

Summary
In Drosophila, the 330 kb bithorax complex regulates cellular differentiation along the anterio-posterior axis during development in the thorax and abdomen and is comprised of three homeotic genes: Ultrabithorax, abdominal-A, and Abdominal-B. The expression of each of these genes is in turn controlled through interactions between transcription factors and a number of cis-regulatory modules in the neighboring intergenic regions. In this study, we examine how the sequence architecture of transcription factor binding sites mediates the functional activity of one of these cis-regulatory modules. Using computational, mathematical modeling and experimental molecular genetic approaches we investigate the IAB7b enhancer, which regulates Abdominal-B expression specifically in the presumptive seventh and ninth abdominal segments of the early embryo. A cross-species comparison of the IAB7b enhancer reveals an evolutionarily conserved signature motif containing two FUSHI-TARAZU activator transcription factor binding sites. We find that the transcriptional repressors KNIRPS, KRUPPEL and GIANT are able to restrict reporter gene expression to the posterior abdominal segments, using different molecular mechanisms including short-range repression and competitive binding. Additionally, we show the functional importance of the spacing between the two FUSHI-TARAZU binding sites and discuss the potential importance of cooperativity for transcriptional activation. Our results demonstrate that the transcriptional output of the IAB7b cis-regulatory module relies on a complex set of combinatorial inputs mediated by specific transcription factor binding and that the sequence architecture at this enhancer is critical to maintain robust regulatory function.
Keywords: transcription, Drosophila, bithorax, enhancer, fushi-tarazu, knirps
Introduction
The bithorax complex (BX-C) is a 330 kb genomic region (Lewis et al., 1995; Martin et al., 1995) that harbors three of the Drosophila homeotic (Hox) genes: abdominal-A (abd-A), Abdominal-B (Abd-B), and Ultrabithorax (Ubx) (Lewis, 1978). Expression of these Hox genes during embryonic development is regulated by a cascade of transcription factors (TFs) (Borok et al., 2010; Sauer et al., 1996). Maternal mRNAs and protein factors deposited into the egg prior to fertilization give rise to the initial TF gradients along the anterio-posterior (A–P) axis of the embryo (Berleth et al., 1988; Driever and Nüsslein-Volhard, 1988). For example, the BICOID (BCD) and HUNCHBACK (HB) TFs are concentrated at the anterior pole, but spread posteriorly, creating a concentration gradient throughout the anterior half of the embryo (Berleth et al., 1988; Driever and Nüsslein-Volhard, 1988; Sauer and Tjian, 1997) (Fig. 1). The maternal TFs activate expression of the Gap genes — including Kruppel, knirps, and giant — in a concentration-dependent manner, resulting in distinct spatial patterns of Gap gene expression throughout the developing embryo (Borok et al., 2010; Driever and Nüsslein-Volhard, 1988; Papatsenko and Levine, 2008; Struhl et al., 1989) (Fig. 1). Gap genes, in turn, activate downstream pair-rule genes including even-skipped (Small et al., 1991a; Small et al., 1996; Small et al., 1991b) and fushi-tarazu, which are expressed in seven stripes along the anterio-posterior axis (Hoch and Jackle, 1993; Sauer et al., 1996) (Fig. 1). Together, these TFs define the spatial expression patterns of Hox genes (Maeda and Karch, 2006; Qian et al., 1991).
Fig. 1. Spatial expression patterns of transcription factors in early Drosophila development.

The mRNA expression profiles 70 mins into development for the maternal TFs (BCD and HB), gap TFs (KR, KNI, and GT), and pair-rule TFs (EVE and FTZ) are shown on a virtual embryo obtained via projection of the BDTNP data set along the anterior-posterior (AP) axis. Positions indicate the percentage of embryo length (%EL).
