

Abstract
Insulin regulates glucose uptake by controlling the subcellular location of GLUT4 glucose transporters. GLUT4 is sequestered within fat and muscle cells during low-insulin states, and is translocated to the cell surface upon insulin stimulation. The TUG protein is a functional tether that sequesters GLUT4 at the Golgi matrix. To stimulate glucose uptake, insulin triggers TUG endoproteolytic cleavage. Cleavage accounts for a large proportion of the acute effect of insulin to mobilize GLUT4 to the cell surface. During ongoing insulin exposure, endocytosed GLUT4 recycles to the plasma membrane directly from endosomes, and bypasses a TUG-regulated trafficking step. Insulin acts through the TC10α GTPase and its effector protein, PIST, to stimulate TUG cleavage. This action is coordinated with insulin signals through AS160/Tbc1D4 and Tbc1D1 to modulate Rab GTPases, and with other signals to direct overall GLUT4 targeting. Data support the idea that the N-terminal TUG cleavage product, TUGUL, functions as a novel ubiquitin-like protein modifier to facilitate GLUT4 movement to the cell surface. The C-terminal TUG cleavage product is extracted from the Golgi matrix, which vacates an “anchoring” site to permit subsequent cycles of GLUT4 retention and release. Together, GLUT4 vesicle translocation and TUG cleavage may coordinate glucose uptake with physiologic effects of other proteins present in the GLUT4-containing vesicles, and with potential additional effects of the TUG C-terminal product. Understanding this TUG pathway for GLUT4 retention and release will shed light on the regulation of glucose uptake and the pathogenesis of type 2 diabetes.
Keywords: GLUT4, TUG, insulin, glucose uptake, proteolysis, Golgi, protein trafficking, unconventional secretion
1. Introduction
Mammals have evolved a finely tuned system to control the storage and mobilization of nutrients. Insulin is the main anabolic hormone, which promotes the uptake and storage of carbohydrates, as well as synthesis of lipid and protein. During low insulin states or in response to counter-regulatory hormones, these stores are mobilized to provide sustenance during periods of limited food availability. Much research during the past decades has focused on understanding how insulin exerts its metabolic effects. Here, we focus on recent advances in understanding a hallmark insulin action: stimulation of glucose uptake into fat and muscle.
To promote glucose uptake, insulin causes the movement of glucose transporters from intracellular membranes to the cell surface. Pioneering work during the 1980s discovered this translocation mechanism in adipocytes [1,2], identified GLUT1, the first facilitative glucose transporter [3], and led to the molecular cloning of GLUT4, the main glucose transporter present in muscle and fat [4-8]. When GLUT4 was identified, it was shown that insulin causes the translocation of this protein, similar to its earlier-described effect on glucose transport activity. Fig. 1 presents an example of this effect, using cultured 3T3-L1 adipocytes. GLUT4 translocation in muscle and adipose is impaired during insulin resistant states, spurring investigation of how translocation normally occurs and how it is affected during the pathogenesis of type 2 diabetes [9].
Figure 1. Insulin stimulated GLUT4 glucose transporter translocation.
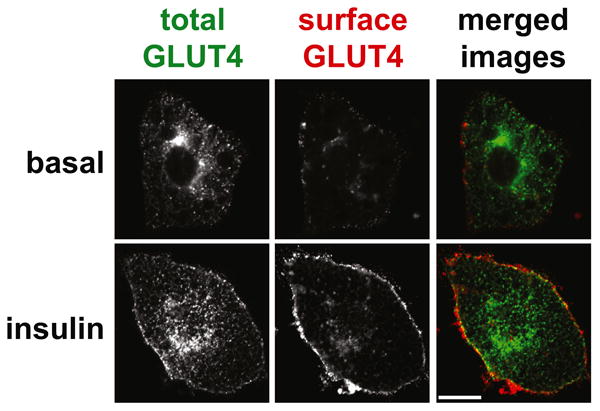
The images show cultured 3T3-L1 adipocytes that express a GLUT4 reporter protein, which contains a 7Myc epitope tag in its first extracellular loop as well as GFP fused at the C-terminus. Cells were serum starved, treated with or without insulin as indicated, then stained to detect the externalized 7Myc epitope tag. Images were acquired by confocal microscopy of GFP (total GLUT4, shown in green in the merged images) and Myc epitope (surface GLUT4, shown in red in the merged images). Scale bar, 10 μM. Reproduced from Yu, C., et al., J Biol Chem (2007) 282, 7710-7722.
Ongoing research has elucidated the main signaling pathways by which insulin acts to redistribute GLUT4 in fat and muscle. Binding of insulin activates the tyrosine kinase activity of its receptor, which phosphorylates insulin receptor substrate (IRS) proteins (particularly IRS1) and activates phosphatidylinositol-3-kinase (PI3K) activity and other downstream effectors [10,11]. PI3K signals through the serine-threonine kinases Akt2 [12,13] and PKCλ [14] to control GLUT4 translocation. Major downstream targets of Akt2 are AS160/Tbc1D4 and Tbc1D1, which modulate the activities of particular Rab proteins (discussed below) to control GLUT4 targeting and glucose uptake [15-18]. In muscle, downstream action through Rac1 mediates effects on cortical actin [19-22]. At least two IRS- and PI3K- independent insulin signals control GLUT4 trafficking. The activated insulin receptor phosphorylates Munc18c directly to promote the fusion of GLUT4-containing vesicles at the cell surface [23]. Insulin also signals through adaptor proteins linking the insulin receptor to the Rho-family GTPase TC10α, which acts on multiple targets to control GLUT4 movement [24-26]. In particular, TC10α acts through its effector, PIST, to stimulate endoproteolytic cleavage of TUG proteins and to mobilize intracellular GLUT4-containing vesicles. This mechanism and how it contributes to overall insulin action are the main topics of this review.
Understanding of the intracellular trafficking itinerary utilized by GLUT4 has lagged behind understanding of the signaling pathways activated by insulin. Beginning in the 1990s, studies by Holman and others defined three main compartments through which GLUT4 transits, based on the kinetics of GLUT4 movement in basal and insulin-stimulated adipocytes [27,28]. These compartments included the plasma membrane, endosomes, and an ill-defined insulin-responsive compartment. Insulin acts acutely to mobilize GLUT4 from the insulin-responsive compartment, and has less effect on rates at other trafficking steps. This result suggested that the bulk of insulin's acute effect is mediated by action at a single trafficking step, and it focused subsequent work to characterize this step in molecular terms. The insulin-responsive compartment is cell type-specific, develops early during the differentiation of 3T3-L1 fibroblast-like cells into adipocytes, and causes intracellular sequestration of GLUT4 within cells not stimulated by insulin [29]. Yet, it remained unclear how developmentally regulated proteins that form this compartment intersect with insulin-regulated proteins involved in GLUT4 targeting. In addition, insulin acts at multiple sites to control GLUT4 movement [25]. Thus, it remained to determine which of these sites is quantitatively most important for insulin-regulated glucose uptake in vivo, and to understand if the same or different sites control the targeting of GLUT4 during acute and sustained insulin actions.
2. GLUT4 Storage Vesicles
The cellular correlate of the kinetically-defined, insulin-responsive compartment is the pool of GLUT4 Storage Vesicles (GSVs). These 50-70 nm diameter carriers are thought to exist as a preformed pool in unstimulated adipocytes; upon insulin stimulation, the pool is depleted concomitant with GLUT4 insertion into the plasma membrane [30,31]. The GSVs have sedimentation and density properties that are not unique, and they have been purified most effectively using immunoadsorption to remove intracellular transport vesicles that are not insulin-responsive [29]. This approach has facilitated proteomic characterization, which complemented earlier studies of GSV cargos [32-34]. Together, the data imply that GSVs have a limited protein composition. As well as GLUT4, the most abundant cargos include IRAP, an insulin-regulated aminopeptidase with a single transmembrane domain. IRAP is thought to cotraffic with GLUT4 throughout its itinerary, and when translocated to the cell surface it cleaves and inactivates vasopressin [35]. GSVs also contain LRP1, a low-density lipoprotein receptor-related protein, which has many functions including uptake of particular lipoproteins as well as Wnt signaling [34]. Sortilin is a developmentally regulated, transmembrane sorting adaptor required for GSV formation, which may participate in lipoprotein uptake; the related SorL1 protein is also present [36-39,34]. Finally, data indicate that GSVs contain SNARE proteins such as syntaxin 6 and VAMP2, which likely participate in their formation and fusion, and the p75 neurotrophin receptor, which recruits particular Rab GTPases involved in trafficking [40,24,29]. Because proteins that affect vasopressin action (IRAP) and lipid metabolism (LRP1, sortilin, SorL1) are coregulated with GLUT4, defective GSV translocation may contribute not only to insulin-resistant glucose uptake, but also to hypertension and dyslipidemia in the metabolic syndrome [25].
