

Abstract
Introduction
The mechanism of intestinal atresia formation remains undefined. Atresia in Fgfr2IIIb−/− mutant mouse embryos is preceded by endodermal apoptosis and involution of the surrounding mesoderm. We have observed that involution of the atretic segment is preceded by down regulation of Sonic hedgehog (SHH) in the endoderm which is a critical organizer of the intestinal mesoderm. We hypothesized that supplementation of Fgfr2IIIb−/− intestinal tracts with exogenous SHH protein prior to atresia formation would prevent involution of the mesoderm and rescue normal intestinal development.
Methods
In situ hybridization were performed on control and Fgfr2IIIb−/− intestinal tracts for Shh or FoxF1 between embryonic (E) day 11.5 and E12.0. Control and Fgfr2IIIb−/− intestinal tracts were harvested at E10.5 and cultured in media supplemented with FGF10 + SHH, or FGF10 with a SHH-coated bead. In situs were performed at E12.5 for Foxf1.
Results
Shh and Foxf1 expression were down-regulated during intestinal atresia formation. Media containing exogenous FGF10 + SHH did not prevent colonic atresia formation (involution). A SHH protein point source bead did induce Foxf1 expression in controls and mutants.
Discussion
Shh and Foxf1 expression are disrupted in atresia formation of distal colon, thereby serving as potential markers of atretic events. Application of exogenous SHH (in media supplement or as a point source bead) is sufficient to induce Foxf1 expression but insufficient to rescue development of distal colonic mesoderm in Fgfr2IIIb−/− mutant embryos. Shh signal disruption is not the critical mechanism by which loss of Fgfr2IIIb function results in atresia formation.
Keywords: Shh, intestinal atresia, expression, Foxf1, Fgfr2IIIb−/−, organ culture
Introduction
The mechanism of intestinal atresia remains undefined. Homozygous mutation of Fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) early in intestinal development causes atresia formation in the colon of mice (1). Atresia formation is preceded by endodermal apoptosis in the areas where the atresia will form (1, 2). Fgfr2IIIb expression is limited to the intestinal endoderm at early stages of development (3) indicating that loss of the receptor in the endoderm causes endodermal apoptosis, loss of intestinal endoderm, and involution of the affected segment of intestine as a result of a loss of instructive signal to the surrounding mesoderm.
Shh is expressed in the early intestinal endoderm. It is a critical organizer of the intestine during development, instructing both radial and longitudinal growth (4, 5). Loss of Shh expression results in impaired organization of the intestinal mesoderm, although the intestinal tube remains continuous and does not form atresias. Tissue specific and spatial-temporal overlap is seen in the expression of Shh and Fgfr2IIIb, each performing a critical role in instructing intestinal development. Therefore, we hypothesized that following endodermal apoptosis, Shh signaling would be disrupted in the atretic precursor region (where the atresia will form).
We set out to investigate the role of Shh signaling pathway disruptions during atresia formation by examining expression patterns of Shh and its downstream mesodermal target, Foxf1, (6–8) by in situ hybridization prior to involution of the atretic segment. Further, we tested whether the addition of exogenous SHH protein to cultured embryonic intestines prior to atresia formation would rescue mesodermal development, prevent involution of the intestinal tube, and halt intestinal atresia formation.
Materials and Methods
Animals
IACUC approval for these studies was obtained from the University of Wisconsin School of Medicine and Public health (P.F.N. protocol # M02258). All animals were maintained in a clean facility with ad libitum access to fresh food and water under a 12-hour alternating light/dark cycle.
Generation of mutant fetuses
Fgfr2IIIb−/− mutant and Fgfr2IIIb+/− littermate control embryos were generated using the HprtCre breeding strategy (9) as has been described previously (2).
Whole mount in situ hybridization
Fgfr2IIIb+/− and Fgfr2IIIb−/− embryos were harvested at Embryonic Day (E) 11.5 and E12.0 into cold PBS and fixed overnight in 4% PFA at 4°C. Fixed samples were dissected and dehydrated to 100% MeOH through a series of escalating Methanol/PBS-Tween steps and stored at −20°C. The in situ hybridization protocol has been published elsewhere (10) and included incubation with antisense riboprobes, at 68°C for Shh and 70°C for Foxf1 (constructs kindly provided by H. Hamada and Y. Saijoh). Photographs were taken using a dissecting light microscope.
