) group, and to the DMBA, TPA and PLX4720 (DTP) group, the addition of celecoxib to the chow significantly delayed the appearance of tumors, and resulted in a greater than 90% decrease in the number of lesions (Figure 1b and c). The reduction in the mean number of tumors by celecoxib was highly significant in mice given DMBA, TPA, PLX4720 and celecoxib (DTPC group, p < 0.001 by t‐test). As expected, the skin papillomas and cuSCCs from all groups of mice contained the HRAS Q61L mutation (Supplemental Figure 1).
Figure 1.
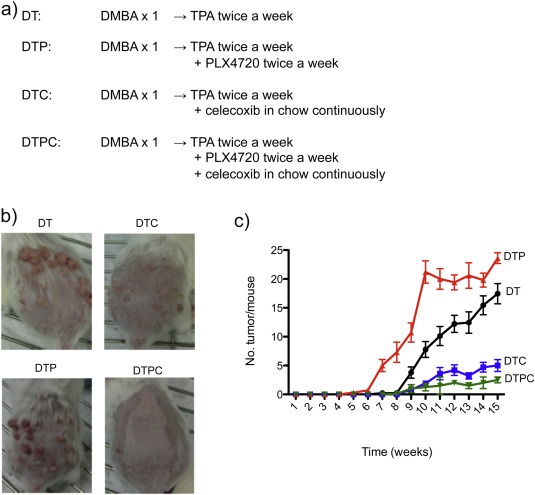
Celecoxib inhibits skin tumor induction in response to DMBA/TPA/PLX4720 treatment. (a) Study groups. DT: FVB/N mice subjected to DMBA/TPA tumor induction. DTP: DMBA/TPA treatment plus intraperitoneal PLX4720. DTC: DMBA/TPA treatment plus oral celecoxib. DTPC: DMBA/TPA treatment plus PLX4720 and celecoxib. (b) Mice from each group at week 13. (c) Tumors per mouse during 15 weeks of treatment (10 mice/group).
3.2. COX‐2 expression in mouse skin tumors
Tumors were harvested and processed for COX‐2 immunohistochemical (IHC) and Western blot analysis 16 weeks after the start of the study. Skin tumors from mice in three of the groups (DT, DTC and DTP) expressed COX‐2 protein (Figure 2a and b). Western blotting suggests that addition of celecoxib to the diet during the tumor development period may slightly increase COX‐2 protein accumulation (Figure 2b). However, the striking effects of celecoxib on tumor formation are consistent with the effects of celecoxib inhibiting COX‐2 enzymatic function as opposed to protein expression. Tumors formed in DTPC‐treated mice were very small, and had a higher stromal component (Figure 2a). As a result, we were unable to obtain tumor samples satisfactory for Western blot testing of COX‐2 protein expression.
Figure 2.
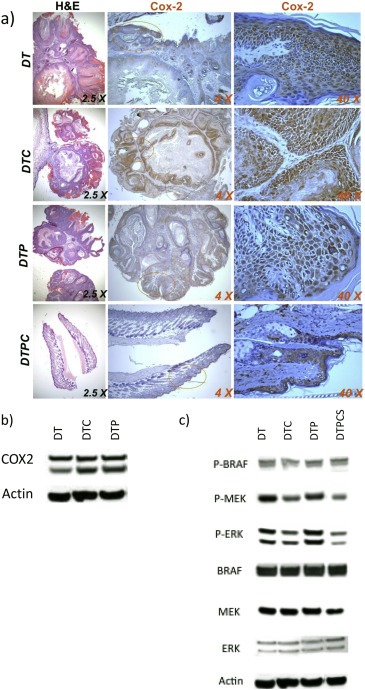
COX‐2 expression and MAPK signaling in tumors from DT, DTC, DTP and DTPC treated mice. (a) Tumor sections from mice at 16 weeks. (b) Western blots of tumor COX‐2 expression. (c) Western blots for BRAF, pBRAF, MEK, pMEK, ERK, pERK and actin. Western blots in panels (b) and (c) were analyzed on a single gel. The blot was successively stained, stripped and re‐stained with different antibodies. For clarity, data are separated into two panels. The same actin loading control is shown for both panels.
3.3. MAPK signaling in mouse skin tumors
Tumor extracts from the samples described in the preceding section were analyzed by Western blot for indications of altered MAPK signaling (Figure 2c). The tumors from mice treated with TPA + celecoxib exhibited decreases both in pMEK and in pERK. However, we were unable to demonstrate a paradoxical effect of BRAFi inhibition of tumor growth on MAP kinase signaling in this set of tumors, and thus were unable to determine if celecoxib modulated the BRAFi induced “paradoxical effect”, as well as tumor growth. As described above, we were unable to obtain extracts suitable for Western blotting from the infrequent, small DTPC tumors that had extensive stromal components.