GSVs are thought to form by mass action, in which the main cargos coalesce through interactions among their luminal domains [38,39]. This excludes other proteins from the nascent vesicle, and forms a cytosolic scaffold for the recruitment of membrane budding components, such as clathrin adaptors (ACAP1, GGA2) and GTPases (ARF6) [41]. Transient modification of GLUT4 by ubiquitin is required for its sorting into GSVs, and may recruit clathrin adaptors [42]. Proteins that are peripherally associated with GSV membranes, such as TUG and AS160, are likely recruited during vesicle budding by interacting with GSV cargos [32,43,34,44]. In cells not stimulated with insulin, these proteins mediate intracellular sequestration of GSVs. Efficient recruitment to the GSVs during vesicle budding may be required to capture the vesicles, and thus to confer insulin-responsive mobilization.
Whether GLUT4 itself is important for regulated GSV trafficking has been debated. An alternative is that GLUT4 contains signals that target it to the GSVs, but that it then functions only as a passenger. Interpretation of studies of IRAP trafficking in the absence of GLUT4, and of GLUT4 trafficking in the absence of IRAP, has been complicated by 1) effects of impaired sequestration in GSVs to reduce protein stability, and 2) incomplete sequestration of GSVs within 3T3-L1 adipocytes, as compared to primary adipocytes [24]. In aggregate, the data show that IRAP, and possibly LRP1, are more important than GLUT4 for insulin-responsive trafficking of the GSVs [45-47,34]. However, GLUT4 also contains signals that contribute to insulin-responsive IRAP trafficking, particularly in primary adipocytes [48,49,46,50]. Newly synthesized GLUT4 enters GSVs prior to traversing the plasma membrane and endosomal system, and sequences in the GLUT4 N-terminus and large cytosolic loop are required for its insulin-responsive translocation [51,52]. An additional peptide within the cytosolic N-terminus of IRAP has been implicated in insulin-responsive trafficking [53,54]. These sequences bind TUG and ACAP1, which may account for their role in insulin-responsive GSV trafficking [44,41, and B.R. Rubin, E.N. Habtemichael, and J.S. Bogan, unpublished observations]. Each binary interaction of a GSV protein and cytosolic regulator may be weak, but if several such interactions occur then in aggregate they may account for efficient vesicle recognition, entrapment, and insulin-stimulated release.
3. Two distinct types of vesicles carry GLUT4 to the cell surface
In adipocytes, insulin can increase glucose uptake by 10- to 30- fold. The remarkable magnitude of this effect reflects the efficient sequestration of GLUT4 away from the plasma membrane in unstimulated cells. In cultured 3T3-L1 adipocytes, sequestration is less complete, and maximal insulin stimulation typically produces a 4- to 8- fold increase in GLUT4 at the cell surface. Kinetic modeling suggested that GLUT4 translocated within ∼5 min. of acute insulin stimulation originates from GSVs, but that subsequently exocytosed GLUT4 may recycle from endosomes [55,56]. This idea has now received experimental support [57]. Exocytosis of GSVs and endosomes were distinguished, based on the different diameters of these vesicles (∼60 vs. ∼150 nm, respectively), using total internal reflection fluorescence microscopy (TIRFM). The data show that a distinct population of GLUT4-containing vesicles, characteristic of GSVs, develops during 3T3-L1 adipocyte differentiation and is regulated by TUG. In fully differentiated, unstimulated 3T3-L1 adipocytes, the bulk of GSV cargo arriving at the surface was present in vesicles with characteristic of endosomes. After 3-6 min. of insulin, the exocytosis rate increased ∼4-fold and most of the exocytic vesicles had features of GSVs. After >15 min. insulin exposure, the increased exocytosis rate was largely maintained, but the exocytic vesicles were again endosome-sized. Thus, acute insulin stimulation causes a switch in the exocytic carrier that is used (Figure 2). As discussed below, this arrangement obviates the requirement for ongoing TUG destruction during sustained insulin action.
Figure 2. A model of GLUT4 trafficking pathways.
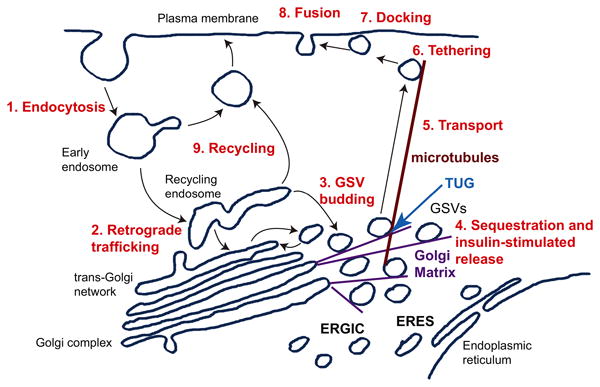
GLUT4 undergoes endocytosis from the plasma membrane (1), and is targeted by retrograde trafficking to the recycling endosome and trans-Golgi network (2). Specialized, insulin-responsive GLUT4 Storage Vesicles (GSVs) likely bud from one or both of these locations (3), and become trapped at the Golgi matrix near the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) and endoplasmic reticulum exit sites (ERES). The TUG protein is required for GSV sequestration at the Golgi matrix, and is cleaved upon insulin stimulation to mobilize the GSVs (4). The released GSVs are transported on microtubules to the cell periphery (5), and undergo insulin-stimulated tethering (6), docking (7), and fusion (8) at the plasma membrane. During the continued presence of insulin, endocytosed GLUT4 recycles directly to the plasma membrane from endosomes (9), and bypasses the TUG-regulated mechanism for GLUT4 sequestration and release. Adapted from Bogan, J.S. (2012) Annu Rev Biochem, 81, 507-532.
In the physiological setting, cells are not taken so rapidly from an unstimulated state to a maximally insulin-stimulated state. Yet, the idea that GLUT4 participates in two distinct exocytic pathways fits well with a physiologic role of insulin to stimulate a “quantal release” mechanism [56]. Increasing concentrations of insulin cause the release of discrete amounts of GLUT4 from a sequestered compartment (i.e. GSVs) into a plasma membrane recycling pathway [58]. The mobilization of discrete numbers of GSVs can be accomplished by TUG cleavage, which is proposed as a critical mechanism to liberate these sequestered vesicles. Then, after a larger meal, more insulin is secreted and a larger number of TUG proteins are cleaved, so that a greater number of GSVs are released to the cell surface. This action also implies that there is a nonlinear signaling action of insulin, which converts an “analog” input (insulin concentration) to a “digital” output (protein cleavage). As discussed further below, the upstream signal that activates TUG cleavage may involve a feed-forward circuit [59,60]. This may explain how signaling may respond to the rate of change of an insulin input signal, and to generate a pulse output that cleaves a number of proteins proportional to this rate.
Once released, the GLUT4 cycles between the plasma membrane and endosomes in the steady-state presence of insulin (Figure 2). As noted above, the idea that it bypasses the GSV compartment during sustained insulin action is compatible with earlier kinetic models, and is supported by TIRFM data from 3T3-L1 adipocytes [57]. Recent data from transgenic mice are also consistent with this view [59]. Mice with constitutive and unregulated TUG cleavage in skeletal muscle had increased glucose turnover and quadriceps-specific glucose uptake during fasting, but there was no effect in these parameters during hyperinsulinemic-euglycemic clamp experiments. Together, the data support a model in which 1) TUG is required to sequester the GSVs away from the cell surface during low-insulin states, and 2) TUG cleavage mediates the bulk of the effect of acute insulin action, but 3) GLUT4 recycles at the cell surface without passing through a TUG-regulated compartment during ongoing, steady-state insulin exposure.
The TIRFM data (together with other data) also support the idea that TUG is not involved in sorting of GLUT4 to the GSVs, but that it regulates intracellular retention and release of the GSVs themselves [26,57]. Specifically, in TUG-depleted 3T3-L1 adipocytes, not only was the basal rate of vesicle fusion at the cell surface similar to the insulin-stimulated rate observed in control cells, but the size of the exocytic vesicles was characteristic of GSVs, not endosomes. Thus, sorting of GSV cargos (VAMP2 and GLUT4) into these vesicles does not require TUG. Additionally, the GSVs fuse directly at the plasma membrane, and are not translocated by first fusing with another endomembrane system such as endosomes. Finally, the small size of the GSVs makes it difficult to detect these vesicles and their exocytosis using TIRFM [61,62,57]. In primary adipocytes, the rapidly moving vesicles that fuse at the plasma membrane after insulin stimulation may in fact be the “second wave” of GLUT4 exocytosis, which derives from endosomes. Of note, most GLUT4 in muscle was not mobile in the unstimulated state [63].
