

Cyclopropanes are a unique class of structure elements found in a number of biologically important compounds and have been demonstrated for a wide range of fundamental and practical applications.[1] One particularly attractive strategy for enantioselective synthesis of chiral cyclopropanes is based on transition metal-catalyzed asymmetric olefin cyclopropanation with diazo reagents.[2] In principle, chiral cyclopropane derivatives with all substitution patterns may be accessible in enantioenriched form by asymmetric cyclopropanation due to the diverse availability of both alkenes and diazo reagents. Among different classes of diazo reagents with various combinations of α-substituents, many acceptor- and donor/acceptor-substituted diazo reagents have been successfully employed as effective carbene sources for metal-catalyzed asymmetric cyclopropanation.[2] In contrast, the capacity of catalytic asymmetric cyclopropanation has not been fully explored with acceptor/acceptor-substituted diazo reagents, which would afford synthetically useful cyclopropane compounds bearing geminal electron-withdrawing functionalities.[3,4] Although there have been some recent successes in this area,[5–9] several important types of acceptor/acceptor-substituted diazo reagents remain challenging for asymmetric olefin cyclopropanation.
The dicarbonyl diazo reagents α-ketodiazoacetates (KDA) represent one type of acceptor/acceptor-substituted diazo reagents that have not been effectively utilized for asymmetric cyclopropanation (Scheme 1). This catalytic process would be highly attractive as the resulting chiral 1,1-cyclopropaneketoesters, which bear both ketone and ester functionalities, can serve as versatile synthons for a wide range of useful asymmetric transformations. In addition to their ready conversion to chiral cyclopropane derivatives having different geminal functionalities,[5a,10] 1,1-cyclopropaneketoesters can be transformed to other valuable chiral molecules through various ring-opening and ring-expanding reactions, which are greatly facilitated by the presence of two electron-withdrawing groups at the geminal position (Scheme 1).[1c,1d,3b] While its non-asymmetric protocols have already been fruitfully applied to natural product synthesis,[11] there have been only a few previous reports on catalytic systems for asymmetric cyclopropanation with KDA.[5a,12,13] Among them, the most notable example is the Rh2-based system developed recently by Charette and coworkers.[5a] It was shown that the PMP-substituted KDA could be successfully used for asymmetric cyclopropanation, producing the corresponding (Z)-1,1-cyclopropaneketoesters in high diastereo- and enantioselectivity when conducted at low temperature (−40 °C). Aside from the fact that PMP was required as stereoselectivity controlling groups,[4b,5d] this Rh2-catalyzed system, however, was limited to aromatic olefins and generally gave low yields (10–70%) even using excess olefins (5.0 equiv). It would be synthetically desirable if effective catalytic systems could be developed for asymmetric cyclopropanation with simple KDA for substrates beyond styrene derivatives. Furthermore, would it be achievable to access both (E)- and (Z)-1,1-cyclopropaneketoesters in highly asymmetric manner?
Scheme 1.
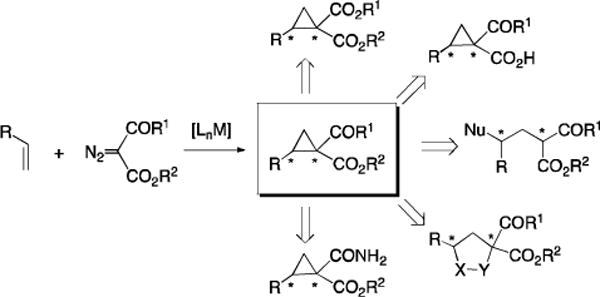
Synthesis of 1,1-Cyclopropaneketoesters by Metal-Catalyzed Asymmetric Cyclopropanation with α-Ketodiazoacetates and Further Transformations.