To gain further insight into the role of COX‐2 activity on PLX4720‐accelerated enhancement of tumor progression and on the BRAFi‐induced “paradoxical” effect in this system, at week 16 we switched a group of mice bearing florid DTP tumors to chow with celecoxib for five days (DTP Celecoxib Switched tumors, or DTPCS tumors). Extracts from the DTPCS tumor group were compared by Western blot with the other three groups (DT, DTC and DTP tumors) for indications of celecoxib‐mediated alterations in MAPK signaling (Figure 2c); The DTPCS tumors, in this second cohort of mice, exhibited celecoxib‐dependent reductions in pMEK and pERK.
3.4. BRAF‐inhibitor mediated changes in gene expression in DMBA/TPA‐induced skin tumors
PLX4720 exposure accelerates the appearance of TPA‐promoted tumors in DT‐treated mice and increases the frequency of DT‐induced tumors (Ref. 4 and Figure 1). We compared transcriptome profiling of DTP‐induced tumors versus DT‐induced tumors, to identify potential candidates for PLX4720 mediated gene expression differences that contribute to the accelerated tumor formation and increased tumor frequency in DTP‐ treated mice. Supplemental Figure 2 summarizes differences observed in the transcription profiles of DTP‐ and DT‐induced tumors; Supplementary Table 1 provides a list of the top 1000 microarray probes that are more highly expressed in DTP‐induced tumors versus DT‐induced tumors and the top 1000 whose expression is suppressed in DTP‐induced tumors versus DT‐induced tumors. The most parsimonious interpretation of these differences is that the former (more highly expressed) genes might include genes whose PLX4720‐driven expression enhances TPA‐driven tumor progression. The latter (PLX‐suppressed expression) genes might include genes whose expression normally retards TPA‐driven tumor progression. PLX4720 suppression of (some) genes in this group might consequently enhance TPA‐driven tumor progression.
3.5. Celecoxib reversal of BRAF inhibitor‐induced properties in skin tumors
To determine the influence of COX‐2 mediated gene expression on PLX4720‐accelerated skin tumor progression, we compared transcriptome analyses of three tumor groups, DT, DTP and DTPCS (Figure 3). Cluster analysis of the differentially expressed genes across the DT, DTP and DTPCS samples resulted in three main groups of genes (Figure 3a and Supplemental Table 2). Group 1 includes genes that have relatively low expression in DT samples, have relatively high expression in DTP samples, and have relatively low to intermediate expression in DTPCS samples (Figure 3b, top panel). Thus, these genes are elevated in the tumors that develop in the presence of TPA plus the BRAF inhibitor, compared to tumors that develop only in the presence of TPA, and whose elevated expression in mice bearing PLX4720 accelerated tumors is reversed when the mice are treated with celecoxib. Based on their expression patterns, these are candidate genes that are likely to contribute to PLX4720‐accelerated tumor progression and to celecoxib‐repressed tumor progression.
Figure 3.
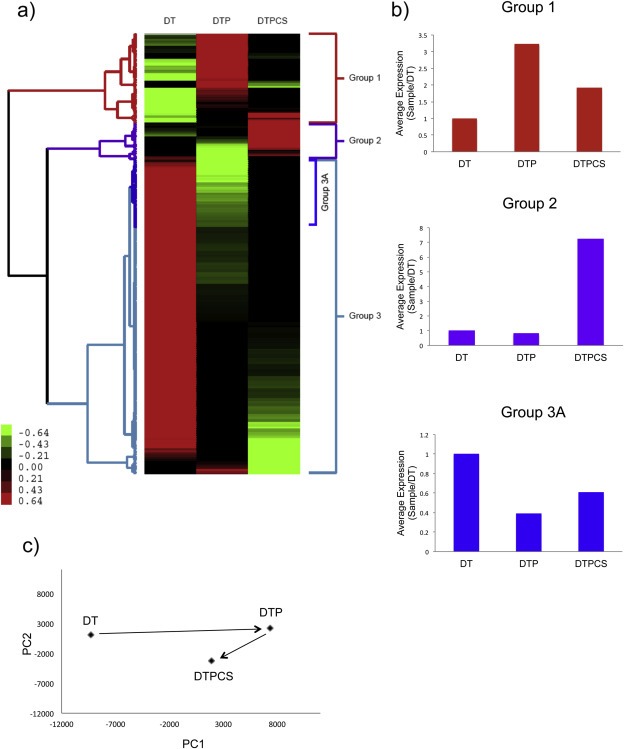
Celecoxib reversal of BRAF inhibitor‐induced transcriptional changes. (a) Hierarchical clustering/grouping of genes differentially expressed in the DT, DTP and DTPCS (in which DTP mice were switched to celecoxib) tumor groups. Each gene is normalized across the samples; green indicates lower relative expression and red higher expression compared to the mean across all three samples. Color scale bar values indicate normalized intensities. (b) Average expression patterns of genes in panel A sub‐groups. (c) Principal component analysis of changes in gene expression in tumors from mice in the DT, DTP and DTPCS group. The first principal component (PC1) reflects the major change in gene expression induced by PLX4720 and partially reversed by celecoxib. The second principal component (PC2) reflects changes induced by celecoxib that are not part of the PLX4720 response.