The two distinct exocytic pathways are likely regulated by distinct Rab GTPases that are activated by insulin signaling through AS160/Tbc1D4 and Tbc1D1. In adipocytes, Rab10 is likely present on GSVs, whereas Rab14 controls GLUT4 in an endosomal compartment [64-66]. In muscle, Rab8a or Rab13 (which are closely related to Rab10) may be on GSVs, and Rab14 may regulate other GLUT4-containing compartments [66-69]. Insulin regulation of these Rab GTPases is likely one mechanism by which insulin switches the targeting of endocytosed GLUT4 between the two exocytic circuits.
4. Sequestration of GSVs at the Golgi matrix
The Golgi matrix includes several long, coiled-coil proteins, golgins, which serve as tentacles to entrap vesicles in the vicinity of the Golgi complex [70]. These proteins are typically anchored at one end to Golgi membranes, and they can extend away from these membranes by ∼100-400 nm. Along this length, golgins are studded with binding sites for particular Rab GTPases, which permit them to recognize and capture particular vesicles carrying cognate, activated Rab isoforms. The TUG carboxyl terminus binds to Golgin-160 and the associated proteins PIST (PDZ protein that interacts specifically with TC10) also known as CAL, GOPC, or FIG) and ACBD3 (Acyl-coenzyme A binding domain containing 3, also known as GPC60 or PAP7) [58, J.P. Belman and J.S. Bogan, unpublished observations]. As the TUG amino terminus binds GSV components, TUG may act similarly to an activated Rab protein to enable the selective capture of GSVs by these Golgi matrix proteins. In unstimulated cells, the vesicles may be held in a relatively static configuration, or they may cycle into and out of a GSV donor membrane compartment. Either way, the vesicles are retained at the Golgi matrix in a configuration from which they can be acutely mobilized upon receipt of an insulin signal.
Data indicate that the GSVs are retained at a pre-Golgi location. Golgin-160 likely acts together with p115, another Golgi matrix protein that binds IRAP and LRP1, to sequester GSVs within unstimulated cells [71,72,34]. Like TUG, Golgin-160 and p115 are present at the cis-Golgi, endoplasmic reticulum-Golgi intermediate compartment (ERGIC), and endoplasmic reticulum exit sites (ERES) [73]. Golgin-160 is linked to the microtubule cytoskeleton through dynein, whereas ACBD3 and p115 each bind giantin, the largest golgin, which may facilitate scaffolding [74-77]. Because the GSVs are mobilized directly to the cell surface from a pre-Golgi location, we proposed that GSV translocation follows an unconventional secretion pathway [25,58].
Unconventional secretion was first hypothesized as a mechanism for the export of soluble cargos that lack a signal sequence, and that do not enter the classical endoplasmic reticulum-Golgi complex secretory pathway [78,79]. More recently, it has been appreciated that membrane proteins such as CFTR and β1-integrin can be translocated to the cell surface by a similar, unconventional Golgi-bypass pathway [80-82]. Like the targeting of CFTR, TUG-regulated GLUT4 translocation is controlled by signaling through the TC10α GTPase and its effector, PIST [58,59,83-85]. In yeast, the secretion of Acb1, an ortholog of ACBD3, occurs by an unconventional pathway in which pre-autophagosomal vesicles are targeted to the cell surface [78,86,87]. It is plausible that such a pathway could be regulated by PIST, which binds Beclin 1, a regulator of the Vps34 phosphatidylinositol-3-kinase complex that controls autophagic flux [88]. The site of GSV sequestration near the ERES and ERGIC is also compatible with the idea that autophagosome biogenesis occurs at these locations [89,90]. Rab10 is localized to ERES, and Rab8A regulates an autophagy-based unconventional secretory pathway for IL-1β [91,92]. The targeting of GLUT4 to such a secretory pre-autophagosomal compartment in unstimulated cells would show how a fundamental and evolutionarily conserved, nutritionally-regulated pathway can be adapted to mediate insulin action in fat and muscle.
4. TUG proteolytic processing and GSV mobilization
The TUG protein (encoded by the ASPSCR1 gene) was identified in a functional screen as a Tether, containing a ubiquitin-like UBX domain, for GLUT4 [93]. Because TUG is subject to proteolysis when bound to GLUT4, it is likely that this genetic approach was critical; indeed, biochemical approaches including proteomic analyses of GLUT4-containing vesicles have failed to detect this protein. Based initially on studies of the kinetics of GLUT4 movement, TUG was proposed to control the main insulin-responsive trafficking step. Subsequent data further support the view that intact TUG sequesters GSVs within unstimulated cells, and that insulin stimulates TUG endoproteolytic cleavage to liberate these vesicles for translocation to the plasma membrane [44,57-59,94]. Depletion of TUG using RNAi mimics much or all of insulin's acute action to redistribute GLUT4, to increase the GSV exocytosis rate, and to increase glucose uptake in 3T3-L1 adipocytes [44,57]. As noted above, insulin signals through TC10α and PIST to stimulate TUG cleavage. Depletion of TC10α using RNAi blocks insulin stimulated GLUT4 translocation and glucose uptake, and prevents the generation of TUG cleavage products in 3T3-L1 adipocytes [58,95]. The neuronal splice form of the TC10α effector, PIST (sometimes called nPIST), is present in muscle, binds directly to TUG, and can participate in a trimeric complex in which TUG links PIST with GLUT4. PIST also binds Golgin-160 and syntaxin-6, which is likely present on membranes that give rise to GSVs. Insulin signaling through TC10α–PIST–TUG is likely coordinated with signals through Akt2 and AS160/Tbc1D4 (in adipocytes) and Tbc1D1 (in muscle) (Figure 3A). TUG cleavage occurs in a site-specific manner at the bond linking residues 164-5, and separates an N-terminal region of TUG that binds GLUT4 from a C-terminal region of TUG that binds Golgin-160 (Figure 3B). Finally, a cleavage-resistant form did not support highly-insulin-responsive GLUT4 translocation, implying that cleavage is required for GSV mobilization [58]. Together, the data imply that TUG cleavage is a defining characteristic of insulin-responsive GSVs.
Figure 3. Insulin signaling and TUG endoproteolytic cleavage.

A. Insulin signals through at least two pathways to mobilize GLUT4 to the plasma membrane. One pathway comprises IRS-1, phosphatidylinositol-3-kinase (PI3K), and Akt2, which phosphorylates and inactivates the Rab GTPase Activating Proteins AS160/Tbc1D1 and/or Tbc1D4 to modulate Rab GTPase isoforms and direct vesicle trafficking. A second pathway is less well studied, and is proposed to involve signaling through APS/CAP/c-Cbl to CrkII and the Rho-family GTP Exchange Factor C3G, which activates the TC10α GTPase. TC10α then signals through its effector, PIST, to trigger TUG endoproteolytic cleavage. Because conflicting data have been reported for the TC10α pathway, upstream components are shown in gray. Insulin-stimulated cleavage separates regions of TUG that bind the Golgi matrix, including Golgin-160 and possibly other proteins, from those that bind GSV proteins, including GLUT4 and likely other cargos. Cleavage thus liberates GLUT4 for exocytic translocation to the plasma membrane. B. The domain structure of the TUG protein is depicted, with residues numbered according to the sequence of the mouse protein. Tandem ubiquitin-like domains (UBL1 and UBL2) are present at the N-terminus, and interact with GLUT4 and possibly other proteins present in GLUT4 vesicles. A third ubiquitin-like region, specifically a UBX domain, is also indicated (UBL3/UBX), and C-terminal regions of TUG bind to Golgin-160 and associated proteins at the Golgi matrix. Insulin stimulates cleavage of the bond linking residues Gly164 and Ser165. This cleavage site follows a Gly-Gly sequence, which is typical of ubiquitin-like precursor proteins. Proteolysis produces TUGUL, an 18 kD N-terminal product thought to function as a ubiquitin-like protein modifier, as well as a 42 kD C-terminal product. Cleavage separates regions of TUG that bind GSVs from regions that bind the Golgi matrix, and thus can liberate GSVs that are sequestered intracellularly at the Golgi matrix. Adapted from Bogan, J.S. et al., (2012) J Biol Chem 287, 23932-23947.