The activity of the TFs in this regulatory cascade is primarily mediated through binding at cis-regulatory modules (CRMs) (Akbari et al., 2006; Maeda and Karch, 2006). When a TF binds at a CRM it can act to either activate or repress transcription of the target gene (Arnone and Davidson, 1997; Borok et al., 2010). TFs can act synergistically in CRMs to activate transcription directly by recruiting RNA polymerase II to the promoter of target genes (Kadonaga, 2004; Ptashne and Gann, 1997). Transcriptional activation can also be achieved indirectly through TF interaction with chromatin-modifying enzymes, such as histone acetyltransferases and nucleosome remodeling factors (Kadonaga, 2004; Mannervik et al., 1999; Gaston & Jayaraman, 2003). TFs can also repress, or quench, the activation activities of other TFs by competition for DNA binding sites in an enhancer CRM or by disrupting protein-protein interactions necessary for transcriptional activation (Gray et al., 1994; Han et al., 1989; Levine and Manley, 1989).
Expression of the Abd-B gene is controlled by CRMs located in the intergenic infraabdominal (iab) regions (Akbari et al., 2006; Maeda and Karch, 2006; Sanchez_Herrero, 1991). The iab-7 chromosomal domain is responsible for directing expression of Abd-B specifically in the presumptive seventh abdominal (A7) segment of the developing Drosophila embryo (Mihaly et al., 2006; Zhou et al., 1999). The full length IAB7b enhancer is a CRM located within this chromosomal domain and is capable of directing reporter gene expression only in two stripes in the posterior of the embryo, A7 and A9 (Mihaly et al., 2006; Starr et al., 2011; Zhou et al., 1999). Bioinformatic analyses, coupled with transgenic assays, have identified a highly-conserved 154 bp signature motif within the IAB7b enhancer (Starr et al., 2011). This signature motif contains two putative FUSHI-TARAZU (FTZ) and two putative KRUPPEL (KR) TF binding sites (Fig. 2a). Constructs containing the two FTZ and two KR binding sites (2F2K) or the two FTZ sites with only the KR site proximal to the FTZ sites (2F1K) are sufficient to drive reporter gene expression in a three stripe pattern in the A5, A7, and A9 segments of transgenic D. melanogaster embryos (Starr et al., 2011). In the context of the IAB7b enhancer, FTZ therefore appears to be the activator TF and KR appears to be a repressor (Carroll and Scott, 1986). At the endogenous BX-C, KR represses Abd-B expression in the central part of the embryo, including T2, A1 and A3 (Casares and Sanchez-Herrero, 1995) (Fig. 1).
Fig. 2. Organization of transcription factor binding sites at IAB7b across Drosophila species.

(A) PATSER was used to computationally predict BCD (pink), HB (purple), KR (red), KNI (brown), FTZ (green), EVE (yellow) and GT (blue) transcription factor binding sites. The height of bars indicates the similarity of the given site to the true binding sites used to construct the PWM, and the top or bottom of the scale bar indicates on which strand the predicted binding site is found. The previously discovered signature motif, consisting of at least one KR and two FTZ sites, is seen across species (yellow box) (Starr et al., 2011). The fragments of IAB7b used in reporter constructs are shown (grey boxes). (B) Transgenic embryos at stage 5 of development. The 2F2K signature motif drives a clear three stripe pattern of lacZ reporter gene expression in presumptive abdominal segments A5, A7 and A9. In comparison, the 2F2K SDM and 2F2K +14 constructs show no reporter gene expression, indicating a lack of activation by FTZ. (C) Transgenic embryos at stage 5 of development. The full length (FL) IAB7b CRM drives a two stripe pattern of lacZ reporter gene expression in presumptive abdominal segments A7 and A9. In comparison, the 2F2K construct drives an additional stripe in A5. The inclusion of three KNI repressor binding sites (2F2K3N) on the construct is sufficient to restrict expression to only A7 and A9.
In this study, we address the internal regulatory architecture of TF binding sites in the IAB7b enhancer. To do this we develop a simple quantitative thermodynamic-based mathematical model to predict the functional output of interactions between TFs and the IAB7b enhancer and generate transgenic lines containing a lacZ reporter gene driven by various segments of the enhancer to investigate three specific questions: 1) the necessity of both FTZ binding sites in the signature motif for activation, 2) the importance of spacing and potential cooperativity between the two putative FTZ binding sites, and 3) the role of KNIRPS and GIANT in limiting the expression driven by the signature motif. Our results indicate that the complex combinatorial TF inputs utilize distinct molecular mechanisms of activation and repression, but can be mediated through just eight highly conserved binding sites.