Group 2 includes a set of genes highly expressed in DTPCS tumors compared to DT and DTP tumors; increased expression of these genes may be a direct result of inhibiting COX‐2 by celecoxib (Figure 3b, middle panel). Group 3 contains genes highly expressed in DT tumor samples compared to the DTPCS and DTP tumor samples; these genes are apparently inhibited by exposure to the BRAF inhibitor PLX4720. A sub‐cluster of Group 3 genes (Group 3A) shows an expression pattern that is the reverse of the genes in Group 1; Group 3A genes are repressed by PLX4720 in DT tumors and de‐repressed by subsequent celecoxib treatment (Figure 3b, lower panel).
Principal component analysis (PCA) of gene expression profiles from PLX4720‐accelerated lesions demonstrated that the addition of celecoxib to PLX4720‐accelerated tumors partially reversed the gene signature induced by exposure to PLX4720 (Figure 3c). In sum, the clustering and PCA analyses demonstrate a signature of TPA plus BRAF inhibitor‐modulated genes that is partially reversed by celecoxib.
3.6. Squamous cell carcinomas and keratoacanthomas from vemurafenib‐treated patients have intense COX‐2 staining
To assess if COX‐2 is expressed in vemurafenib‐induced cuSCC/KA tumors in patients, we analyzed COX‐2 expression in 10 cuSCCs and KAs obtained from 10 patients with BRAF mutant melanoma who were treated with vemurafenib, as well as histologically similar skin tumors arising spontaneously in 12 patients not exposed to vemurafenib. COX‐2 expression was not detectable by our IHC analysis in the human skin samples from individuals not exposed to vemurafenib. Strong COX‐2 expression is easily detectable in vemurafenib‐elicited human SCC tumors (Figure 4).
Figure 4.
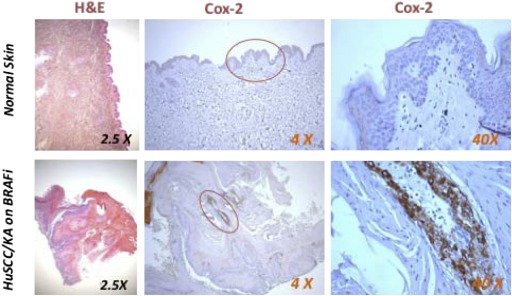
Immunohistochemical COX‐2 analysis of a normal skin sample and a cuSCC/KA obtained from a patient with BRAFV600E mutant melanoma receiving vemurafenib therapy.
3.7. Vemurafenib‐stimulated growth of PDV cells is inhibited by celecoxib
The PDV cell line is a DMBA‐transformed mouse cuSCC cell line containing the HRAS Q61L mutation (Caulin et al., 1993) found in nearly all DMBA/TPA induced mouse cuSCCs. To investigate the cellular effects of vemurafenib and celecoxib on skin epithelial cells harboring this mutation, we assessed cell growth, MAPK signaling, and both COX‐2 and prostaglandin E2 (PGE2) levels in PDV cells.
We examined the effects of celecoxib, vemurafenib, and the combination of the two agents on the growth of PDV cells in a three‐dimensional agar colony growth assay. Vehicle‐treated PDV cells formed relatively few colonies of relatively small size (Figure 5a and b). Celecoxib alone had little or no effect on either the number of PDV colonies or their size. In contrast, vemurafenib greatly increased both the frequency of PDV soft agar colony formation and the size of the colonies. Celecoxib substantially reduced the vemurafenib‐stimulated increase in PDV colony numbers (p < 0.0001 by t‐test) and in the size of the soft‐agar colonies.
Figure 5.