The N-terminal TUG cleavage product is thought to function as a novel 18 kDa ubiquitin-like protein modifier, termed TUGUL (for TUG Ubiquitin-Like). Ubiquitin and most ubiquitin-like proteins are produced by endoproteolytic cleavage of larger precursors [96-98]. Cleavage typically occurs after a conserved diglycine sequence, present in TUG at residues 163-164, and is catalyzed by proteases that are members of the family of deubiquitylating enzymes. Nearly 100 members of this family are encoded in the human genome, and it remains unknown which of these enzymes functions to cleave TUG. Cleavage of a ubiquitin-like precursor generates the mature ubiquitin-like modifier as the N-terminal cleavage product. This protein can be covalently attached to protein or lipid substrates by the actions of activating (E1), conjugating (E2), and (usually) ligating (E3) enzymes. Attachment to target proteins occurs between the C-terminal glycine of the modifier and (usually) the amino side chain of a lysine residue present in the target substrate. Modification can affect the enzymatic activity, subcellular targeting, or interactions of substrate proteins; classical attachment of ubiquitin chains targets substrate proteins to the proteasome for degradation. Similar to protein phosphorylation, ubiquitin-like modification is reversible. The same deubiquitylating enzymes that produce the mature modifier from the precursor protein can also remove the modifier from target substrates.
Similar to two other ubiquitin-like modifiers, ISG15 and FAT10, TUGUL contains tandem ubiquitin-like β-grasp domains. The structure of the first (UBL1) of these has been determined, but the second (UBL2) likely does not fold stably in the absence of an interacting partner, possibly GLUT4 or IRAP [44,99]. This may account for the observation that in 3T3-L1 adipocytes, TUG that is not membrane associated does not undergo proteolytic processing [58]. Like highly insulin-responsive GLUT4 translocation, TUG proteolysis occurs in a cell type –specific manner in fat and muscle cells [58,59]. The number of TUG proteins cleaved corresponds approximately to the number of GSVs that are mobilized to the cell surface. In 3T3-L1 adipocytes, insulin stimulated processing results in the attachment of TUGUL to a single ∼110 kDa target substrate. This TUGUL-modified (“tugulated”) protein cofractionates with GSVs and plasma membranes. Some data suggest it may be a kinesin motor, which has previously been found to facilitate microtubule-based movement of GSVs from the perinuclear region to the cell periphery [100,101, J.S. Bogan, unpublished observations]. Thus, insulin-stimulated TUG cleavage may both release a tether and also activate vesicle translocation to the cell surface.
The carboxyl-terminal TUG cleavage product is a 42 kDa protein that is generated exclusively on membranes, presumably still bound at the site to which intact TUG links GLUT4 in unstimulated cells [58]. The stability of this fragment is determined by its initial residue, Ser165, according to the mammalian N-end rule [102,103]. Serine is not a classical destabilizing residue. Yet neither is it fully stabilizing, because changing it to methionine increased the abundance of the TUG C terminal product [58]. In 3T3-L1 adipocytes, this fragment is modified to produce a 54 kDa form and simultaneously removed to the cytosol. Although the identity of this modification is not yet known, one possibility is that the modification acts to remove the cleavage product from the Golgi matrix. Removal would be required to vacate an “anchoring site” to which intact TUG links GSVs in unstimulated cells, so that this site will be available for subsequent cycles of GLUT4 retention and release. The modified form of the C-terminal was not observed in skeletal muscle [59]. One possibility is that the modification does not occur in muscle, yet it seems more likely that the modified fragment was not well solubilized in muscle tissue lysates. TUG C-terminal fragments accumulate in the nucleus in transfected cells, and contain a putative nuclear localization signal [73]. It is possible that the endogenously produced C-terminal fragment also enters the nucleus. Further work will be required to determine if this is the case.
TUG does not undergo proteolysis in fibroblast cells, and proteolytic products were observed in differentiated 3T3-L1 adipocytes, but not in preadipocytes [58]. Indeed, even in mature 3T3-L1 adipocytes, the degree to which insulin accelerated TUG proteolysis is variable because of the presence of cleavage products in unstimulated cells. By contrast, insulin-stimulated cleavage is robust and marked in primary tissues, including skeletal muscle [59] and fat (J.S. Bogan, unpublished observations). Thus, insulin-stimulated TUG cleavage is cell type –specific, and the TUG-regulated GSV trafficking pathway is likely an adaptation of a general membrane trafficking pathway that is present in most cells. Understanding this pathway will be important to shed light on GLUT4 translocation and how it is regulated by insulin.
A major binding partner for TUG in most cells is p97 (also known as VCP or, in yeast, cdc48), a chaperone that is a member of the family of ATPases associated with diverse cellular activities [73]. Unlike most other UBX-domain-containing proteins, TUG may not serve as an adaptor to couple p97 ATPase activity to various targets. Rather, TUG can disassemble the hexameric p97 ATPase into its monomeric subunits. This action is evolutionarily conserved, and inactivates ATPase activity in vitro [104,105]. Very recently, it was shown that TUG recruits the methyltransferase METTLD21 to a complex containing p97 and, probably, UBXD1 [106-108]. This results in methylation of p97, which negatively regulates its ATPase activity. This effect may regulate p97 ATPase activity at or near the ERES/ERGIC, and control the targeting of ubiquitylated cargos in endosomal and autophagy pathways [109-111]. Possibly, the ubiquitin-dependent sorting of GLUT4 may involve this mechanism [112].
5. TUG proteolysis in muscle, glucose homeostasis, and energy metabolism
Expression of an artificial, truncated C-terminal fragment of TUG, termed UBX-Cter, causes GLUT4 targeting to the cell surface and increases glucose uptake in 3T3-L1 adipocytes [44]. These actions are similar to the effects of RNAi-mediated TUG depletion or of insulin action and, together with other data, suggested that UBX-Cter binds and occupies the “anchoring site” at the Golgi matrix. According to this model, it cannot retain the vesicles intracellularly because it lacks sequences that interact with GSV proteins. Thus, UBX-Cter was considered to act as a dominant negative inhibitor of the action of intact TUG.
In fact, the UBX-Cter fragment mimics insulin action to stimulate endoprotolytic cleavage of the endogenous TUG protein [59]. The UBX-Cter protein contains degradation signals, including a ubiquitin-like UBX domain and PEST sequence, and data support the idea that it turns over rapidly [58]. It can effectively redistribute GLUT4, despite its very low abundance, implying that it acts irreversibly. We therefore considered that UBX-Cter may recruit an interacting protein for degradation, and examined PIST, Golgin-160, and ACBD3. Among these, PIST protein abundance (but not mRNA abundance) is markedly reduced [59]. The data support a model in which PIST normally binds TUG and inhibits TUG cleavage in unstimulated cells. Then, in transgenic muscle, the selective degradation of PIST by UBX-Cter permits the enzymatic activity of the TUG protease to cleave the endogenous TUG protein, similar to normal insulin action. This model also suggests how insulin normally acts though GTP-TC10α, which binds PIST and may then displace or reposition it within a PIST-TUG complex to enable the activity of the TUG protease. Definitive testing of this model will require identification of the protease, which is likely a deubiquitylating enzyme family member, as noted above.
Mice with muscle-specific transgenic expression of UBX-Cter have constitutive and unregulated TUG proteolysis during fasting in muscle [59]. GLUT4 is translocated to T-tubules and glucose uptake is increased during fasting. The mice have reduced fasting glucose and insulin concentrations, and increased whole-body glucose turnover during fasting. Although the transgene is likely not fully effective in all muscles, effects in quadriceps, where it is most highly expressed, are quite marked. The fraction of GLUT4 present in T-tubules is increased ∼5.7-fold. Total GLUT4 protein abundance is slightly decreased, likely because of reduced GLUT4 protein stability (which is also known to result from insulin action) [44,113,114]. The absolute increase in GLUT4 in T-tubule – enriched membrane fractions is ∼3.6-fold, which corresponds well with the ∼2.7-fold increase in quadriceps-specific glucose uptake observed during fasting. This rate of tissue-specific glucose uptake, in transgenic animals during fasting, is similar to that observed in wildtype animals during hyperinsulinemic-euglycemic clamp studies. Together, the data emphasize that TUG cleavage is a major action by which insulin stimulates glucose uptake acutely in muscle.