Materials and Methods
Bioinformatic analysis
The relevant region of the bithorax complex (BX-C) in the Drosophila melanogaster genome was determined using the University of California Santa Cruz (UCSC) Genome Bioinformatics Genome Browser (http://genome.ucsc.edu). The full length 728 bp IAB7b region was identified, from the annotated U31961 sequence, to have coordinates chr3R:12741380-12742107 in the assembly last updated April 2006 (Starr et al., 2011). Putative transcription factor (TF) binding sites were determined using Patser (http://rsat.ulb.ac.be/rsat/patser_form.cgi) (Hertz and Stormo, 1999) with previously-assembled consensus Position Weight Matrices (PWMs) for BICOID (BCD), HUNCHBACK (HB), KRUPPEL (KR), KNIRPS (KNI), GIANT (GT), FUSHI-TARAZU (FTZ), EVEN-SKIPPED (EVE) and FUSHI-TARAZU FACTOR 1 (FTZ-F1) using ln(p-value) cutoff values described in previous studies (Bergman et al., 2005; Ho et al., 2009).
Transgenic reporter constructs
PCR primers were designed to amplify IAB7b regions containing the 2F2K signature motif (Starr et al., 2011) and additional sequences from the defined CRM (see table below). PCR amplicons were cloned into pGEM®-T Easy vector (Promega), and sub-cloned as a NotI or XhoI fragment into the placZattB transformation vector (Bischof et al., 2007). The putative FTZ binding site proximal to abdominal-A (abd-A) in the IAB7b signature motif was mutagenized using a QuikChange II XL Site Directed Mutagenesis Kit (Stratagene) and site-directed mutagenesis primers: CACTCTTTATTTCTTTCTTTTTGCCCTTGCCTAGGCACTGTCAGCGATTCTGTGATTTGACTCAGCAAACG CGTTTGCTGAGTCAAATCACAGAATCGCTGACAGTGCCTAGGCAAGGGCAAAAAGAAAGAAATAAAGAGTG on a previously constructed plasmid containing the 2F2K region in the pGEM®-T Easy vector (Promega). Once successful mutagenesis had been confirmed by sequencing, the 2F2K SDM region was sub-cloned into the placZattB vector as previously described (Starr et al., 2011). To separate the putative FTZ binding sites by an additional 14 bp, the portion of the 2F2K region distal from Abd-B, including the distal FTZ site, and the portion of the 2F2K region proximal to Abd-B, including the proximal FTZ site, were separately cloned into pGEM®-T Easy. These constructs were then inserted into the XhoI and NotI sites in the placZattB vector, which created fourteen base pairs of extra space between the two FTZ TF binding sites. The additional base pairs between the two FTZ TF binding sites do not result in any additional putative TF binding sites for FTZ, EVE, GT, HB, KR or KNI.
Table 1.
| IAB7b Sub-region |
Forward Primer (5′-3′) Reverse Primer (5′-3′) |
Coordinates amplified (chr3R) | Product length (bp) |
|---|---|---|---|
| FL | CGTCTTTTGTGTGTATTGGC TTATGAATGGGGCAGGTAGC |
12,741,292 – 12,742,119 | 828 |
| 2F2K3N | CGTCTTTTGTGTGTATTGGC CTAACTCGACTTGCTAACCTT |
12,741,292 – 12,741,800 | 509 |
| 2F2K | GTGCGTTTTCCTTTTAAGCCT CTAACTCGACTTGCTAACCTT |
12,741,647 – 12,741,800 | 154 |
| 2F2K +14 Proximal | CTCGAGAAACGGCGAGCTAA CTCGAGAACTCGACTTGCTAACCT |
12,741,758 – 12,741,798 | 53 |
| 2F2K +14 Distal | GCGGCCGCGTTTTCCT GCGGCCGCTGAGTCAAAT |
12,741,649 – 12,741,757 | 121 |
Transgenesis
Constructs containing IAB7b sub-regions were introduced into the D. melanogaster germline via φC31-mediated integration (Bischof et al., 2007). All microinjections were carried out by BestGene Inc., using the attB 68E site for targeted integration of the reporter constructs.