Cobalt(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] have emerged as a new class of effective catalysts for asymmetric cyclopropanation.[6a,6b,6d,14] Increasing evidence supports that the Co(II)-based metalloradical cyclopropanation proceeds with stepwise radical mechanism and possesses a distinct reactivity and selectivity profile from electrophilic cyclopropanation by the widely studied Rh2- and Cu-based closed-shell systems.[15] To date, Co(II)-based metalloradical catalysis (MRC) has been shown to be effective for asymmetric cyclopropanation reactions of different types of olefins with several classes of diazo reagents, including acceptor/acceptor-substituted diazo reagents such as α-cyanodiazoacetates (CDA)[6b] and α-nitrodiazoacetates (NDA)[6d]. To further exploit the unique potential of Co(II)-MRC, we began to investigate the possibility of [Co(D2-Por*)]-based catalysts for asymmetric cyclopropanation using acceptor/acceptor-substituted diazo reagents beyond CDA and NDA. Considering the aforesaid challenges in the area, we embarked on a specific project to investigate the use of dicarbonyl diazo reagents such as KDA for asymmetric intermolecular cyclopropanation via Co(II)-MRC. In view of the radical nature of Co(II)-based metalloradical cyclopropanation,[15] it was unclear at the onset of this project if [Co(D2-Por*)] could allow for achieving high control of enantioselectivity and diastereoselectivity through discriminating two similar carbonyl groups of common KDA. As the result of this investigation, herein we wish to report the first catalytic system for asymmetric cyclopropanation with simple α-acetodiazoacetates (ADA) that is effective for different kinds of olefins, leading to high-yielding synthesis of 1,1-cyclopropaneketoesters as (E)-diastereomers in high enantiomeric purity. In addition to using olefins as limiting substrates, this highly asymmetric catalytic system can be operated at room temperature without the need of slow addition of the diazo reagents. Furthermore, we describe an unprecedented epimerization process promoted by NaI that allows for conversion of the resulting (E)-1,1-cyclopropaneketoesters to their (Z)-isomers with complete retention of the enantiopurity.
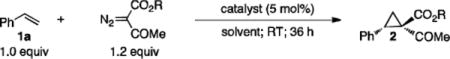
The cobalt(II) complex of the D2-symmetric chiral porphyrin 3,5-DitBu-ChenPhyrin, [Co(P1)] (Figure 1a), was shown to be an effective catalyst for asymmetric cyclopropanation with NDA[6d] and CDA.[6b] Its catalytic effectiveness toward these two acceptor/acceptor-substituted diazo reagents was attributed to the double hydrogen bonding interactions between the amide N–H donors on the P1 ligand and two acceptors on the carbene moiety in the postulated metallocarbene radical intermediate.[6b,6d] Since the C=O unit of ketone groups is also known as a suitable hydrogen bond acceptor, we hypothesized the potential existence of similar double hydrogen bonding interactions in the resulting cobalt-carbene radical intermediate from the reaction with KDA (Figure 1b). This hypothesis prompted us for a systematic effort to examine asymmetric cyclopropanation with common α-acetodiazoacetates using styrene (1a) as a model substrate (Table 1). We were pleased to observe that the simple methyl acetodiazoacetate (MADA) could be effectively activated by [Co(P1)] to cyclopropanate styrene under mild conditions with a practical protocol (at room temperature with the alkene as the limiting reagent without slow addition of the diazo reagent), affording the desired cyclopropane in near quantitative yield (entry 2). In addition to the excellent yield, it is important to note that the (E)-diastereoselectivity exhibited by the Co(II)-based system is the opposite of that previously reported for Rh2-based catalytic systems (entry 1).[4b,5d,16] Furthermore, the (E)-selectivity allowed us to improve the diastereoselectivity of the catalytic system significantly by employing bulkier tert-butyl acetodiazoacetate (t-BADA) while increasing its enantioselectivity at the same time (entry 3). Among solvents screened, toluene was found to be the solvent of choice, producing 1,1-cyclopropaneketoester 2a in 88% yield with 92% de and 94% ee (entries 3–6). The relative and absolute configurations of 2a were established as (E) and [1R,2S], respectively, by anomalous-dispersion effects in X-ray diffraction measurement of its single crystal (Figure S1).
Figure 1.