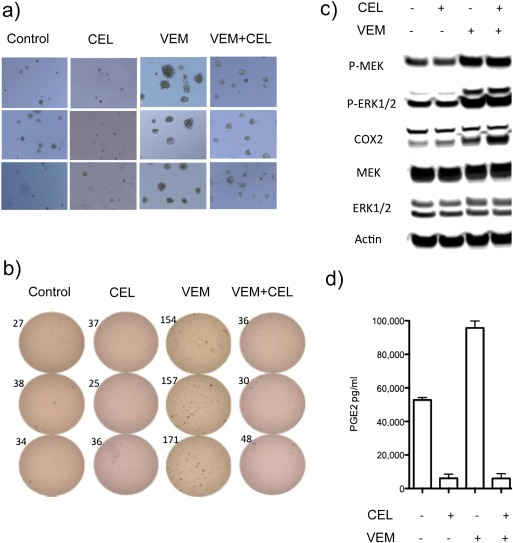
Soft‐agar colony formation, COX‐2 expression, MAPK signaling and PGE2 production by PDV cells in the presence or absence of vemurafenib (VEM) and/or celecoxib (CEL). (a) PDV cells were treated in triplicate for seven days with vehicle, CEL (32 μM), VEM (2 μM) or VEM + CEL. (b) Colony quantification: colony numbers are shown next to each plate. (c). Western blot analyses of VEM‐ and CEL‐mediated COX‐2 expression and MAPK signaling. (d) PGE2 in culture media.
We also performed Western blot analyses of COX‐2 expression and MAPK signaling in the PDV cell line in the presence or absence of vemurafenib and celecoxib. Exposure to the BRAF inhibitor vemurafenib induced the now well‐known paradoxical increase in pMEK and pERK in cultured PDV cells (Figure 5c). The vemurafenib‐induced pMEK and pERK elevation did not change in the presence of celecoxib (Figure 5c).
To more closely resemble the conditions in the two‐stage tumor induction paradigm in mice, we also tested the addition of TPA to celecoxib‐ and vemurafenib‐treated PDV cells. Like vemurafenib, TPA alone increased pMEK and pERK levels. Again, celecoxib had no effect either on this induction, or on the elevation in pMEK and pERK levels observed in the presence of TPA and vemurafenib (data not shown).
One immediate consequence of COX‐2 expression in epithelial cells is an increase in PGE2 production, which promotes epithelial cell proliferation (Menter and Dubois, 2012). Untreated PDV cells produce substantial amounts of PGE2 (Figure 5d). Nevertheless, vemurafenib treatment induced significant increases in PGE2 levels in PDV cell supernatants (p < 0.05 by one‐way Anova). Both “basal” (i.e., vehicle‐treated) and vemurafenib‐induced PGE2 accumulation were significantly decreased with exposure to celecoxib (p < 0.0001 by one‐way Anova, Figure 5d). TPA administration also induced additional PGE2 accumulation above “basal levels” in PDV cells (data not shown). This induction was enhanced by vemurafenib. Celecoxib again reduced the drug‐induced PGE2 accumulation to well below the basal levels produced by PDV cells.
3.8. Trametinib, a MEK inhibitor, also reduces vemurafenib‐stimulated PDV soft‐agar colony formation
A MEK inhibitor (MEKi), administered in combination with a BRAF inhibitor, can increase the therapeutic efficacy of BRAF kinase inhibitors in patients with melanoma by inhibiting the paradoxical activation of the MAPK pathway induced by BRAF inhibitor (Su et al., 2012; McArthur and Ribas, 2013), but the incidence of cuSCC and keratinocytic proliferative skin lesions was not fully avoided in the clinic (Flaherty et al., 2012). To determine whether the PDV soft agar colony formation assay could reflect this tendency for MEKi to reduce BRAFV600 kinase inhibitor‐stimulated proliferation of RAS‐initiated epithelial cells, we examined trametinib inhibition of soft agar colony formation by vemurafenib‐treated PDV cells (Figure 6). While there was a clear trametinib dose–response related reduction in soft‐agar colony formation by PDV cells, the ability of trametinib to suppress PDV soft‐agar colony formation was not complete.
Figure 6.
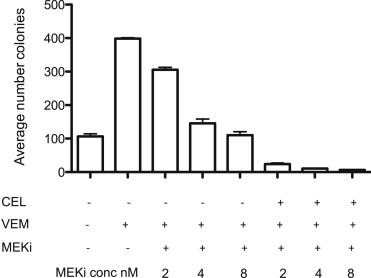
Combined MEK and COX‐2 inhibition optimally suppresses vemurafenib‐stimulated PDV colony formation. PDV cells were treated in duplicate with VEM (2 μM), trametinib (MEKi, at concentrations shown) and/or CEL (32 μM). Colony quantification was determined at seven days.
3.9. Increased anti‐proliferative activity by trametinib and celecoxib in vemurafenib‐stimulated PDV soft‐agar colony formation
Although MEK inhibition increased the efficacy of dabrafenib therapy for BRAF V600 metastatic melanoma, and appeared to reduce darafenib‐induced skin tumors, reduction in skin non‐melanoma epithelial tumors was only marginally effective (Flaherty et al., 2012). We reasoned that the growth of PDV paradoxically stimulated by vemurafenib may be best inhibited with the addition of celecoxib to the MEK inhibitor therapy, serving as a surrogate indicator of the possible effects of this combination on the formation of skin tumors in patients treated with the combination of a BRAFV600 kinase inhibitor and a MEK inhibitor. Indeed, this was the case; for all concentrations of trametinib tested, the addition of celecoxib substantially decreased vemurafenib‐stimulated, partially trametinib‐inhibited PDV colony forming efficacy (Figure 6).