Organ culture
For the media supplemented SHH protein experiments, Fgfr2IIIb+/− and Fgfr2IIIb−/− embryos were harvested at E10.5 and the developing intestinal tracts were isolated. Intestinal tracts were cultured in Matrigel (BD Biosciences, Bedford, MA) and allowed to polymerize at 37°C for 30 minutes within Millicell EZslide wells (Millipore, Billerica, MA). Matrigel embedded tracts were overlaid with a base media of DMEM/F-12 (HyClone, Logan, UT) containing L-Glutamine, penicillin/streptomycin, and fetal bovine serum. In this set of experiments, the base culture media was supplemented with FGF10 (PeproTech, Rocky Hill, NJ) and SHH (R&D Systems, Minneapolis, MN) to a final concentration of 500 ng/mL each. Media-supplemented organ culture were conducted 5 times following previously published protocols (10), with 12 normal (Fgfr2IIIb+/−) littermate controls and 8 mutant (Fgfr2IIIb−/−) tracts.
For the point source SHH protein experiments, E10.5 intestinal tracts were harvested as described above. These 3 additional culture experiments included 17 littermate controls and 9 mutant intestinal tracts. Intestinal explants were cultured in Matrigel with SHH-laden Affi-Gel beads (Bio-Rad, Hercules, CA, 153–7302) at 80 to 150 μm diameter. Beads were incubated with approximately 50 μg/mL of SHH protein before being polymerized within Matrigel and overlaid with media containing 500 ng/mL of FGF10 within EZslide wells. Each well contained at least 1 control tract, 1 mutant tract and 1 SHH-laden bead. All organ cultures were maintained at 37°C and 5% CO 2 conditions. Media was refreshed after 24 hours. After 48 hours of culture, tissues were fixed with 4% PFA within culture wells and then processed for in situ hybridization with a Foxf1 antisense riboprobe at 70°C. Photographs were take n at time of culture (E10.5 + 0 hours), E10.5 + 24 hours, and E10.5 + 48 hours, under a dissecting light microscope.
Results
At E11.5 on littermate Shh expression was down regulated in the colons of mutant Fgfr2IIIb−/− embryos compared to controls but expressed equaling the small intestine of Fgfr2IIIb+/− and Fgfr2IIIb−/− embryos (Figure 1A and B).
Figure 1. Shh Expression is absent in Fgfr2IIIb−/− colon at E 11.5.
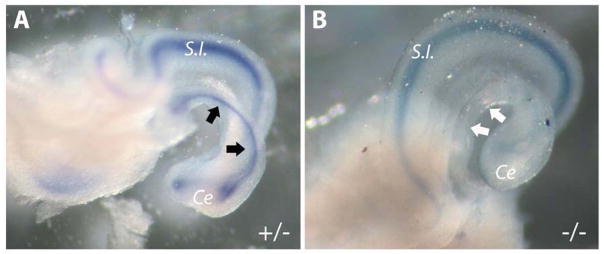
Whole mount in situ hybridization for Shh in (A) control Fgfr2IIIb+/− and (B) mutant Fgfr2IIIb−/− colons. Presence of staining in control colon is indicated by black arrows, while absence of staining in mutant colon is indicated by white arrows. (Ce) cecum, (S.I.) small intestine.
To determine if the above colonic Shh expression loss was associated with a downstream signaling disruption, Foxf1 expression was examined by whole mount in situ hybridization at E12.0. Fgfr2IIIb−/− embryos exhibited a complete absence of Foxf1 expression in the atretic precursor region (mid and distal colon) in contrast to controls (Figure 2A and B), indicating disruptions in the Shh signaling cascade in the atretic precursor region of the colon.
Figure 2. Foxf1 Expression is absent in the mid and distal colon of Fgfr2IIIb−/− embryos E 12.0.