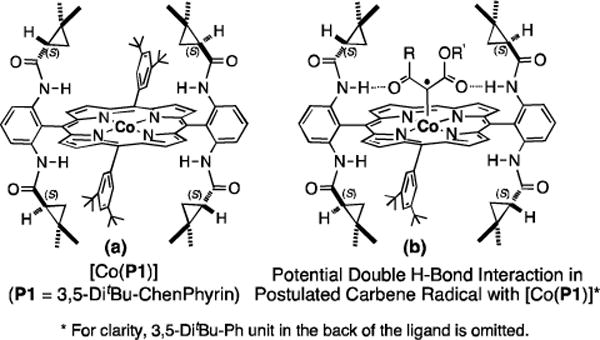
Structures of D2-symmetric Chiral Co(II) Porphyrins.
Table 1.
Asymmetric Cyclopropanation of Styrene with ADA by Metalloradical Catalyst [Co(P1)].[a]
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | CO2R | catalyst | solvent | yield (%)[b] | (E):(Z) | ee(%)[c] |
| 1 | CO2Me | Rh2(Oct)4 | DCM | 62 | 22:78 | – |
| 2 | CO2Me | Co(P1) | DCM | 99 | 79:21 | 63 |
| 3 | CO2tBu | Co(P1) | DCM | 86 | >99:1 | 90 |
| 4 | CO2tBu | Co(P1) | PhCl | 81 | >99:1 | 92 |
| 5 | CO2tBu | Co(P1) | Hexanes | 71 | 96:4 | 82 |
| 6 | CO2tBu | Co(P1) | Toluene | 88 | 96:4 | 94[d] |
Reactions were carried out in a one-time protocol using 5 mol % [Co(P1)] under N2 at RT for 36 h with [olefin] = 0.20 M.
Isolated yields.
Enantiomeric excess of major (E)-diastereomer determined by chiral HPLC.
[1R,2S] Absolute configuration determined by X-ray diffraction measurement on single crystal.
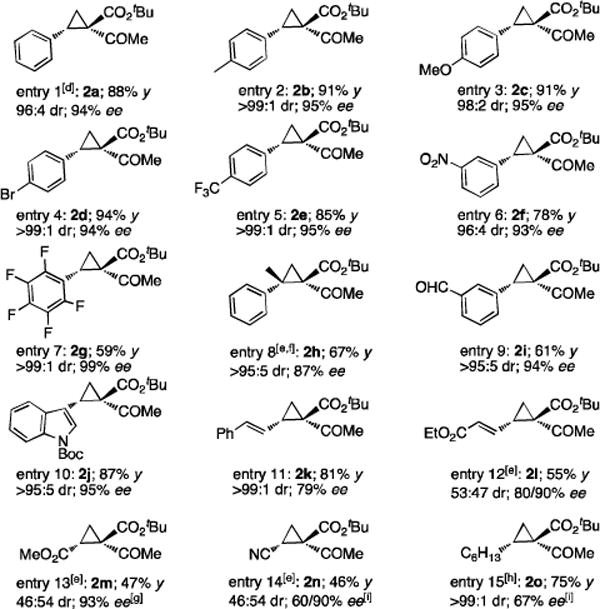
The [Co(P1)]-catalyzed asymmetric cyclopropanation could be successfully applied for a wide range of olefins under similar reaction conditions (Table 2). For example, styrene derivatives, regardless of the position and electronic property of the substituent (including electron-donating Me and MeO as well as electron-withdrawing Br, CF3, and NO2 groups), could be reliably cyclopropanated with t-BADA, generating the corresponding cyclopropaneketoesters 2a–f in high yields with both high diastereoselectivity and enantioselectivity (entries 1–6). As a further highlight of the unique catalytic property of the Co(II)-MRC, cyclopropanation of even the extremely electron-deficient pentafluorostyrene could be positively performed to form the desired cyclopropane 2g with complete control of stereoselectivities, albeit in a lower yield (entry 7). The use of α-methylstyrene allowed for the stereoselective construction of cyclopropane 2h with two contiguous all-carbon quaternary stereogenic centers (entry 8). Furthermore, the Co(II)-based asymmetric cyclopropanation could be effectively applied for styrenes containing sensitive functionalities such as aldehyde group (entry 9) and olefins derived from heteroarenes such as indole (entry 10) without complications arising from potential ylide-mediated reactions. When conjugated alkenes were used as substrates, regioselective cyclopropanation of the terminal double bonds was observed, affording the corresponding vinyl cyclopropanes in moderate to good yields with varied stereoselectivities (entries 11 and 12). Electron-deficient olefins such as α,β-unsaturated esters and nitriles, which have been known to be challenging substrates for cyclopropanation, could be also asymmetrically cyclopropanated by [Co(P1)] with t-BADA, but with diminished diastereoselectivity (entries 13 and 14). Under a neat condition, aliphatic alkenes, which represent another type of problematic substrates for metal-catalyzed asymmetric cyclopropanation, were also successfully converted into the desired cyclopropanes as exemplified with 1-octene for highly diastereoselective formation of cyclopropane 2o, despite with moderate enantioselectivity (entry 15).