The increased muscle-specific glucose uptake resulted in a ∼17% increase in the rate of whole-body glucose turnover during fasting [59]. This effect would likely have been more marked, but for the limited action of the transgene in some tissues. It was initially surprising that the UBX-Cter was less effective in transgenic muscles, compared to 3T3-L1 adipocytes. Yet, it is now clear that efficient binding of TUG to ACBD3 requires acetylation of residues at the TUG C-terminus, which appears to be nearly complete in 3T3-L1 adipocytes, but which is probably more limited in muscle (J.P. Belman and J.S. Bogan, unpublished observations). Thus, one possibility is that efficient UBX-Cter-induced degradation of PIST requires an interaction between UBX-Cter and ACBD3. The transgenic mice had no increase in tissue-specific or whole-body glucose uptake during hyperinsulinemic-euglycemic clamp studies. This result is consistent with the idea that TUG acts in the unstimulated state to sequester GLUT4 and, as it is cleaved, to mobilize GLUT4 acutely, but that it does not participate in GLUT4 recycling through endosomes during steady-state insulin action (Fig. 2). Additionally, there was no additive biochemical effect of insulin and the UBX-Cter transgene to stimulate TUG proteolysis in quadriceps, which may also account for the absence of an effect of the transgene in hyperinsulinemic-euglycemic clamp studies.
Mice with transgenic expression of UBX-Cter in muscle have a 12-13% increase in their metabolic rate, compared to wildtype controls, as assessed by indirect calorimetry [59]. This appears not to result solely from effects of GLUT4 at the cell surface. Plasma lactate is decreased, not decreased as was observed in GLUT1 or GLUT4 transgenic mice [115-120]. Respiratory exchange ratio is unchanged, not increased as in GLUT4 transgenic mice [115]. Many earlier studies on GLUT-transgenic mice were done before the widespread use of indirect calorimetry for phenotyping. Nonetheless, it is likely that the increased metabolic rate in muscle UBX-Cter transgenic mice results from effects of proteins other than GLUT4. As noted above, GSVs contain IRAP, LRP1, sortilin, and other proteins that may act physiologically when translocated together with GLUT4 to the cell surface [25]. As well, proteolytic cleavage generates the TUG C-terminal product, which may have subsequent effects independent of its action as part of intact TUG. As noted above, this product is targeted to the nucleus in transfected cells, and it may participate in the control of genes regulating energy metabolism. Other proteins implicated in GLUT4 trafficking are better known as modulators of gene expression, including RIP140 (which interacts with PGC-1α and may regulate energy expenditure) and Daxx [121,122,101]. We speculate that these proteins function together with the TUG C-terminal product to coordinate gene expression and GSV translocation, and possibly to contribute to the thermic effect of food [123,124].
6. Regulated translocation and insulin resistance
In principle, impairment of insulin-regulated glucose uptake could result from defects in insulin signaling or vesicle trafficking (or both). Insulin signaling defects have been better characterized, and can occur in response to excess diacylglycerols and sphingolipids including ceramides [125]. Data indicate that insulin-independent defects in vesicle trafficking also occur. Intracellular targeting of GLUT4 and IRAP is altered during fasting in insulin-resistant individuals, compared to controls [126-128]. GLUT4 targeting defects are not well understood, but they may also result from effects of excess lipids. Other data show that IRS-1-independent defects are major nodes of insulin resistance, consistent with effects on non-IRS-1 signaling or insulin-independent trafficking [129]. TC10α signaling is cell type-specific and, in 3T3-L1 adipocytes, requires the association of TC10α with lipid raft membrane domains [130,131]. The upstream signaling components (e.g. CAP, flotillin) that mediate insulin-stimulated TC10α signaling are upregulated during 3T3-L1 adipocyte differentiation, and it is possible that excess lipids alter their function [25]. TC10α signaling has not been well studied, and the observation that APS and c-Cbl knockout mice had enhanced, rather than impaired, insulin action has led to uncertainty about the relevance of this pathway [132,133]. Yet, together with the idea that TC10α signaling results in quantized release of discrete amounts of GLUT4, the data are consistent with the idea that these proteins are components of a feed-forward circuit [59,60]. Understanding how this process may be defective in insulin resistant states will require further study.
To conclude, why translocate GLUT4? Wouldn't it have been simpler to activate transporters already resident in plasma membranes of fat and muscle cells? Possibly, the answer to this question is that translocation is a mechanism for the coordinate regulation of distinct physiologic outputs. Work from several laboratories implies that GSV translocation may control not only glucose uptake, but may also contribute to the regulation of blood pressure, lipid metabolism, and energy expenditure. Impaired, insulin-stimulated GSV translocation may therefore contribute to multiple aspects of the metabolic syndrome. As well, GSV-like vesicles are present in a range of differentiated cell types, and regulated exocytic translocation is likely a conserved mechanism that can respond to a variety of extracellular stimuli [25,112,134]. For example, the translocation of AQP2 water channels in the renal collecting duct, and of H+-pumps in gastric parietal cells, may employ a similar vesicle trafficking mechanism. Understanding this mechanism will thus have broad relevance for physiology and pathophysiology, and will inform the pathogenesis of type 2 diabetes and its complications.
Acknowledgments
The authors acknowledge support from the NIH (grants R01DK075772, R01DK092661, and R21AG041383 to J.S.B) and the American Diabetes Association (grant 1-12-BS-16 to J.S.B.). E.N.H. was supported by NIH T32DK007058, and J.P.B was supported by NIH F30DK093198.
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest relevant to the content of this review article.
Contributor Information
Jonathan P. Belman, Section of Endocrinology and Metabolism, Department of Internal Medicine, and Department of Cell Biology, Yale University School of Medicine, 333 Cedar Street, Box 208020, New Haven CT 06520-8020 USA
Estifanos N. Habtemichael, Section of Endocrinology and Metabolism, Department of Internal Medicine, Yale University School of Medicine, 333 Cedar Street, Box 208020, New Haven CT 06520-8020 USA
Jonathan S. Bogan, Section of Endocrinology and Metabolism, Department of Internal Medicine, and Department of Cell Biology, Yale University School of Medicine, 333 Cedar Street, Box 208020, New Haven CT 06520-8020 USA
References
- 1.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255(10):4758–4762. [PubMed] [Google Scholar]
- 2.Suzuki K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci U S A. 1980;77(5):2542–2545. doi: 10.1073/pnas.77.5.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueckler M, Caruso C, Baldwin SA, Panico M, Blench I, Morris HR, et al. Sequence and structure of a human glucose transporter. Science. 1985;229(4717):941–945. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- 4.Birnbaum MJ. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell. 1989;57(2):305–315. doi: 10.1016/0092-8674(89)90968-9. [DOI] [PubMed] [Google Scholar]
- 5.James DE, Strube M, Mueckler M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature. 1989;338(6210):83–87. doi: 10.1038/338083a0. [DOI] [PubMed] [Google Scholar]
- 6.Charron MJ, Brosius FC, 3rd, Alper SL, Lodish HF. A glucose transport protein expressed predominately in insulin-responsive tissues. Proc Natl Acad Sci U S A. 1989;86(8):2535–2539. doi: 10.1073/pnas.86.8.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumoto H, Kayano T, Buse JB, Edwards Y, Pilch PF, Bell GI, et al. Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. J Biol Chem. 1989;264(14):7776–7779. [PubMed] [Google Scholar]
- 8.Kaestner KH, Christy RJ, McLenithan JC, Braiterman LT, Cornelius P, Pekala PH, et al. Sequence, tissue distribution, and differential expression of mRNA for a putative insulin-responsive glucose transporter in mouse 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 1989;86(9):3150–3154. doi: 10.1073/pnas.86.9.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel ED, Shepherd PR, Kahn BB. Glucose transporters and pathophysiologic states. In: Leroith D, Olefsky JM, Taylor S, editors. Diabetes Mellitus: A Fundamental and Clinical Text. Third. Philadelphia: J. B. Lippincott; 2003. pp. 917–938. [Google Scholar]