To investigate whether celecoxib has any antagonistic effect on the inhibition of melanoma cell growth by vemurafenib + trametinib, we tested growth inhibition of two BRAFV600E mutant melanoma cell lines, M262 and M249. Both cell lines were treated with vemurafenib and trametinib, either in the presence or absence of celecoxib. Concomitant celecoxib treatment of the melanoma cells does not antagonize the growth inhibition of vemurafenib and trametinib combination (Supplemental Figure 3).
3.10. Reduced COX‐2 expression and consequent PGE2 modulation as a possible mode of action in trametinib inhibition of PDV soft‐agar colony formation
Since celecoxib inhibition of COX‐2 activity can eliminate vemurafenib‐enhanced skin tumor induction (Figure 1) and vemurafenib‐stimulated PDV soft agar colony formation (Figure 5), one possible mechanism for trametinib inhibition of PDV soft agar colony formation might be inhibition of RAS*‐driven, MEK‐dependent increase in COX‐2 production and consequent prostaglandin production. Consistent with the suggestion, trametinib reduced vemurafenib‐stimulated COX‐2 protein accumulation (Figure 7a) and vemurafenib‐stimulated PGE2 accumulation (Figure 7b) in PDV cells.
Figure 7.
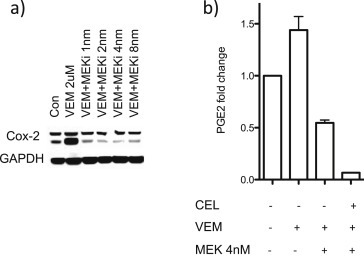
Trametinib effects on vemurafenib (VEM)‐mediated COX‐2 protein levels and PGE2 production by PDV cells. PDV cells were treated in triplicate with vehicle, CEL (32 μM), VEM (2 μM), and/or trametinib (MEKi) as indicated. Cells were harvested twenty‐four hours after treatment. (a) COX‐2 and GAPDH Western blots. (b) PGE2 in culture media.
4. Discussion
Both pharmacologic enzyme inhibition studies and genetic gene deletion studies have demonstrated that DMBA/TPA mouse skin tumor induction is nearly completely dependent on COX‐2 expression. Here we demonstrate that continuous oral dosing with the COX‐2 inhibitor celecoxib in the chow also nearly completely inhibits the development of PLX4720‐accelerated papillomas and cuSCCs in the two‐stage DMBA/TPA skin carcinogenesis model.
Our data expand on prior observations using COX‐2 inhibitors as chemopreventive agents for cuSCC formation. Data generated in Cox2 knock‐out mice demonstrated the critical requirement for COX‐2 expression both in the development of skin lesions in the two‐stage DMBA/TPA skin carcinogenesis model and in ultraviolet light‐induced skin carcinogenesis (Tiano et al., 2002; Fischer et al., 2007). In addition, two prior reports tested the use of pharmacological COX‐2 inhibitors as a local therapy (Muller‐Decker et al., 1998; Chun et al., 2006). Both studies demonstrated a decrease in the incidence of skin tumors in the DMBA/TPA two‐stage skin carcinogenesis model when a COX‐2 inhibitor was topically applied. Our study demonstrates, for the first time, that orally administered celecoxib has a chemopreventive effect in this mouse model, with a striking effect seen in the BRAF inhibitor‐accelerated skin tumors. Supporting this chemoprotective interpretation, gene expression profiling of BRAF inhibitor‐accelerated tumors treated, post‐tumor development, with celecoxib demonstrated that the principal effect of celecoxib was to reverse the gene expression changes caused by BRAF inhibition. mRNA microarray comparisons of tumors from DMBA/TPA induced tumors, DMBA/TPA/PLX induced tumors, and tumors from mice treated with DMBA/TPA/PLX initially and then switched to a diet that also included celecoxib suggests that BRAF inhibitors elicit expression of genes whose functions may accelerate tumor formation, while celecoxib treatment elicits expression of genes whose functions may retard TPA and PLX4720‐driven tumor progression. The concept of using celecoxib to prevent the development of non‐melanoma cutaneous cancers has been tested in the clinic within a double‐blind placebo‐controlled randomized clinical trial (Elmets et al., 2010). This study demonstrated that celecoxib at 200 mg twice daily for 9 months decreased the incidence of cuSCC in subjects who had prior extensive actinic sun damage.