Table 2.
[Co(P1)]-Catalyzed Diastereo- and Enantioselective Cyclopropanation of Various Alkenes with t-BADA.[a–c]

|
|---|
|
See Table 1.
[1R,2S] Absolute configuration determined by X-ray diffraction measurements on single crystal.
5.0 equiv. of olefin.
At 40 ºC.
Enantiomeric excess of minor.
Neat reaction.
Determined by chiral GC.
The Co(II)-based metalloradical cyclopropanation displayed a different sense of diastereoselectivity from the reported Rh2-based catalytic systems,[4b,5d,16] allowing for high-yielding production of (E)-1,1-cyclopropaneketoesters in high enantiopurity. In an effort to gain accesses of enantiopure 1,1-cyclopropaneketoesters in both (E)-and (Z)-diastereomeric forms, we explored the possibility of obtaining the corresponding (Z)-diastereomers through stereospecific epimerization of the resulting (E)-diastereomers 2. Two major methods have been previously developed for the epimerization of cyclopropane carbonyl derivatives (Scheme 2). The first method involved a pathway of ring cleavage and reformation mediated by in situ generated methoxy dimethyl sulfonium iodide (Scheme 2A).[17] The sulfonium ion was proposed to function as a Lewis acid to facilitate the cleavage of cyclopropane ring by coordinating with the carbonyl group of the formylcyclopropanes. Due to the destruction of both chiral centers in the acyclic intermediate, racemization was observed for this type of epimerization.[17b] The second method proceeded via a cyclopropane enol intermediate by employing a sequence of deprotonation and protonation under basic condition (Scheme 2B).[4b,17b,18] Although racemization could be avoided through the use of suitable bases, this pathway was not compatible for the epimerization of 1,1-cyclopropaneketoesters due to lack of the required acidic C–H bonds on the cyclopropane ring at the position adjacent to the carbonyl group. In search for a suitable epimerization method, we postulated a stereocontrolled epimerization pathway through ring-opening and -closing steps by a succession of double SN2 reactions (Scheme 2C) This proposal is in part based on the increased electrophilicity of 1,1-cyclopropaneketoesters as a result of the two geminal electron-withdrawing groups.[3b,19]
Scheme 2.
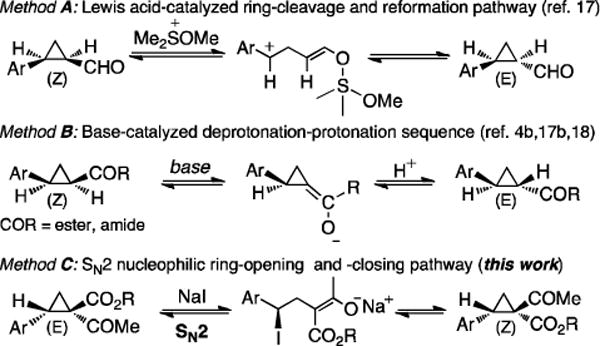
Epimerization of Cyclopropane Carbonyls.