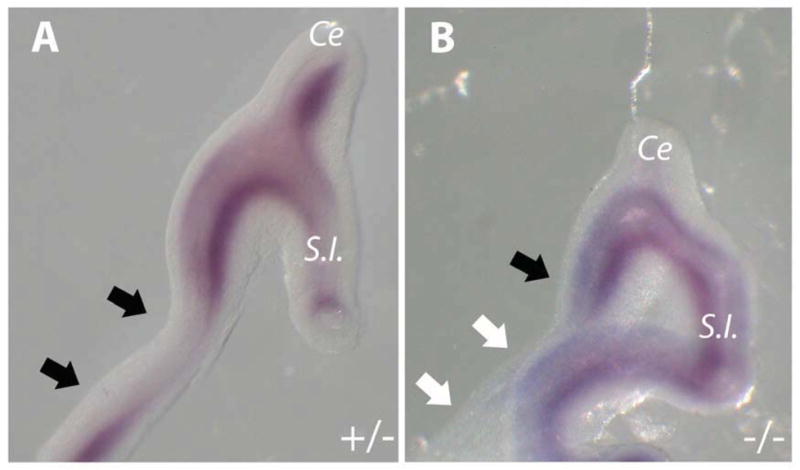
Whole mount in situ hybridization for Foxf1 in (A) control Fgfr2IIIb+/− and (B) mutant Fgfr2IIIb−/− colons. Presence of staining in colon is indicated by black arrows, while absence of staining is indicated by white arrows. (Ce) cecum, (S.I.) small intestine.
To determine whether exogenous SHH protein could rescue Fgfr2IIIb−/− colons from an atretic fate, intestinal explants were cultured in the presence of SHH, either as a media supplement or as a point source on an Affi-Gel bead. In the presence of SHH protein, control Fgfr2IIIb+/− intestines developed normally, exhibiting cecal bud formation and presence of an intact colon (Figure 3A–C). In contrast, Fgfr2IIIb−/− colons failed to develop normally with the colons ending in a blind end (Figure 3). Exogenous SHH alone could not rescue colonic development from an atretic fate. When SHH was supplied on an Affi-Gel bead, Foxf1 expression was induced at the boundary of the endoderm and mesoderm of the control intestines and along the surface of the mesoderm of the Fgfr2IIIb−/− intestine. Control intestines maintained normal architecture with both the colon and the proximal intestine maintaining an open tube at either end. In contrast, Fgfr2IIIb−/− colons ended blindly at the cecal region indicating the formation of an atresia (Figure 4).
Figure 3. Media supplemented with exogenous SHH protein does not rescue cultured Fgfr2IIIb−/− colons from an atretic fate.

Whole mount photographs of control (A–C) and mutant (D–F) colons at E10.5 (A and D), E10.5 + 24 hour culture (B and E) and E10.5 + 48 hours (C and F) in the presence of SHH protein. Colonic development (Black arrows) proceeds normally in Fgfr2IIIb+/− controls. In Fgfr2IIIb−/− mutants the colon ends blindly (White arrows).
Figure 4. Exogenous SHH protein provided on an Affi-Gel bead does not rescue cultured Fgfr2IIIb−/− colons from an atretic fate.
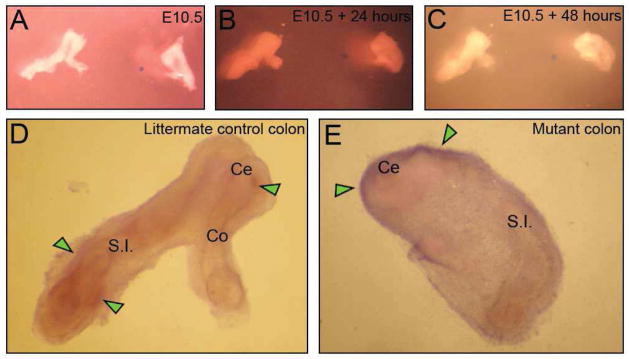
Representative whole mount photographs of littermate control and mutant colon at E10.5 (A), E10.5 + 24 hours (B), E10.5 + 48 hours (C), in the presence of a SHH-laden bead (blue circle to the right of center), and Foxf1 in situ after 48 hours of culture (D and E). Control intestine and colon display normal architecture and defined Foxf1 expression in the mesoderm adjacent to the endoderm (Green arrows indicate Foxf1 staining at the boundary between the endoderm and mesoderm) (D). Fgfr2IIIb−/− mutant colon ends blindly and expresses Foxf1 on the surface of the mesoderm (Green arrows) (E). (Ce) cecum, (Co) colon, (S.I.) small intestine.