- 10.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 12.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292(5522):1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 13.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. The Journal of clinical investigation. 2003;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farese RV, Sajan MP, Yang H, Li P, Mastorides S, Gower WR, Jr, et al. Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. The Journal of clinical investigation. 2007;117(8):2289–2301. doi: 10.1172/JCI31408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab. 2007;32(3):557–566. doi: 10.1139/H07-026. [DOI] [PubMed] [Google Scholar]
- 16.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5(4):237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Wang HY, Ducommun S, Quan C, Xie B, Li M, Wasserman DH, et al. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. The Biochemical journal. 2013;449(2):479–489. doi: 10.1042/BJ20120702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lansey MN, Walker NN, Hargett SR, Stevens JR, Keller SR. Deletion of Rab GAP AS160 modifies glucose uptake and GLUT4 translocation in primary skeletal muscles and adipocytes and impairs glucose homeostasis. American journal of physiology Endocrinology and metabolism. 2012;303(10):E1273–1286. doi: 10.1152/ajpendo.00316.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cellular signalling. 2011;23(10):1546–1554. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Chiu TT, Sun Y, Koshkina A, Klip A. Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance. The Journal of biological chemistry. 2013;288(24):17520–17531. doi: 10.1074/jbc.M113.467647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nozaki S, Takeda T, Kitaura T, Takenaka N, Kataoka T, Satoh T. Akt2 regulates Rac1 activity in the insulin-dependent signaling pathway leading to GLUT4 translocation to the plasma membrane in skeletal muscle cells. Cellular signalling. 2013;25(6):1361–1371. doi: 10.1016/j.cellsig.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 22.Sylow L, Jensen TE, Kleinert M, Hojlund K, Kiens B, Wojtaszewski J, et al. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes. 2013;62(6):1865–1875. doi: 10.2337/db12-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jewell JL, Oh E, Bennett SM, Meroueh SO, Thurmond DC. The tyrosine phosphorylation of Munc18c induces a switch in binding specificity from syntaxin 4 to Doc2beta. J Biol Chem. 2008;283(31):21734–21746. doi: 10.1074/jbc.M710445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubin BR, Bogan JS. Intracellular retention and insulin-stimulated mobilization of GLUT4 glucose transporters. Vitamins and Hormones. 2009;80:155–192. doi: 10.1016/S0083-6729(08)00607-9. [DOI] [PubMed] [Google Scholar]
- 25.Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Annual review of biochemistry. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- 26.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nature reviews Molecular cell biology. 2012;13(6):383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 27.Holman GD, Lo Leggio L, Cushman SW. Insulin-stimulated GLUT4 glucose transporter recycling. A problem in membrane protein subcellular trafficking through multiple pools. J Biol Chem. 1994;269(26):17516–17524. [PubMed] [Google Scholar]
- 28.Muretta JM, Mastick CC. How insulin regulates glucose transport in adipocytes. Vitamins and Hormones. 2009;80:245–286. doi: 10.1016/S0083-6729(08)00610-9. [DOI] [PubMed] [Google Scholar]
- 29.Bogan JS, Kandror KV. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Current opinion in cell biology. 2010;22(4):506–512. doi: 10.1016/j.ceb.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kandror KV, Coderre L, Pushkin AV, Pilch PF. Comparison of glucose-transporter-containing vesicles from rat fat and muscle tissues: evidence for a unique endosomal compartment. The Biochemical journal. 1995;307(Pt 2):383–390. doi: 10.1042/bj3070383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Z, Kandror KV. Translocation of small preformed vesicles is responsible for the insulin activation of glucose transport in adipose cells. Evidence from the in vitro reconstitution assay. J Biol Chem. 2002;277(50):47972–47975. doi: 10.1074/jbc.C200486200. [DOI] [PubMed] [Google Scholar]
- 32.Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. The Journal of biological chemistry. 2005;280(45):37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 33.Foster LJ, Rudich A, Talior I, Patel N, Huang X, Furtado LM, et al. Insulin-dependent interactions of proteins with GLUT4 revealed through stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2006;5(1):64–75. doi: 10.1021/pr0502626. [DOI] [PubMed] [Google Scholar]
- 34.Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, et al. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285(1):104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallis MG, Lankford MF, Keller SR. Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. American journal of physiology Endocrinology and metabolism. 2007;293(4):E1092–1102. doi: 10.1152/ajpendo.00440.2007. [DOI] [PubMed] [Google Scholar]
- 36.Lin BZ, Pilch PF, Kandror KV. Sortilin is a major protein component of Glut4-containing vesicles. The Journal of biological chemistry. 1997;272(39):24145–24147. doi: 10.1074/jbc.272.39.24145. [DOI] [PubMed] [Google Scholar]
- 37.Morris NJ, Ross SA, Lane WS, Moestrup SK, Petersen CM, Keller SR, et al. Sortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytes. The Journal of biological chemistry. 1998;273(6):3582–3587. doi: 10.1074/jbc.273.6.3582. [DOI] [PubMed] [Google Scholar]
- 38.Pilch PF. The mass action hypothesis: formation of Glut4 storage vesicles, a tissue-specific, regulated exocytic compartment. Acta Physiol (Oxf) 2008;192(1):89–101. doi: 10.1111/j.1748-1716.2007.01788.x. [DOI] [PubMed] [Google Scholar]
- 39.Kandror KV, Pilch PF. The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic. 2011;12(6):665–671. doi: 10.1111/j.1600-0854.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- 40.Baeza-Raja B, Li P, Le Moan N, Sachs BD, Schachtrup C, Davalos D, et al. p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(15):5838–5843. doi: 10.1073/pnas.1103638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Peters PJ, Bai M, Dai J, Bos E, Kirchhausen T, et al. An ACAP1-containing clathrin coat complex for endocytic recycling. The Journal of cell biology. 2007;178(3):453–464. doi: 10.1083/jcb.200608033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamb CA, McCann RK, Stockli J, James DE, Bryant NJ. Insulin-regulated trafficking of GLUT4 requires ubiquitination. Traffic. 2010;11(11):1445–1454. doi: 10.1111/j.1600-0854.2010.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, et al. Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Molecular endocrinology. 2006;20(10):2576–2583. doi: 10.1210/me.2005-0476. [DOI] [PubMed] [Google Scholar]
- 44.Yu C, Cresswell J, Loffler MG, Bogan JS. The glucose transporter 4-regulating protein TUG is essential for highly insulin-responsive glucose uptake in 3T3-L1 adipocytes. The Journal of biological chemistry. 2007;282(10):7710–7722. doi: 10.1074/jbc.M610824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gross DN, Farmer SR, Pilch PF. Glut4 storage vesicles without Glut4: transcriptional regulation of insulin-dependent vesicular traffic. Mol Cell Biol. 2004;24(16):7151–7162. doi: 10.1128/MCB.24.16.7151-7162.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jordens I, Molle D, Xiong W, Keller SR, McGraw TE. Insulin-regulated aminopeptidase is a key regulator of GLUT4 trafficking by controlling the sorting of GLUT4 from endosomes to specialized insulin-regulated vesicles. Mol Biol Cell. 2010;21(12):2034–2044. doi: 10.1091/mbc.E10-02-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeh TY, Sbodio JI, Tsun ZY, Luo B, Chi NW. Insulin-stimulated exocytosis of GLUT4 is enhanced by IRAP and its partner tankyrase. The Biochemical journal. 2007;402(2):279–290. doi: 10.1042/BJ20060793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang H, Li J, Katz EB, Charron MJ. GLUT4 ablation in mice results in redistribution of IRAP to the plasma membrane. Biochem Biophys Res Commun. 2001;284(2):519–525. doi: 10.1006/bbrc.2001.4994. [DOI] [PubMed] [Google Scholar]
- 49.Carvalho E, Schellhorn SE, Zabolotny JM, Martin S, Tozzo E, Peroni OD, et al. GLUT4 overexpression or deficiency in adipocytes of transgenic mice alters the composition of GLUT4 vesicles and the subcellular localization of GLUT4 and insulin-responsive aminopeptidase. J Biol Chem. 2004;279(20):21598–21605. doi: 10.1074/jbc.M312269200. [DOI] [PubMed] [Google Scholar]
- 50.Liao W, Nguyen MT, Imamura T, Singer O, Verma IM, Olefsky JM. Lentiviral short hairpin ribonucleic acid-mediated knockdown of GLUT4 in 3T3-L1 adipocytes. Endocrinology. 2006;147(5):2245–2252. doi: 10.1210/en.2005-1638. [DOI] [PubMed] [Google Scholar]
- 51.Khan AH, Capilla E, Hou JC, Watson RT, Smith JR, Pessin JE. Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is dependent upon both the amino terminus and the large cytoplasmic loop. J Biol Chem. 2004;279(36):37505–37511. doi: 10.1074/jbc.M405694200. [DOI] [PubMed] [Google Scholar]