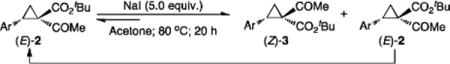
The key to the realization of the proposed SN2-type epimerization pathway is the identification of a suitable reagent that could function as both nucleophile and leaving group. Given that I− ion is known as a good leaving group as well as a nucleophile, ionic iodide reagents were targeted for such dual role (Scheme 2C). While cyclopropane ring-closing via intramolecular SN2 reaction with I− as leaving group is unknown, iodides have been previously employed as nucleophiles for ring-opening of cyclopropanes.[20] Among various iodides having different counter cations that were evaluated, NaI was shown to be the best reagent for promoting epimerization of 1,1-cyclopropaneketoester (Table S1). As demonstrated with (E)-cyclopropane 2a, treatment of its acetone solution with 5 equiv NaI at 80 °C resulted in the formation of (Z)-cyclopropane 3a with preservation of the high enantiomeric excess (eq 1). The epimerization was found to be reversible and could reach equilibrium within 20 h, with a diastereomeric ratio of 38:62 for (E)-2a:(Z)-3a. When isolated (Z)-3a was treated with NaI under the identical conditions, the same distribution of (E)-2a and (Z)-3a was obtained. Although counterintuitive, these results indicate that the (Z)-isomer is relatively more stable than the (E)-isomer (eq 1). To the best of our knowledge, this represents the first example of cyclopropane epimerization allowing conversion of (E)- to sterically encumbered (Z)-diastereomer without loss of optical purity.
 |
(1) |
The NaI-promoted epimerization process appeared to be general and could be applied to different (E)-1,1-cyclopropaneketoesters for the synthesis of corresponding (Z)-1,1-cyclopropaneketoesters with retention of their original enantiopurity (Table 3). Taking advantage of the reversibility of the epimerization process, the overall yield for conversion to (Z)-1,1-cyclopropaneketoesters could be improved by subjecting the remaining (E)-isomers to a second round of epimerization after their isolation from the first round. For example, the isolated yield of (Z)-3a from (E)-2a was increased to 70% without racemization by using this double epimerization procedure (entry 1). Similarly, (Z)-1,1-cyclopropaneketoesters 3d and 3e could be obtained in 65% and 70% yields, respectively, with maintenance of high enantiopurity from the corresponding (E)-isomers (entries 2 and 3). The epimerization procedure was also successfully applied for the indole-based 1,1-cyclopropaneketoester 2j, affording (Z)-3j in 68% yield and 95% ee (entry 4).
Table 3.
Stereospecific Epimerization of 1,1-Cyclopropaneketoesters Promoted by Sodium Iodide.
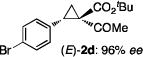
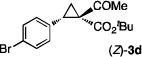
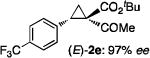
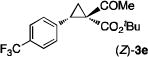
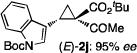
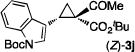
Combined isolated yields from two succesive epimerization processes.
Enantiomeric excess determined by chiral GC.
Enantiomeric excess determined by chiral HPLC.
In summary, we have demonstrated that metalloradical catalyst [Co(P1)] is highly effective for asymmetric olefin cyclopropanation with α-ketodiazoacetates (KDA). This represents the first successful application of Co(II)-based metalloradical catalysis (MRC) for asymmetric cyclopropanation with acceptor/acceptor-substituted diazo reagents bearing two α-carbonyl groups. In addition to high enantioselectivity, the [Co(P1)]-catalyzed cyclopropanation displays a distinct sense of diastereoselectivity, permitting for the first time the direct synthesis of chiral (E)-1,1-cyclopropaneketoesters from a broad range of alkenes with simple KDA. Furthermore, we have uncovered an iodide-promoted stereospecific epimerization process that allows for the conversion of enantioenriched (E)-1,1-cyclopropaneketoesters to their (Z)-diastereomers with retention of optical purity. Together, these two processes provide practical access to both (E)- and (Z)-1,1-cyclopropaneketoesters in a highly asymmetric manner.