To develop a model in which mechanistic interactions between BRAF inhibitors and COX‐2 inhibitors on tumor cell proliferation could be evaluated, we optimized a cell culture soft‐agar colony growth model, using the PDV cell line. PDV cells, derived from mouse epidermal keratinocytes transformed in culture by DMBA, contain the characteristic HRAS Q61L mutation found in 90% of DMBA/TPA induced skin papillomas and SCCs (Caulin et al., 1993). PDV cells form soft‐agar colonies very poorly; however, vemurafenib treatment strongly promotes colony formation. Like PLX4720‐accelerated DMBA/TPA tumor induction, co‐administration of celecoxib with the BRAF inhibitor substantially suppressed PDV soft‐agar colony formation. However, we did not observe celecoxib inhibition of the BRAF inhibitor‐induced paradoxical MAPK activation in the PDV cell culture studies. We attempted to more closely recapitulate inhibition of the paradoxical effect observed in mice by adding TPA to the PDV cell culture system, but did not observe an effect of celecoxib on vemurafenib‐stimulated pERK accumulation (data not shown). The celecoxib‐induced decrease in vemurafenib‐dependent pERK expression seen in skin tumors obtained from mice may be due to a longer exposure to celecoxib, or to differences in cellular composition of the tumor cells and stroma in the mouse tumors versus the PDV cultures.
Recent studies have demonstrated that the combination of BRAF inhibitors and MEK inhibitors may be more effective in combination therapy of patients with BRAF V600 metastatic melanoma than either inhibitor alone. A randomized phase II study using the combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib demonstrated a significant improvement in progression‐free survival (Flaherty et al., 2012). Although there was a decrease in proliferative skin lesions in patients receiving the combined therapy, the authors concluded that the rate of these lesions was “nonsignificantly reduced”. Given (i) the apparent ability of PDV soft agar colony formation to reflect BRAF inhibitor‐driven stimulation of cutaneous tumor growth, and (ii) the ability of celecoxib to suppress BRAF inhibitor‐stimulated colony formation, we compared the efficacy of trametinib either alone, or in combination with celecoxib, in suppressing vemurafenib‐driven PDV cell soft agar colony formation. Although trametinib did reduce vemurafenib‐driven PDV colony formation in a dose‐dependent manner, its effect was not as robust as that of celecoxib inhibition of soft agar growth. Moreover, at all concentrations tested, the combination of trametinib and celecoxib was more effective at suppressing PDV cell colony formation than the MEK inhibitor alone. Based on these results from this vemurafenib‐driven PDV colony formation surrogate assay for BRAF inhibitor‐driven cutaneous tumor appearance, we suggest that the combination of a MEK inhibitor and celecoxib may be an optimal therapeutic combination to reduce cutaneous tumor induction by BRAF inhibitors.
Both the reduction in DMBA/TPA‐induced skin tumors and the reduction in PLX4720‐accelerated skin tumor occurrence by celecoxib, and the reduction by celecoxib in vemurafenib‐driven PDV soft agar colony formation, are unlikely to be caused by any mechanism other than reduction of COX‐2 dependent prostaglandin synthesis. Trametinib is able to reduce COX‐2 protein expression, PGE2 production and soft agar colony formation of vemurafenib‐treated PDV cells. These data suggest that one pathway by which MEK inhibition may reduce BRAF inhibitor‐driven cutaneous tumor formation in BRAFV600 melanoma patients may be by blocking COX‐2 dependent, prostanoid driven proliferation of RAS‐initiated skin epithelial cells. However, at present, this suggestion is based on a correlation between soft agar colony formation and reduced activity of the MAPK/COX‐2/prostaglandin pathway; verification of this potential (perhaps partial) explanation for the ability of MEK inhibitors to suppress BRAF inhibitor‐driven cutaneous tumor formation in patients will require additional analyses of tumors from patients receiving individual versus combined BRAF and MEK inhibitors.
In conclusion, our data demonstrate that celecoxib prevents the development of BRAF inhibitor‐induced epithelioid skin lesions in mice and inhibits the proliferative effects of vemurafenib in the PDV mouse skin epithelial RAS transformed cell line. Together, these observations provide strong support for the conduct of a clinical trial using celecoxib as a chemopreventive agent for cuSCC/KAs in patients who are taking BRAF inhibitors, either alone or in combination with a MEK inhibitor, for the treatment of cancer.
Supporting information
The following are the supplementary data related to this article:
Table S1 Gene expression signature for BRAF inhibitor (PLX4720) acceleration of DMBA/TPA tumors. List of the top microarray probes differentially expressed by gene expression profiling in skin tumors resected from mice as depicted in Supplemental Figure 2. The upregulated and downregulated probes are listed in separate tabs. Each probe is annotated with feature number, probe number, accession number, Unigene accession number, gene symbol, and gene description. The DTP/DT fold change (log2 ratio) and DT and DTP intensities are listed. Fold change values are background adjusted (see Materials and Methods).
