

Abstract
Diastereoselective aza-Wacker cyclization of O-allyl hemiaminals under aerobic conditions enables efficient access to 1,2-aminoalcohol derivatives from allylic alcohols. The scope of this method is presented and its utility is highlighted in a streamlined synthesis of the biologically important aminosugar (–)-acosamine.
Keywords: palladium, oxidation, stereoselective catalysis, amination, Wacker oxidation, organic synthesis
The stereoselective synthesis of vicinal aminoalcohols from simple starting materials is a prominent challenge in organic chemistry. The prevalence of the vicinal aminoalcohol moiety in biologically active molecules, as well as the difficulty with which the 1,2-oxidation pattern can be synthesized, has justified the development of a diverse array of approaches to access these molecules.[1] Intermolecular oxidative difunctionalization of alkenes is an appealing strategy for the generation of the 1,2-aminooxygenation pattern, but methods with a combination of high stereo- and regioselectivity and diverse scope remain elusive. Examples include Sharpless asymmetric aminohydroxylation,[2] metal-catalyzed activation of oxaziridines,[3] and palladium-catalyzed aminoacetoxylation reactions that employ hypervalent iodine oxidants.[4] Our interest in palladium-catalyzed aerobic oxidation of alkenes (Wacker-type reactions) prompted us to investigate new methods for the synthesis of vicinal aminoalcohols from readily available, stereochemically defined starting materials. Here, we employ a detachable, tethered nitrogen nucleophile to generate the 1,2-aminooxygenation pattern from allylic alcohols via an aza-Wacker cyclization (Scheme 1a).[5 , 6 , 7] The cyclization step forms 5-membered oxazolidine products and exhibits high levels of diastereoselectivity. This strategy is amenable to the de novo synthesis of aminosugars, which are key substructures of several antibiotic and anticancer natural products (Scheme 1b).[8] Implementation of a redox-relay approach9 enables rapid synthesis of (−)-acosamine and highlights the utility of this method.
Scheme 1.

a) Detachable tethered nucleophile approach for the synthesis of vicinal aminoalcohols from allylic alcohols and b) aminosugars in natural product antibiotic and anticancer agents.
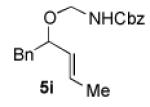
We began our studies with an assessment of the reactivity and diastereoselectivity of Wacker cyclization when using distinct tethering units for the attachment of oxygen or nitrogen nucleophiles to secondary allylic amine or allylic alcohol substrates (Table 1). These efforts included the allylic N-tosyl carbamate 1 (entries 1-3) and N-allyl hemiaminal 3 (entries 4-6) substrates, reported previously by Bäckvall[5f] and Hiemstra,[5a] as well as aminal 5a. Assessment of a variety of catalyst conditions for aerobic oxidative cyclization revealed PdII/dimethylsulfoxide-based catalysts to be the most promising.[10] Effective catalyst systems included Pd(DMSO)2-(TFA)2 in THF,[10c] which is effective at ambient temperature (condition A), Pd(TFA)2 in DMSO, which is compatible with higher reaction temperatures (condition B), and Pd(OAc)2 in DMSO, resembling conditions originally discovered by Larock[10a] and Hiemstra[10b] (condition C). Allylic N-tosyl carbamate substrate 1 was susceptible to decomposition under these direct aerobic reoxidation conditions.[11] When cyclization to oxazolidinone 2 occurred (e.g., entry 2), only modest diastereoselectivity was observed. N-Allyl hemiaminal 3 cyclized to the corresponding oxazolidine 4 in moderate yields, but the diastereoselectivity of the transformation was poor.[12] O-Allyl hemiaminals are an appealing class of substrates because they may be synthesized directly from allylic alcohols, but they have not been tested previously in Wacker-type cyclizations. The benzyl carbamate (Cbz) derivative 5a, underwent cyclization in excellent yield and good diastereoselectivity to afford the trans oxazolidine 6a (entries 7-9). Conditions A and B were particularly effective.
Table 1.
Evaluation of diastereoselective oxidative cyclization of substrates derived from an allylic alcohol or an allylic amine.
| entry | substrate | major product |
conditions[a] | yield (%) / d.r.[b] |
|---|---|---|---|---|
| 1 |

|
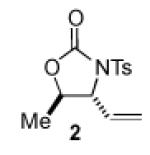
|
A | 0 / nd |
| 2 | B | 11 / 3.5:1[c] | ||
| 3 | C | 0 / nd | ||
| 4 |