Supplementary Material
Acknowledgments
We are grateful for financial support by NSF (CHE-1152767) and NIH (R01-GM098777).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Carson CA, Kerr MA. Chem Soc Rev. 2009;38:3051–3060. doi: 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]; b) Reichelt A, Martin SF. Acc Chem Res. 2006;39:433–442. doi: 10.1021/ar030255s. [DOI] [PubMed] [Google Scholar]; c) Yu M, Pagenkopf BL. Tetrahedron. 2005;61:321–347. [Google Scholar]; d) Reissig HU, Zimmer R. Chem Rev. 2003;103:1151–1196. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]
- 2.a) Pellissier H. Tetrahedron. 2008;64:7041–7095. [Google Scholar]; b) Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; c) Davies HML, Antoulinakis EG. Org React. 2001;57:1–326. [Google Scholar]; d) Doyle MP, Forbes DC. Chem Rev. 1998;98:911–935. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- 3.For selected reviews on applications of cyclopropanes bearing geminal electron-withdrawing groups, see ref. 1a and; a) Campbell MJ, Johnson JS, Parsons AT, Pohlhaus PD, Sanders SD. J Org Chem. 2010;75:6317–6325. doi: 10.1021/jo1010735. [DOI] [PubMed] [Google Scholar]; b) Danishefsky S. Acc Chem Res. 1979;12:66–72. [Google Scholar]
- 4.For recent examples on applications of cyclopropanes bearing geminal electron-withdrawing groups see:; a) Cerat P, Gritsch PJ, Goudreau SR, Charette AB. Org Lett. 2010;12:564–567. doi: 10.1021/ol902766f. [DOI] [PubMed] [Google Scholar]; b) Marcoux D, Goudreau SR, Charette AB. J Org Chem. 2009;74:8939–8955. doi: 10.1021/jo902066y. [DOI] [PubMed] [Google Scholar]; c) Pohlhaus PD, Sanders SD, Parsons AT, Li W, Johnson JS. J Am Chem Soc. 2008;130:8642–8650. doi: 10.1021/ja8015928. [DOI] [PubMed] [Google Scholar]; d) Perreault C, Goudreau SR, Zimmer LE, Charette AB. Org Lett. 2008;10:689–692. doi: 10.1021/ol702414e. [DOI] [PubMed] [Google Scholar]; e) Lifchits O, Charette AB. Org Lett. 2008;10:2809–2812. doi: 10.1021/ol8009286. [DOI] [PubMed] [Google Scholar]; f) Lifchits O, Alberico D, Zakharian I, Charette AB. J Org Chem. 2008;73:6838–6840. doi: 10.1021/jo8010705. [DOI] [PubMed] [Google Scholar]; g) Jackson SK, Karadeolian A, Driega AB, Kerr MA. J Am Chem Soc. 2008;130:4196–4201. doi: 10.1021/ja710289k. [DOI] [PubMed] [Google Scholar]; h) Young IS, Kerr MA. J Am Chem Soc. 2007;129:1465–1469. doi: 10.1021/ja068047t. [DOI] [PubMed] [Google Scholar]; i) Pohlhaus PD, Johnson JS. J Am Chem Soc. 2005;127:16014–16015. doi: 10.1021/ja055777c. [DOI] [PubMed] [Google Scholar]
- 5.For recent examples on asymmetric cyclopropanation with acceptor/acceptor-substituted diazo reagents by Rh-based catalysts, see:; a) Lindsay VNG, Nicolas C, Charette AB. J Am Chem Soc. 2011;133:8972–8981. doi: 10.1021/ja201237j. [DOI] [PubMed] [Google Scholar]; b) Nishimura T, Maeda Y, Hayashi T. Angew Chem Int Ed. 2010;49:7324–7327. doi: 10.1002/anie.201003775. [DOI] [PubMed] [Google Scholar]; c) Marcoux D, Azzi S, Charette AB. J Am Chem Soc. 2009;131:6970–6972. doi: 10.1021/ja902205f. [DOI] [PubMed] [Google Scholar]; d) Marcoux D, Charette AB. Angew Chem Int Ed. 2008;47:10155–10158. doi: 10.1002/anie.200804586. [DOI] [PubMed] [Google Scholar]; e) Doyle MP, Davies SB, Hu WH. Org Lett. 2000;2:1145–1147. doi: 10.1021/ol005730q. [DOI] [PubMed] [Google Scholar]