In situ hybridization
Embryos from transgenic D. melanogaster were collected, fixed, and hybridized with a digoxigenin-labeled lacZ probe as previously described (Bae et al., 2002).
Mathematical modeling of transcription
To mathematically model expression levels as a function of TF concentrations, the concentration gradients of each TF were needed. For this study, the TFs we were interested in were BCD, HB, KR, KNI, GT, EVE and FTZ. Data were taken from the BDTNP, which contains three-dimensional (3D) measurements of relative mRNA concentration for each gene (bcd, hb, Kr, kni, ftz, eve and gt) 110 mins into development, corresponding to 6078 nuclei on the periphery of a reference “virtual embryo” (Fowlkes et al., 2008). To create one-dimensional concentration gradients corresponding to one half of a laterally dissected virtual embryo, we first projected the 3D coordinates into 2-dimensional (2D) coordinates. We then took a strip from 10 – 90 % along the A–P axis, 31 pixels wide, and averaged concentration values at each coordinate along the A–P axis. Once these 1D mRNA concentration gradients were generated, modeling was performed under the assumption that protein concentration gradients are directly proportional to these mRNA gradients.
Mathematical modeling of the IAB7b enhancer was done using a thermodynamic-based (fractional occupancy) model as previously described (Dresch and Drewell, 2012). Since minimal information is available regarding the interactions taking place between TFs bound to the IAB7b enhancer and no quantitative data was available to fit model parameters, the model did not include any distance-dependence in the parameters representing cooperative interactions and quenching. However, the model did include competitive binding where overlapping binding sites are predicted bioinformatically, quenching with a 100% efficiency when short-range repressors and activators are bound simultaneously and a constant cooperativity term for homotypic FTZ interactions when FTZ binding sites are bound simultaneously. Since stripes of expression are observed in a binary fashion (either present or not), we used a threshold of 0.70 to convert continuous model output (in the range [0,1]) to a more appropriate discrete output (Ilsley et al., 2013). This threshold allows us to assign each spatial location along the A–P axis a binary number representing gene expression (i.e. either expression is observed (1) or not (0)). Once this threshold was set, values for TF binding affinities were chosen to avoid a predicted saturation in expression, corrected for different relative levels of protein concentrations and provide realistic qualitative predictions. Parameter values for TF binding affinities were held constant (KF=5.5, KR=3.3, KN=0.05, KG=3.5, KH=0.07) for all segments modeled and all cooperativity values tested. This approach allowed us to provide insight into the qualitative differences in the expression patterns arising under different hypotheses addressing the TFs involved in the functional regulation of the output driven by various segments of the IAB7b enhancer. Cooperativity values ranging from 0 to 2.2 were tested. These values cover a large range of hypotheses regarding the cooperative nature of FTZ binding sites; 0 corresponding to a scenario in which the state of the enhancer with both FTZ sites bound has absolutely no impact on the transcriptional output, values between 0 and 1 corresponding to a scenario in which this state has an impact less than that which would be observed if the proteins were activating independently, and values above 1 corresponding to a scenario in which this state has a greater impact on the transcriptional output than that which would be observed if the proteins were activating independently. Competitive binding is also included in the model, but with no additional parameters. The basic assumption is that although the same stretch of DNA may be capable of binding two different TFs, they cannot bind simultaneously. So, for a binding site of this type (referred to as an overlapping binding site), instead of having two possible states for that binding site, bound or unbound, the two TFs compete for binding, leading to three possible states in the model: the site is bound by the first TF, the site is bound by the other TF, or the site is unbound. Virtual embryo illustrations of model output were created using TF gradients obtained from the projected BDTNP data.