|
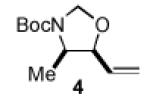
|
A | 34 / 1.6:1 |
| 5 | B | 51 / 1.6:1 | ||
| 6 | C | 67 / 1.8:1 | ||
| 7 |

|
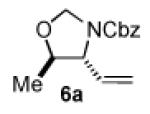
|
A | 93 / 9:1 |
| 8 | B | 94 / 8:1 | ||
| 9 | C | 77 / 5:1 |
Catalyst condition A: 5 mol% Pd(TFA)2, 20 mol% DMSO, 20 mol% NaOBz, 3Å M.S., THF (0.1 M), 25 °C, 24 h, 1 atm O2. Catalyst condition B: 5 mol% Pd(TFA)2, 20 mol% NaOBz, 3Å M.S., DMSO (0.1 M), 60 °C, 24 h, 1 atm O2. Catalyst condition C: 5 mol% Pd(OAc)2, DMSO (0.1 M), 60 °C, 24 h, 1 atm O2.
Yield / diastereomeric ratio based on 1H NMR spectroscopic analysis of the crude reaction mixture with phenyltrimethylsilane as the internal standard.
Isolated yield. Diastereomeric ratio based on 1H NMR analysis of the purified products.
The three-step detachable tethered nucleophile approach for the conversion of trans crotyl alcohol to vicinal aminoalcohol derivative 8 is illustrated in Scheme 2. Trans-crotyl alcohol is readily converted to the corresponding O-allyl hemiaminal, and aza-Wacker cyclization proceeds smoothly at ambient temperature (condition A). The oxazolidine ring is unmasked under acidic conditions. Several additional primary O-allyl hemiaminal substrates were similarly effective. Comparison of benzyl and tert-butyl carbamate (Cbz- and Boc-) O-allyl hemiaminals revealed that the Cbz-derived nitrogen nucleophile is more effective (cf. 6c and 6d). A propyl-substituted alkene produced oxazolidine 6e with negligible alkene isomerization.[13] Finally, reaction with a trisubstituted alkene affords a tertiary C–N bond (cf. 6f); condition B was more effective than condition A in this reaction.
Scheme 2.
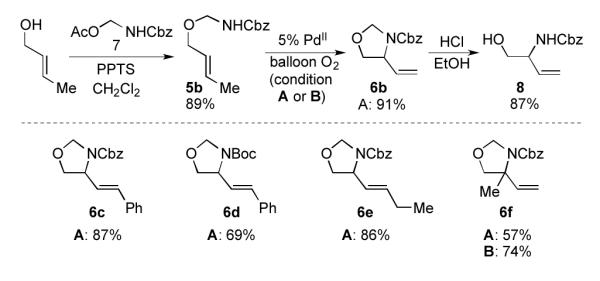
Transformation of primary allylic alcohols to vicinal aminoalcohols. (PPTS = pyridinium p-toluenesulfonate)
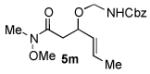
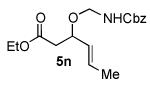
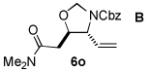
We next explored the scope of the diastereoselective cyclization of O-allyl hemiaminals derived from secondary alcohols (Table 2). Both trans and cis allylic alcohols are good substrates and afford the same trans-4,5-disubstituted oxazolidine (entries 2-3). The effectiveness of the trans-allylic alcohol substrate is noteworthy because such substrates are more readily accessible than the cis analog; however, higher diastereoselectivity can be achieved with the cis substrate. Increasing the size of the secondary allylic substituent improves the diastereoselectivity, and condition B provides higher reactivity with these more sterically encumbered substrates (entries 4-6). Cyclic O-allyl hemiaminals 5j and 5k yield the cis ring-fused products in good yields (entries 7-8). Allylic alcohols derived from aldol additions to crotonaldehyde also provide effective O-allyl hemiaminal substrates (entries 9-12). Ketone adduct 5l is prone to decomposition under the reaction conditions and provided only modest yield of oxazolidine 6l, but the Weinreb amide 5m, ethyl ester 5n, and dimethylamide 5o are well-tolerated.
Table 2.
Diastereoselective synthesis of oxazolidines from cyclization of secondary O-allyl hemiaminals.
| entry | substrate | product | conditions[a] | yield (%) / d.r.[b] |
|---|---|---|---|---|
| 1 |
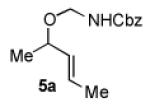
|