- 6.For recent examples on asymmetric cyclopropanation with acceptor/acceptor-substituted diazo reagents by Co-based catalysts, see:; a) Xu X, Lu HJ, Ruppel JV, Cui X, de Mesa SL, Wojtas L, Zhang XP. J Am Chem Soc. 2011;133:15292–15295. doi: 10.1021/ja2062506. [DOI] [PubMed] [Google Scholar]; b) Zhu SF, Xu X, Perman JA, Zhang XP. J Am Chem Soc. 2010;132:12796–12799. doi: 10.1021/ja1056246. [DOI] [PubMed] [Google Scholar]; c) Doyle MP. Angew Chem Int Ed. 2009;48:850–852. doi: 10.1002/anie.200804940. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhu SF, Perman JA, Zhang XP. Angew Chem Int Ed. 2008;47:8460–8463. doi: 10.1002/anie.200803857. [DOI] [PubMed] [Google Scholar]
- 7.For recent examples on asymmetric cyclopropanation with acceptor/acceptor-substituted phenyliodonium ylides by Cu-based catalysts, see:; a) Deng C, Wang L-J, Zhu J, Tang Y. Angew Chem Int Ed. 2012;51:11620–11623. doi: 10.1002/anie.201206376. [DOI] [PubMed] [Google Scholar]; b) Moreau B, Charette AB. J Am Chem Soc. 2005;127:18014–18015. doi: 10.1021/ja056192l. [DOI] [PubMed] [Google Scholar]
- 8.For asymmetric cyclopropanation with other acceptor/acceptor-substituted phenyliodonium ylides, see:; a) Ghanem A, Gardiner MG, Williamson RM, Muller P. Chem-Eur J. 2010;16:3291–3295. doi: 10.1002/chem.200903231. [DOI] [PubMed] [Google Scholar]; b) Muller P, Ghanem A. Org Lett. 2004;6:4347–4350. doi: 10.1021/ol048159u. [DOI] [PubMed] [Google Scholar]; c) Muller P, Allenbach YF, Ferri M, Bernardinelli G. Arkivoc. 2003:80–95. [Google Scholar]; d) Muller P, Allenbach Y, Robert E. Tetrahedron-Asymmetr. 2003;14:779–785. [Google Scholar]
- 9.Lindsay VNG, Fiset D, Gritsch PJ, Azzi S, Charette AB. J Am Chem Soc. 2013;135:1463–1470. doi: 10.1021/ja3099728. [DOI] [PubMed] [Google Scholar]
- 10.a) Chen GQ, Tang XY, Shi M. Chem Commun. 2012;48:2340–2342. doi: 10.1039/c2cc17581a. [DOI] [PubMed] [Google Scholar]; b) Zhang DY, Zhang R, Xiang DX, Zhang N, Liang YJ, Dong DW. Synthesis-Stuttgart. 2012;44:705–710. [Google Scholar]; c) Zhao QW, Curran DP, Malacria M, Fensterbank L, Goddard JP, Lacote E. Synlett. 2012:433–437. [Google Scholar]
- 11.a) Nicolaou KC, Ding HF, Richard JA, Chen DYK. J Am Chem Soc. 2010;132:3815–3818. doi: 10.1021/ja9093988. [DOI] [PubMed] [Google Scholar]; b) Campbell MJ, Johnson JS. J Am Chem Soc. 2009;131:10370–10371. doi: 10.1021/ja904136q. [DOI] [PubMed] [Google Scholar]; c) Ito H, Takeguchi S, Kawagishi T, Iguchi K. Org Lett. 2006;8:4883–4885. doi: 10.1021/ol061947u. [DOI] [PubMed] [Google Scholar]