Discussion
The atretic precursor region of the colon exhibits loss of Shh signaling prior to involution in Fgfr2IIIb−/− mutants. The absence of Shh expression is nearly pan colonic at this stage (Figure 1). Further, Foxf1 expression is absent in the mid and distal colon 12 hours after the loss of Shh expression (Figure 2). Interestingly, there is some preservation of Foxf1 expression in the proximal colon of Fgfr2IIIb−/− mutants. We suspect that the expression of Foxf1 in the proximal colon may be a result of Shh expression arising from the distal small intestine (precursor of the terminal ileum) and acting longitudinally over a short distance.
Exogenous SHH protein, however, fails to rescue colon explants from an atretic fate in organ culture (Figures 3–4) regardless of method of supplementation: addition to culture media or via a SHH impregnated bead in proximity to the intestinal tract. The data suggest that the loss of intestinal endoderm through apoptosis leads to the absence of Shh expression and downstream Foxf1 expression, which is critical for mesodermal development (4, 5). However, simply restoring SHH protein availability is insufficient to rescue development. The data also suggest that exogenous SHH protein does not prevent endoderm loss, directly or indirectly, via a feedback loop from the mesoderm to the endoderm.
In summary, atresia formation arising from homozygous mutation of Fgfr2IIIb is associated with endodermal apoptosis as reported previously (1,2) and with a loss of Shh expression and signaling. However, our data indicate that disruptions in Shh signaling are not the critical event that leads to the involution of the atretic precursor. In fact, supplementation with exogenous Shh protein can restore some level of Shh signaling in the mesoderm as evidenced by induction of Foxf1 expression in the cultured intestinal tracts of Fgfr2IIIb−/− mutants. Yet this is insufficient to rescue intestinal development. The findings indicate that disruptions in Shh signaling may serve as a marker, but do not support them as a critical event in atresia formation.
Acknowledgments
The authors would like to acknowledge the following funding agencies that supported this work: the Society for Surgery of the Alimentary Tract, The American Pediatric Surgical Association, The NIDDK (1K08DK087854-03) and the University of Wisconsin-SMPH Department of Surgery.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Amy L. Reeder, Division of Section of Pediatric Surgery Department of Surgery, University of Wisconsin SMPH Madison, WI.
Krzysztof M. Zaremba, Division of Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Rebeca M. Liebl, Division of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Anna Kowalkowski, Division of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Peter F. Nichol, Email: nichol@surgery.wisc.edu, Division of Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI, (O) (608) 263-9419, (F) (608) 261-1876.
References
- 1.Fairbanks TJ, Kanard RC, Del Moral PM, Sala FG, De Langhe SP, Lopez CA, et al. Colonic atresia without mesenteric vascular occlusion. The role of the fibroblast growth factor 10 signaling pathway. J Pediatr Surg. 2005 Feb;40(2):390–6. doi: 10.1016/j.jpedsurg.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 2.Nichol PF, Botham R, Saijoh Y, Reeder AL, Zaremba KM. A more efficient method to generate null mutants using Hprt-Cre with floxed alleles. J Pediatr Surg. 2011 Sep;46(9):1711–9. doi: 10.1016/j.jpedsurg.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, et al. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993 Aug;158(2):475–86. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 4.Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000 Jun;127(12):2763–72. doi: 10.1242/dev.127.12.2763. [DOI] [PubMed] [Google Scholar]
- 5.Sukegawa A, Narita T, Kameda T, Saitoh K, Nohno T, Iba H, et al. The concentric structure of the developing gut is regulated by Sonic hedgehog derived from endodermal epithelium. Development. 2000 May;127(9):1971–80. doi: 10.1242/dev.127.9.1971. [DOI] [PubMed] [Google Scholar]
- 6.Madison BB, McKenna LB, Dolson D, Epstein DJ, Kaestner KH. FoxF1 and FoxL1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J Biol Chem. 2009 Feb 27;284(9):5936–44. doi: 10.1074/jbc.M808103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001 Jun;128(12):2397–406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 8.Ormestad M, Astorga J, Landgren H, Wang T, Johansson BR, Miura N, et al. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006 Mar;133(5):833–43. doi: 10.1242/dev.02252. [DOI] [PubMed] [Google Scholar]
- 9.Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002 Mar;32(3):199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 10.Reeder AL, Botham RA, Franco M, Zaremba KM, Nichol PF. Formation of intestinal atresias in the Fgfr2IIIb−/− mice is not associated with defects in notochord development or alterations in Shh expression. J Surg Res. 2012 Sep;177(1):139–45. doi: 10.1016/j.jss.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]