|
A | 86 / 12:1 |
| 2 | B | 88 / 11:1 | ||
| 3 |

|
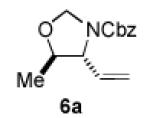
|
B | 73 / >20:1 |
| 4 |

|
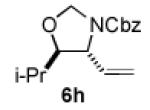
|
A | 72 / >20:1 |
| 5 | B | 85 / >20:1 | ||
| 6 |
|
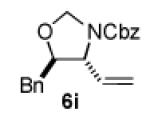
|
B | 75 / >20:1 |
| 7 |

|
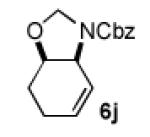
|
B | 78 / >20:1 |
| 8 |

|

|
B | 59 / >20:1 |
| 9 |
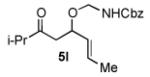
|
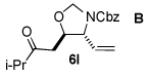
|
B | 53 / >20:1 |
| 10 |
|

|
B | 62 / 9:1 |
| 11 |
|

|
B | 74 / 19:1 |
| 12 |

|
|
B | 75 / 9:1 |
See Table 1 for descriptions of catalyst conditions A and B.
Isolated yields. Diastereomeric ratio based on 1H NMR spectroscopic analysis of the isolated material. Relative configurations of 6a and 6j were assigned by NOESY-1D spectroscopic analysis (other structures assigned by analogy).
The aldol reaction is one of many potential routes to enantioenriched allylic alcohols[14] and thus can provide access to enantioenriched aminoalcohol derivatives in connection with our method. Asymmetric aldol chemistry enabled the synthesis of enantioenriched allylic alcohol 9 from crotonaldehyde,[15] which was converted to the corresponding O-allyl hemiaminal 5p and cyclized cleanly to oxazolidine 6p (Scheme 3). Oxazolidine 6p was obtained with exquisite diastereoselectivity and no epimerization of the methyl stereocenter.
Scheme 3.
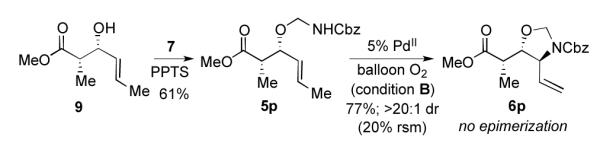
Transformation of enantioenriched allylic alcohol to the oxazolidine with no epimerization.
The utility of this method is illustrated in the synthesis of the 2-deoxy-3-aminosugar (−)-acosamine.[16] 2-Deoxy-3-aminosugars can be obtained via synthesis of the corresponding acyclic 1,3-aminoaldehyde precursor. We envisioned that the aminoaldehyde could be accessed rapidly by employing our cyclization in a redox-relay sequence, whereby cyclization of an O-allyl hemiaminal derived from a bis-allylic alcohol precursor would generate a masked aldehyde intermediate (Scheme 4). Transfer of the olefin redox equivalent via β-hydride elimination from the second allylic alcohol position would form an enol ether and expedite elaboration of the oxazolidine to the desired aminosugar target.
Scheme 4.

Retrosynthetic analysis of 2-deoxy-3-aminosugars with application of aza-Wacker cyclization and redox-relay.
This redox-relay strategy enabled the synthesis of (−)-N-Cbz-O-methylacosamine, a conveniently isolated derivative of acosamine, from TBS-protected (−)-lactaldehyde in five steps (Scheme 5). First, vinyllithium addition of silyl-protected allylic alcohol 11 to lactaldehyde 10 yielded 12 with the desired stereochemistry. Installation of the O-allyl hemiaminal and aza-Wacker cyclization provided the silyl enol ether 14 with good efficiency as a single diastereomer.[17] The silyl enol ether and secondary TBS-ether were removed to unveil the free aldehyde and alcohol. Finally, the oxazolidine ring was opened under acidic conditions with concomitant formation of the methyl-protected cyclic acetal 16 [(−)-N-Cbz-O-methylacosamine]. This rapid and redox economical synthesis of (−)-acosamine illustrates a versatile approach that could be applied to a variety of other aminosugar derivatives.
Scheme 5.