- 12.For intramolecular asymmetric cyclopropanation with dicarbonyl-substituted carbenoids, see ref. 6a and; a) Honma M, Sawada T, Fujisawa Y, Utsugi M, Watanabe H, Umino A, Matsumura T, Hagihara T, Takano M, Nakada M. J Am Chem Soc. 2003;125:2860–2861. doi: 10.1021/ja029534l. [DOI] [PubMed] [Google Scholar]; b) Ida R, Nakada M. Tetrahedron Lett. 2007;48:4855–4859. [Google Scholar]; c) Takeda H, Honma M, Ida R, Sawada T, Nakada M. Synlett. 2007:579–582. [Google Scholar]
- 13.For indirect asymmetric synthesis of 1,1-cyclopropaneketoesters, see:; Muller P, Bernardinelli G, Allenbach YF, Ferri M, Flack HD. Org Lett. 2004;6:1725–1728. doi: 10.1021/ol049554n. [DOI] [PubMed] [Google Scholar]
- 14.a) Zhu SF, Ruppel JV, Lu HJ, Wojtas L, Zhang XP. J Am Chem Soc. 2008;130:5042–5043. doi: 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]; b) Chen Y, Zhang XP. J Org Chem. 2007;72:5931–5934. doi: 10.1021/jo070997p. [DOI] [PubMed] [Google Scholar]; c) Chen Y, Ruppel JV, Zhang XP. J Am Chem Soc. 2007;129:12074–12075. doi: 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]
- 15.a) Lu HJ, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J Am Chem Soc. 2011;133:8518–8521. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]; b) Belof JL, Cioce CR, Xu X, Zhang XP, Space B, Woodcock HL. Organometallics. 2011;30:2739–2746. doi: 10.1021/om2001348. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J Am Chem Soc. 2010;132:10891–10902. doi: 10.1021/ja103768r. [DOI] [PubMed] [Google Scholar]
- 16.Davies HML, Bruzinski PR, Fall MJ. Tetrahedron Lett. 1996;37:4133–4136. [Google Scholar]
- 17.a) Sasaki T, Eguchi S, Ohno M. Bull Chem Soc Jpn. 1980;53:1469–1470. [Google Scholar]; b) Yamaguchi K, Kazuta Y, Abe H, Matsuda A, Shuto S. J Org Chem. 2003;68:9255–9262. doi: 10.1021/jo0302206. [DOI] [PubMed] [Google Scholar]
- 18.a) Ortizdemontellano PR, Dinizo SE. J Org Chem. 1978;43:4323–4328. [Google Scholar]; b) Feit BA, Elser R, Melamed U, Goldberg I. Tetrahedron. 1984;40:5177–5180. [Google Scholar]
- 19.For select examples of stereocontrolled nucleophilic ring-opening of cyclopropanes, see ref. 4f (alcohols as nucleophiles), ref. 4e (amines as nucleophiles), and refs. 4b & 5a (cuprates as nucleophiles).
- 20.a) Alper PB, Meyers C, Lerchner A, Siegel DR, Carreira EM. Angew Chem Int Ed. 1999;38:3186–3189. [PubMed] [Google Scholar]; b) Bertozzi F, Gustafsson M, Olsson R. Org Lett. 2002;4:3147–3150. doi: 10.1021/ol0264814. [DOI] [PubMed] [Google Scholar]; c) Lautens M, Han WS. J Am Chem Soc. 2002;124:6312–6316. doi: 10.1021/ja011110o. [DOI] [PubMed] [Google Scholar]; d) Lautens M, Han W, Liu JHC. J Am Chem Soc. 2003;125:4028–4029. doi: 10.1021/ja028967l. [DOI] [PubMed] [Google Scholar]; e) Ma SM, Lu LH, Zhang JL. J Am Chem Soc. 2004;126:9645–9660. doi: 10.1021/ja0494860. [DOI] [PubMed] [Google Scholar]; f) Scott ME, Han W, Lautens M. Org Lett. 2004;6:3309–3312. doi: 10.1021/ol048769u. [DOI] [PubMed] [Google Scholar]; g) Scott ME, Lautens M. J Org Chem. 2008;73:8154–8162. doi: 10.1021/jo802049u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.