Figure S2 Gene expression signature for BRAF inhibitor (PLX4720) acceleration of DMBA/TPA tumors. Waterfall plot of microarray probes ranked by their differential expression (log fold change) between the DTP and DT tumors. The top and bottom 20 genes are listed here, and the top and bottom 1000 probes are listed in Supplemental Table 1.
Supplementary data
Figure S1 HRAS codon 61 gene sequencing of DMBA/TPA skin tumors from FVB/N mice and the PDV cell line.
Table S2 Reversal of BRAF inhibitor‐induced transcriptional changes in tumors by celecoxib. Lists of the microarray probes and their gene information for the gene groups defined in the clustering analysis of Figure 3. Each group is listed in a separate tab. Group 3 is split into sub‐groups 3A and 3B.
Acknowledgments
This work was funded by NIH grant P50 CA086306 (to H.R.H. and A.R.). Support was also provided by the Seaver Institute, the Dr. Robert Vigen Memorial Fund, the Louise Belley and Richard Schnarr Fund, the Wesley Coyle Memorial Fund, the Garcia‐Corsini Family Fund, the Bila Alon Hacker Memorial Fund, the Fred L. Hartley Family Foundation, the Ruby Family Foundation, the Jonsson Cancer Center Foundation, the Caltech‐UCLA Joint Center for Translational Medicine to A.R and NIH/National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR000124 (T.G.G.). HE‐O was supported by the V Foundation‐Gil Nickel Family Endowed Fellowship in Melanoma Research. The Immuno/BioSpot Core is supported by a grant from the “James B. Pendleton Charitable Trust”, UCLA CFAR grant 5P30 AI028697 and the UCLA AIDS Institute.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.11.005.
Escuin-Ordinas Helena, Atefi Mohammad, Fu Yong, Cass Ashley, Ng Charles, Huang Rong Rong, Yashar Sharona, Comin-Anduix Begonya, Avramis Earl, Cochran Alistair J., Marais Richard, Lo Roger S., Graeber Thomas G., Herschman Harvey R. and Ribas Antoni, (2014), COX‐2 inhibition prevents the appearance of cutaneous squamous cell carcinomas accelerated by BRAF inhibitors, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.11.005.
Contributor Information
Harvey R. Herschman, Email: hherschman@mednet.ucla.edu
Antoni Ribas, Email: aribas@mednet.ucla.edu.
References
- Abel, E.L. , Angel, J.M. , Kiguchi, K. , DiGiovanni, J. , 2009. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat. Protoc.. 4, 1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter, O. , Brown, P.O. , Botstein, D. , 2000. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. U. S A.. 97, 10101–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnault, J.P. , Mateus, C. , Escudier, B. , Tomasic, G. , Wechsler, J. , Hollville, E. , 2012. Skin tumors induced by sorafenib; Paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res.. 18, 263–272. [DOI] [PubMed] [Google Scholar]
- Atefi, M. , von Euw, E. , Attar, N. , Ng, C. , Chu, C. , Guo, D. , 2011. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS One. 6, e28973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmain, A. , Ramsden, M. , Bowden, G.T. , Smith, J. , 1984. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 307, 658–660. [DOI] [PubMed] [Google Scholar]
- Caulin, C. , Bauluz, C. , Gandarillas, A. , Cano, A. , Quintanilla, M. , 1993. Changes in keratin expression during malignant progression of transformed mouse epidermal keratinocytes. Exp. Cell Res.. 204, 11–21. [DOI] [PubMed] [Google Scholar]
- Chapman, P.B. , Hauschild, A. , Robert, C. , Haanen, J.B. , Ascierto, P. , Larkin, J. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New Engl. J. Med.. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, K.S. , Kundu, J.K. , Park, K.K. , Chung, W.Y. , Surh, Y.J. , 2006. Inhibition of phorbol ester-induced mouse skin tumor promotion and COX‐2 expression by celecoxib: C/EBP as a potential molecular target. Cancer Res. Treat.. 38, 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, M.B. , Spellman, P.T. , Brown, P.O. , Botstein, D. , 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci U. S. A.. 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmets, C.A. , Viner, J.L. , Pentland, A.P. , Cantrell, W. , Lin, H.Y. , Bailey, H. , 2010. Chemoprevention of nonmelanoma skin cancer with celecoxib: a randomized, double-blind, placebo-controlled trial. J. Natl. Cancer Inst.. 102, 1835–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S.M. , Pavone, A. , Mikulec, C. , Langenbach, R. , Rundhaug, J.E. , 2007. Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol Carcinog.. 46, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty, K.T. , Puzanov, I. , Kim, K.B. , Ribas, A. , McArthur, G.A. , Sosman, J.A. , 2010. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl. J. Med.. 363, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty, K.T. , Infante, J.R. , Daud, A. , Gonzalez, R. , Kefford, R.F. , Sosman, J. , 2012. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New Engl. J. Med.. 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday, B.B. , Adjei, A.A. , 2008. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res.. 14, 342–346. [DOI] [PubMed] [Google Scholar]