Synthesis of (−)-N-Cbz-O-methylacosamine
In summary, we have demonstrated that aza-Wacker cyclizations can be used to synthesize stereodefined vicinal aminoalcohols from allylic alcohol precursors by employing a detachable tethered nucleophile approach. These oxidative functionalization reactions are operationally simple and the catalytic reactions give clean conversion of starting material to desired product. The diversity of methods for the asymmetric synthesis of allylic alcohols makes the diastereoselective transformation described here particularly apt for application to a variety of contexts.
Supplementary Material
Footnotes
We thank the NIH (R01 GM67163) and Organic Syntheses (ACS Division of Organic Chemistry fellowship for A.B.W.) for financial support of this work. Spectroscopic instrumentation was partially funded by the NSF (CHE-1048642, CHE-0342998, CHE-9208463).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Adam B. Weinstein, Department of Chemistry University of Wisconsin-Madison 1101 University Avenue, Madison, WI 53706 (USA)
David P. Schuman, Department of Chemistry University of Wisconsin-Madison 1101 University Avenue, Madison, WI 53706 (USA)
Zhi Xu Tan, Department of Chemistry University of Wisconsin-Madison 1101 University Avenue, Madison, WI 53706 (USA).
Prof. Shannon S. Stahl, Department of Chemistry University of Wisconsin-Madison 1101 University Avenue, Madison, WI 53706 (USA).
References
- [1].Bergmeier SC. Tetrahedron. 2000;56:2561–2576. [Google Scholar]
- [2] a).For leading references, see: Li GG, Chang HT, Sharpless KB. Angew. Chem. 1996;108:449–452.; Angew. Chem. Int. Ed. 1996;35:451–454.; Reddy KL, Sharpless KB. J. Am. Chem. Soc. 1998;120:1207–1217.; O’Brien P. Angew. Chem. 1998;111:339–342.; Angew. Chem. Int. Ed. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T.; Donohoe TJ, Callens CKA, Flores A, Lacy AR, Rathi AH. Chem. Eur. J. 2011;17:58–76. doi: 10.1002/chem.201002323.
- [3] a).Michaelis DJ, Ischay MA, Yoon TP. J. Am. Chem. Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. [DOI] [PubMed] [Google Scholar]; c) Williamson KS, Yoon TP. J. Am. Chem. Soc. 2012;134:12370–12373. doi: 10.1021/ja3046684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4] a).Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; b) Martínez C, Wu Y, Weinstein AB, Stahl SS, Liu G, Muñiz K. J. Org. Chem. 2013;78:6309–6315. doi: 10.1021/jo400671q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).For related examples of the use of detachable tethered nucleophiles in the synthesis of 1,2-aminoalcohol derivatives, see: van Benthem RATM, Hiemstra H, Speckamp WN. J. Org. Chem. 1992;57:6083–6085.; van Benthem RATM, Hiemstra H, Longarela GR, Speckamp WN. Tetrahedron Lett. 1994;35:9281–9284.; Espino CG, Du Bois J. Angew. Chem. 2001;113:618–620.; Angew. Chem. Int. Ed. 2001;40:598–600.; Lei AW, Liu G, Lu X. J. Org. Chem. 2002;67:974–980. doi: 10.1021/jo0161429.; Fraunhoffer KJ, White MC. J. Am. Chem. Soc. 2007;129:7274–7176. doi: 10.1021/ja071905g.; Joosten A, Persson AKA, Millet R, Johnson MT, Bäckvall J-E. Chem. Eur. J. 2012;18:15151–15157. doi: 10.1002/chem.201202359.
- [6].For related work from our group on the synthesis of diamines, see: McDonald RI, Stahl SS. Angew. Chem. 2010;122:5661–5664. doi: 10.1002/ange.200906342.; Angew. Chem. Int. Ed. 2010;49:5529–5532. doi: 10.1002/anie.200906342.