- 52.Blot V, McGraw TE. Molecular mechanisms controlling GLUT4 intracellular retention. Mol Biol Cell. 2008;19(8):3477–3487. doi: 10.1091/mbc.E08-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson AO, Lampson MA, McGraw TE. A di-leucine sequence and a cluster of acidic amino acids are required for dynamic retention in the endosomal recycling compartment of fibroblasts. Mol Biol Cell. 2001;12(2):367–381. doi: 10.1091/mbc.12.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waters SB, D'Auria M, Martin SS, Nguyen C, Kozma LM, Luskey KL. The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J Biol Chem. 1997;272(37):23323–23327. doi: 10.1074/jbc.272.37.23323. [DOI] [PubMed] [Google Scholar]
- 55.Bogan JS, McKee AE, Lodish HF. Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Molecular and cellular biology. 2001;21(14):4785–4806. doi: 10.1128/MCB.21.14.4785-4806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Govers R, Coster AC, James DE. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol Cell Biol. 2004;24(14):6456–6466. doi: 10.1128/MCB.24.14.6456-6466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Y, Rubin BR, Orme CM, Karpikov A, Yu C, Bogan JS, et al. Dual-mode of insulin action controls GLUT4 vesicle exocytosis. The Journal of cell biology. 2011;193(4):643–653. doi: 10.1083/jcb.201008135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bogan JS, Rubin BR, Yu C, Loffler MG, Orme CM, Belman JP, et al. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. The Journal of biological chemistry. 2012;287(28):23932–23947. doi: 10.1074/jbc.M112.339457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loffler MG, Birkenfeld AL, Philbrick KM, Belman JP, Habtemichael EN, Booth CJ, et al. Enhanced Fasting Glucose Turnover in Mice with Disrupted Action of TUG Protein in Skeletal Muscle. The Journal of biological chemistry. 2013;288(28):20135–20150. doi: 10.1074/jbc.M113.458075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hart Y, Alon U. The Utility of Paradoxical Components in Biological Circuits. Molecular Cell. 2013;49(2):213–221. doi: 10.1016/j.molcel.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 61.Lizunov VA, Matsumoto H, Zimmerberg J, Cushman SW, Frolov VA. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. The Journal of cell biology. 2005;169(3):481–489. doi: 10.1083/jcb.200412069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang L, Fan J, Bai L, Wang Y, Chen Y, Yang L, et al. Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J Biol Chem. 2008;283(13):8508–8516. doi: 10.1074/jbc.M708688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lizunov VA, Stenkula KG, Lisinski I, Gavrilova O, Yver DR, Chadt A, et al. Insulin stimulates fusion, but not tethering, of GLUT4 vesicles in skeletal muscle of HA-GLUT4-GFP transgenic mice. American journal of physiology Endocrinology and metabolism. 2012;302(8):E950–960. doi: 10.1152/ajpendo.00466.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, et al. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. The Journal of cell biology. 2012;198(4):545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Molecular biology of the cell. 2013;24(16):2544–2557. doi: 10.1091/mbc.E13-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reed SE, Hodgson LR, Song S, May MT, Kelly EE, McCaffrey MW, et al. A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. Journal of cell science. 2013;126(Pt 9):1931–1941. doi: 10.1242/jcs.104307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(46):19909–19914. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Randhawa VK, Ishikura S, Talior-Volodarsky I, Cheng AW, Patel N, Hartwig JH, et al. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J Biol Chem. 2008;283(40):27208–27219. doi: 10.1074/jbc.M804282200. [DOI] [PubMed] [Google Scholar]
- 69.Ishikura S, Klip A. Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am J Physiol Cell Physiol. 2008;295(4):C1016–1025. doi: 10.1152/ajpcell.00277.2008. [DOI] [PubMed] [Google Scholar]
- 70.Munro S. The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harbor perspectives in biology. 2011;3(6) doi: 10.1101/cshperspect.a005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hosaka T, Brooks CC, Presman E, Kim SK, Zhang Z, Breen M, et al. p115 Interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol Biol Cell. 2005;16(6):2882–2890. doi: 10.1091/mbc.E05-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams D, Hicks SW, Machamer CE, Pessin JE. Golgin-160 Is Required for the Golgi Membrane Sorting of the Insulin-responsive Glucose Transporter GLUT4 in Adipocytes. Mol Biol Cell. 2006 doi: 10.1091/mbc.E06-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orme CM, Bogan JS. The ubiquitin regulatory X (UBX) domain-containing protein TUG regulates the p97 ATPase and resides at the endoplasmic reticulum-golgi intermediate compartment. The Journal of biological chemistry. 2012;287(9):6679–6692. doi: 10.1074/jbc.M111.284232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sohda M, Misumi Y, Yamamoto A, Yano A, Nakamura N, Ikehara Y. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. The Journal of biological chemistry. 2001;276(48):45298–45306. doi: 10.1074/jbc.M108961200. [DOI] [PubMed] [Google Scholar]
- 75.Lesa GM, Seemann J, Shorter J, Vandekerckhove J, Warren G. The amino-terminal domain of the golgi protein giantin interacts directly with the vesicle-tethering protein p115. J Biol Chem. 2000;275(4):2831–2836. doi: 10.1074/jbc.275.4.2831. [DOI] [PubMed] [Google Scholar]
- 76.Linstedt AD, Jesch SA, Mehta A, Lee TH, Garcia-Mata R, Nelson DS, et al. Binding relationships of membrane tethering components. The giantin N terminus and the GM130 N terminus compete for binding to the p115 C terminus. The Journal of biological chemistry. 2000;275(14):10196–10201. doi: 10.1074/jbc.275.14.10196. [DOI] [PubMed] [Google Scholar]
- 77.Yadav S, Puthenveedu MA, Linstedt AD. Golgin160 recruits the dynein motor to position the Golgi apparatus. Developmental cell. 2012;23(1):153–165. doi: 10.1016/j.devcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malhotra V. Unconventional protein secretion: an evolving mechanism. The EMBO journal. 2013;32(12):1660–1664. doi: 10.1038/emboj.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang M, Schekman R. Cell biology. Unconventional secretion, unconventional solutions. Science. 2013;340(6132):559–561. doi: 10.1126/science.1234740. [DOI] [PubMed] [Google Scholar]
- 80.Wang C, Yoo Y, Fan H, Kim E, Guan KL, Guan JL. Regulation of Integrin beta 1 recycling to lipid rafts by Rab1a to promote cell migration. The Journal of biological chemistry. 2010;285(38):29398–29405. doi: 10.1074/jbc.M110.141440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. The Journal of biological chemistry. 2002;277(13):11401–11409. doi: 10.1074/jbc.M110263200. [DOI] [PubMed] [Google Scholar]
- 82.Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146(5):746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 83.Cheng J, Wang H, Guggino WB. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J Biol Chem. 2004;279(3):1892–1898. doi: 10.1074/jbc.M308640200. [DOI] [PubMed] [Google Scholar]
- 84.Cheng J, Wang H, Guggino WB. Regulation of Cystic Fibrosis Transmembrane Regulator Trafficking and Protein Expression by a Rho Family Small GTPase TC10. J Biol Chem. 2005;280(5):3731–3739. doi: 10.1074/jbc.M410026200. [DOI] [PubMed] [Google Scholar]
- 85.Cheng J, Cebotaru V, Cebotaru L, Guggino WB. Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Molecular biology of the cell. 2010;21(7):1178–1187. doi: 10.1091/mbc.E09-03-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bruns C, McCaffery JM, Curwin AJ, Duran JM, Malhotra V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. The Journal of cell biology. 2011;195(6):979–992. doi: 10.1083/jcb.201106098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. The Journal of cell biology. 2010;188(4):537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35(5):921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 89.Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Graef M, Friedman JR, Graham C, Babu M, Nunnari J. ER exit sites are physical and functional core autophagosome biogenesis components. Molecular biology of the cell. 2013 doi: 10.1091/mbc.E13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nature cell biology. 2012;15(2):169–178. doi: 10.1038/ncb2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. The EMBO journal. 2011;30(23):4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bogan JS, Hendon N, McKee AE, Tsao TS, Lodish HF. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature. 2003;425(6959):727–733. doi: 10.1038/nature01989. [DOI] [PubMed] [Google Scholar]
- 94.Schertzer JD, Antonescu CN, Bilan PJ, Jain S, Huang X, Liu Z, et al. A transgenic mouse model to study glucose transporter 4myc regulation in skeletal muscle. Endocrinology. 2009;150(4):1935–1940. doi: 10.1210/en.2008-1372. [DOI] [PubMed] [Google Scholar]