- Hauschild, A. , Grob, J.J. , Demidov, L.V. , Jouary, T. , Gutzmer, R. , Millward, M. , 2012. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 380, 358–365. [DOI] [PubMed] [Google Scholar]
- Holter, N.S. , Mitra, M. , Maritan, A. , Cieplak, M. , Banavar, J.R. , Fedoroff, N.V. , 2000. Fundamental patterns underlying gene expression profiles: simplicity from complexity. Proc. Natl. Acad. Sci. U. S. A.. 97, 8409–8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, R.R. , Jalil, J. , Economou, J.S. , Chmielowski, B. , Koya, R.C. , Mok, S. , 2011. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin. Cancer Res.. 17, 4101–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa, T.O. , Jain, N.K. , Herschman, H.R. , 2010. COX‐2 gene expression in chemically induced skin papillomas cannot predict subsequent tumor fate. Mol. Oncol.. 4, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa, T.O. , Kumar, I.P. , Machado, H.B. , Wong, K.P. , Kusewitt, D. , Huang, S.C. , 2010. Positron emission tomography imaging of DMBA/TPA mouse skin multi-step tumorigenesis. Mol. Oncol.. 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur, G.A. , Ribas, A. , 2013. Targeting oncogenic drivers and the immune system in melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol.. 31, 499–506. [DOI] [PubMed] [Google Scholar]
- Menter, D.G. , Dubois, R.N. , 2012. Prostaglandins in cancer cell adhesion, migration, and invasion. Int. J. Cell Biol.. 2012, 723419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Decker, K. , Kopp-Schneider, A. , Marks, F. , Seibert, K. , Furstenberger, G. , 1998. Localization of prostaglandin H synthase isoenzymes in murine epidermal tumors: suppression of skin tumor promotion by inhibition of prostaglandin H synthase-2. Mol. Carcinog.. 23, 36–44. [DOI] [PubMed] [Google Scholar]
- Oberholzer, P.A. , Kee, D. , Dziunycz, P. , Sucker, A. , Kamsukom, N. , Jones, R. , 2012. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol.. 30, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla, M. , Brown, K. , Ramsden, M. , Balmain, A. , 1986. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 322, 78–80. [DOI] [PubMed] [Google Scholar]
- Sanger, F. , Nicklen, S. , Coulson, A.R. , 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A.. 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosman, J.A. , Kim, K.B. , Schuchter, L. , Gonzalez, R. , Pavlick, A.C. , Weber, J.S. , 2012. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. New Engl. J. Med.. 366, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, F. , Viros, A. , Milagre, C. , Trunzer, K. , Bollag, G. , Spleiss, O. , 2012. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. New Engl. J. Med.. 366, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiano, H.F. , Loftin, C.D. , Akunda, J. , Lee, C.A. , Spalding, J. , Sessoms, A. , 2002. Deficiency of either cyclooxygenase (COX)-1 or COX‐2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res.. 62, 3395–3401. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Table S1 Gene expression signature for BRAF inhibitor (PLX4720) acceleration of DMBA/TPA tumors. List of the top microarray probes differentially expressed by gene expression profiling in skin tumors resected from mice as depicted in Supplemental Figure 2. The upregulated and downregulated probes are listed in separate tabs. Each probe is annotated with feature number, probe number, accession number, Unigene accession number, gene symbol, and gene description. The DTP/DT fold change (log2 ratio) and DT and DTP intensities are listed. Fold change values are background adjusted (see Materials and Methods).
Figure S2 Gene expression signature for BRAF inhibitor (PLX4720) acceleration of DMBA/TPA tumors. Waterfall plot of microarray probes ranked by their differential expression (log fold change) between the DTP and DT tumors. The top and bottom 20 genes are listed here, and the top and bottom 1000 probes are listed in Supplemental Table 1.
Supplementary data
Figure S1 HRAS codon 61 gene sequencing of DMBA/TPA skin tumors from FVB/N mice and the PDV cell line.
Table S2 Reversal of BRAF inhibitor‐induced transcriptional changes in tumors by celecoxib. Lists of the microarray probes and their gene information for the gene groups defined in the clustering analysis of Figure 3. Each group is listed in a separate tab. Group 3 is split into sub‐groups 3A and 3B.