- [7] a).For examples of intramolecular aminooxygenation reactions that generate heterocyclic products, see: Donohoe TJ, Johnson PD, Cowley A, Keenan M. J. Am. Chem. Soc. 2002;124:12934–12935. doi: 10.1021/ja0276117.; Alexanian EJ, Lee C, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k.; Desai LV, Sanford MS. Angew. Chem. 2007;119:5839–5842.; Angew. Chem. Int. Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454.; Fuller PH, Kim JW, Chemler SR. J. Am. Chem. Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m.; Lovick HM, Michael FE. J. Am. Chem. Soc. 2010;132:1249–1251. doi: 10.1021/ja906648w.; Schmidt VA, Alexanian EJ. J. Am. Chem. Soc. 2011;133:11402–11405. doi: 10.1021/ja204255e.; Donohoe TJ, Callens CKA, Lacy AR, Winter C. Eur. J. Org. Chem. 2012:655–663.; Liu G, Zhang Y, Yuan Y, Xu H. J. Am. Chem. Soc. 2013;135:3343–3346. doi: 10.1021/ja311923z.
- [8] a).For leading references, see: Arcamone F, Penco S, Vigevani A, Redaelli S, Franchi G, Dimarco A, Casazza AM, Dasdia T, Formelli F, Necco A, Soranzo C. J. Med. Chem. 1975;18:703–707. doi: 10.1021/jm00241a013.; Mallams AK. In: Carbohydrate Chemistry. Kennedy JF, editor. Clarendon Press; Oxford: 1988. pp. 73–133.; Myers AG, Kort ME, Hammond MA. J. Am. Chem. Soc. 1997;119:2965–2972.; Croatt MP, Carreira EM. Org. Lett. 2011;13:1390–1393. doi: 10.1021/ol2000765.; Wilcock BC, Endo MM, Uno BE, Burke MD. J. Am. Chem. Soc. 2013;135:8488–8491. doi: 10.1021/ja403255s.
- [9] a).Burns NZ, Baran PS, Hoffmann RW. Angew. Chem. 2009;121:2896–2910. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:2854–2876. [Google Scholar]; b) Werner EW, Mei TS, Burckle AJ, Sigman MS. Science. 2012;338:1455–1458. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mei TS, Werner EW, Burckle AJ, Sigman MS. J. Am. Chem. Soc. 2013;135:6830–6833. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10] a).See reference 6 and: Larock RC, Hightower TR. J. Org. Chem. 1993;58:5298–5300.; van Benthem RATM, Hiemstra H, Michels JJ, Speckamp WN. J. Chem. Soc. Chem. Commun. 1994:357–59.; Diao T, White P, Guzei I, Stahl SS. Inorg. Chem. 2012;51:11898–11909. doi: 10.1021/ic301799p.
- [11].See reference 5f for a report that employs benzoquinone to promote the aza-Wacker cyclization of allylic N-tosyl carbamates.
- [12].These results are consistent with prior results reported in reference 5a by van Benthem et al.
- [13].Reversible β-hydride elimination can allow for alkene isomerization: Ye X, Liu G, Popp BV, Stahl SS. J. Org. Chem. 2011;76:1031–1044. doi: 10.1021/jo102338a.
- [14].For a recent review, see: Lumbroso A, Cooke ML, Breit B. Angew. Chem. 2013;125:1942–1986. doi: 10.1002/anie.201204579.; Angew. Chem. Int. Ed. 2013;52:1890–1932. doi: 10.1002/anie.201204579.
- [15] a).Evans DA, Bartroli J, Shih TL. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]; b) Gage JR, Evans DA. Org. Synth. 1990;68:83–91. [Google Scholar]
- [16] a).For leading references on other syntheses of (−)-acosamine and related aminosugars, see references 5e, 8d, 8e and: Trost BM, Sudhakar AR. J. Am. Chem. Soc. 1987;109:3792–3794.; Hirama M, Shigemoto T, Ito S. J. Org. Chem. 1987;52:3342–3346.; Ager DJ, East MB. Tetrahedron. 1993;49:5683–5765.; Kirschning A, Jesberger M, Schoning KU. Synthesis. 2001:507–540.; Sames D, Polt R. J. Org. Chem. 1994;59:4596–4601.; Myers AG, Liang J, Hammond M, Harrington PM, Wu Y, Kuo EY. J. Am. Chem. Soc. 1998;120:5319–5320.; Nicolaou KC, Baran PS, Zhong Y, Vega JA. Angew. Chem. 2000;112:2625–2629.; Angew. Chem. Int. Ed. 2000;39:2525–2529.; Ginesta X, Pasto M, Pericas MA, Riera A. Org. Lett. 2003;5:3001–3004. doi: 10.1021/ol034843h.; Levy DE, Fugedi P. The Organic Chemistry of Sugars. CRC Press; Florida: 2006.
- [17].Optimization of the concentration and reaction time were necessary to minimize the slow decomposition of the silyl enol ether product under the reaction conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.