- 95.Chang L, Chiang SH, Saltiel AR. TC10alpha is required for insulin-stimulated glucose uptake in adipocytes. Endocrinology. 2007;148(1):27–33. doi: 10.1210/en.2006-1167. [DOI] [PubMed] [Google Scholar]
- 96.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 97.Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458(7237):422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annual review of biochemistry. 2012;81:323–357. doi: 10.1146/annurev-biochem-093010-153308. [DOI] [PubMed] [Google Scholar]
- 99.Tettamanzi MC, Yu C, Bogan JS, Hodsdon ME. Solution structure and backbone dynamics of an N-terminal ubiquitin-like domain in the GLUT4-regulating protein, TUG. Protein science : a publication of the Protein Society. 2006;15(3):498–508. doi: 10.1110/ps.051901806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Semiz S, Park JG, Nicoloro SM, Furcinitti P, Zhang C, Chawla A, et al. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. The EMBO journal. 2003;22(10):2387–2399. doi: 10.1093/emboj/cdg237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lalioti VS, Vergarajauregui S, Tsuchiya Y, Hernandez-Tiedra S, Sandoval IV. Daxx functions as a scaffold of a protein assembly constituted by GLUT4, JNK1 and KIF5B. Journal of cellular physiology. 2009;218(2):416–426. doi: 10.1002/jcp.21614. [DOI] [PubMed] [Google Scholar]
- 102.Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein science : a publication of the Protein Society. 2011 doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tasaki T, Sriram SM, Park KS, Kwon YT. The N-end rule pathway. Annual review of biochemistry. 2012;81:261–289. doi: 10.1146/annurev-biochem-051710-093308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park S, Rancour DM, Bednarek SY. Protein domain-domain interactions and requirements for the negative regulation of Arabidopsis CDC48/p97 by the plant ubiquitin regulatory X (UBX) domain-containing protein, PUX1. The Journal of biological chemistry. 2007;282(8):5217–5224. doi: 10.1074/jbc.M609042200. [DOI] [PubMed] [Google Scholar]
- 105.Rancour DM, Park S, Knight SD, Bednarek SY. Plant UBX domain-containing protein 1, PUX1, regulates the oligomeric structure and activity of arabidopsis CDC48. The Journal of biological chemistry. 2004;279(52):54264–54274. doi: 10.1074/jbc.M405498200. [DOI] [PubMed] [Google Scholar]
- 106.Cloutier P, Coulombe B. Regulation of molecular chaperones through post-translational modifications: decrypting the chaperone code. Biochimica et biophysica acta. 2013;1829(5):443–454. doi: 10.1016/j.bbagrm.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cloutier P, Lavallee-Adam M, Faubert D, Blanchette M, Coulombe B. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS genetics. 2013;9(1):e1003210. doi: 10.1371/journal.pgen.1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kernstock S, Davydova E, Jakobsson M, Moen A, Pettersen S, Maelandsmo GM, et al. Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nature communications. 2012;3:1038. doi: 10.1038/ncomms2041. [DOI] [PubMed] [Google Scholar]
- 109.Haines DS, Lee JE, Beauparlant SL, Kyle DB, den Besten W, Sweredoski MJ, et al. Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Molecular & cellular proteomics : MCP. 2012;11(6):M111 016444. doi: 10.1074/mcp.M111.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bug M, Meyer H. Expanding into new markets--VCP/p97 in endocytosis and autophagy. Journal of structural biology. 2012;179(2):78–82. doi: 10.1016/j.jsb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 111.Ritz D, Vuk M, Kirchner P, Bug M, Schutz S, Hayer A, et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nature cell biology. 2011;13(9):1116–1123. doi: 10.1038/ncb2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shewan AM, McCann RK, Lamb CA, Stirrat L, Kioumourtzoglou D, Adamson IS, et al. Endosomal sorting of GLUT4 and Gap1 is conserved between yeast and insulin-sensitive cells. Journal of cell science. 2013;126(Pt 7):1576–1582. doi: 10.1242/jcs.114371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sargeant RJ, Paquet MR. Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. The Biochemical journal. 1993;290(Pt 3):913–919. doi: 10.1042/bj2900913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim SS, Bae JW, Jung CY. GLUT-4 degradation rate: reduction in rat adipocytes in fasting and streptozotocin-induced diabetes. Am J Physiol. 1994;267(1 Pt 1):E132–139. doi: 10.1152/ajpendo.1994.267.1.E132. [DOI] [PubMed] [Google Scholar]
- 115.Bao S, Garvey WT. Exercise in transgenic mice overexpressing GLUT4 glucose transporters: effects on substrate metabolism and glycogen regulation. Metabolism. 1997;46(11):1349–1357. doi: 10.1016/s0026-0495(97)90243-2. [DOI] [PubMed] [Google Scholar]
- 116.Treadway JL, Hargrove DM, Nardone NA, McPherson RK, Russo JF, Milici AJ, et al. Enhanced peripheral glucose utilization in transgenic mice expressing the human GLUT4 gene. J Biol Chem. 1994;269(47):29956–29961. [PubMed] [Google Scholar]
- 117.Hansen PA, Gulve EA, Marshall BA, Gao J, Pessin JE, Holloszy JO, et al. Skeletal muscle glucose transport and metabolism are enhanced in transgenic mice overexpressing the Glut4 glucose transporter. J Biol Chem. 1995;270(4):1679–1684. doi: 10.1074/jbc.270.5.1679. [DOI] [PubMed] [Google Scholar]
- 118.Deems RO, Evans JL, Deacon RW, Honer CM, Chu DT, Burki K, et al. Expression of human GLUT4 in mice results in increased insulin action. Diabetologia. 1994;37(11):1097–1104. doi: 10.1007/BF00418373. [DOI] [PubMed] [Google Scholar]
- 119.Tsao TS, Burcelin R, Katz EB, Huang L, Charron MJ. Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes. 1996;45(1):28–36. doi: 10.2337/diab.45.1.28. [DOI] [PubMed] [Google Scholar]
- 120.Ren JM, Marshall BA, Mueckler MM, McCaleb M, Amatruda JM, Shulman GI. Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. The Journal of clinical investigation. 1995;95(1):429–432. doi: 10.1172/JCI117673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ho PC, Lin YW, Tsui YC, Gupta P, Wei LN. A negative regulatory pathway of GLUT4 trafficking in adipocyte: new function of RIP140 in the cytoplasm via AS160. Cell metabolism. 2009;10(6):516–523. doi: 10.1016/j.cmet.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lalioti VS, Vergarajauregui S, Pulido D, Sandoval IV. The insulin-sensitive glucose transporter, GLUT4, interacts physically with Daxx. Two proteins with capacity to bind Ubc9 and conjugated to SUMO1. The Journal of biological chemistry. 2002;277(22):19783–19791. doi: 10.1074/jbc.M110294200. [DOI] [PubMed] [Google Scholar]
- 123.Watanabe T, Nomura M, Nakayasu K, Kawano T, Ito S, Nakaya Y. Relationships between thermic effect of food, insulin resistance and autonomic nervous activity. The journal of medical investigation : JMI. 2006;53(1-2):153–158. doi: 10.2152/jmi.53.153. [DOI] [PubMed] [Google Scholar]
- 124.Piaggi P, Krakoff J, Bogardus C, Thearle MS. Lower “Awake and Fed Thermogenesis” Predicts Future Weight Gain in Subjects with Abdominal Adiposity. Diabetes. 2013 doi: 10.2337/db13-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. The Journal of clinical investigation. 1998;101(11):2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maianu L, Keller SR, Garvey WT. Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J Clin Endocrinol Metab. 2001;86(11):5450–5456. doi: 10.1210/jcem.86.11.8053. [DOI] [PubMed] [Google Scholar]
- 128.Vassilopoulos S, Esk C, Hoshino S, Funke BH, Chen CY, Plocik AM, et al. A role for the CHC22 clathrin heavy-chain isoform in human glucose metabolism. Science. 2009;324(5931):1192–1196. doi: 10.1126/science.1171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, et al. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7(5):421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Watson RT, Shigematsu S, Chiang SH, Mora S, Kanzaki M, Macara IG, et al. Lipid raft microdomain compartmentalization of TC10 is required for insulin signaling and GLUT4 translocation. The Journal of cell biology. 2001;154(4):829–840. doi: 10.1083/jcb.200102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bridges D, Chang L, Lodhi IJ, Clark NA, Saltiel AR. TC10 is regulated by caveolin in 3T3-L1 adipocytes. PLoS ONE. 2012;7(8):e42451. doi: 10.1371/journal.pone.0042451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Minami A, Iseki M, Kishi K, Wang M, Ogura M, Furukawa N, et al. Increased insulin sensitivity and hypoinsulinemia in APS knockout mice. Diabetes. 2003;52(11):2657–2665. doi: 10.2337/diabetes.52.11.2657. [DOI] [PubMed] [Google Scholar]
- 133.Molero JC, Turner N, Thien CB, Langdon WY, James DE, Cooney GJ. Genetic ablation of the c-Cbl ubiquitin ligase domain results in increased energy expenditure and improved insulin action. Diabetes. 2006;55(12):3411–3417. doi: 10.2337/db06-0955. [DOI] [PubMed] [Google Scholar]
- 134.Chieregatti E, Meldolesi J. Regulated exocytosis: new organelles for non-secretory purposes. Nat Rev Mol Cell Biol. 2005;6(2):181–187. doi: 10.1038/nrm1572. [DOI] [PubMed] [Google Scholar]