Results and Discussion
Specific spacing of FTZ activator binding sites in IAB7b
The IAB7b signature motif can be identified in orthologs of divergent Drosophila species from D. melanogaster to D. willistoni. In each case, the signature motif contains two putative FUSHI-TARAZU (FTZ) binding sites and at least one putative KRUPPEL (KR) binding site (Fig. 2a). Surprisingly, the architecture of the FTZ binding sites is completely conserved. The two putative FTZ binding sites are always found exactly 43 bp apart and on opposite strands, even across evolutionarily distant Drosophila species (Fig. 3a). Deletion of the distal FTZ site (relative to the KR sites) from the D. melanogaster signature motif results in a complete loss of activation function (Starr et al., 2011) for the enhancer. To further test the functional requirement for these two FTZ sites, we carried out a site directed mutagenesis of the proximal FTZ site. Disruption of this site in the signature motif (2F2K SDM) also results in a complete loss of activation (Fig. 2b). Furthermore, the insertion of 14 bp of additional sequence between the two FTZ sites (2F2K +14) also prevents activation from the CRM (Fig. 2b).
Fig. 3. Binding site conservation in the IAB7b Signature Motif.

(A) The spacing between the predicted FTZ (green), KR (red), and HB (purple) binding sites in the IAB7b signature motif across several Drosophila species. In all species, FTZ (green) binding sites are found on opposite strands and exactly 43 bp apart. (B) Sequence and spacing of PATSER computationally predicted KR (red), FTZ (green) and FTZ-F1 (gray) transcription factor binding sites in the signature motif region. Even in distantly related Drosophila species the organization of these binding sites is highly conserved. The site and sequence of the 14bp insertion in the 2F2K +14 construct is also indicated (blue).
Taken together, the requirement for both FTZ sites and the evolutionarily conserved distance between the sites in divergent species (Tamura et al., 2004) strongly implies that the spacing between these two sites plays a critical functional role. One possible model is that the spacing of the binding sites may allow the FTZ activator to bind, presumably through its DNA-binding homeodomain, to the IAB7b enhancer in a cooperative manner. The potential for cooperativity between FTZ molecules echoes the classic example of the bacteriophage lambda repressor (Ptashne, 2004) and is supported by the recent discovery that the homeodomain-containing TF SEX COMBS REDUCED in Drosophila forms homodimers which result in increased transcriptional specificity (Papadopoulos et al., 2012). Previous in vitro studies indicated that FTZ only binds as a monomer (Florence et al., 1991). However, as these studies used only the homeodomain of FTZ and did not test cooperative binding beyond a 10 bp distance, they do not preclude the possibility of FTZ binding cooperatively in vivo. Similar studies of the Hox TF UBX found a peptide containing the homeodomain did not bind DNA cooperatively, though the full length protein is able to bind in a cooperative manner (Beachy et al., 1993).
FTZ can also be recruited to DNA indirectly by interacting with the FTZ-F1 cofactor (Yoo et al., 2011); this interaction is necessary for FTZ to regulate its own gene expression (Ueda et al., 1990). Therefore, an alternative explanation for FTZ recruitment to the IAB7b enhancer is that two FTZ molecules could be cooperatively binding through another mechanism, such as the FTZ-F1 cofactor (Yoo et al., 2011). To examine this possibility we investigated the sequence in the signature motif for predicted FTZ-F1 binding sites. There is in fact a highly conserved FTZ-F1 site directly adjacent to the proximal FTZ site (Fig. 3b), leaving open the possibility for functional interactions between FTZ and FTZ-F1 at the IAB7b enhancer.
Repression by KRUPPEL, KNIRPS and GIANT
The three stripe expression profile directed by the IAB7b signature motif in the developing embryo (Fig. 2c) (Starr et al., 2011) suggests that additional regulatory inputs, beyond the FTZ activator and KR repressor, are needed to restrict gene expression solely to A7 and A9, as seen when expression is driven by the full length IAB7b enhancer (Fig. 2c). Likely candidate factors for such repression are the TFs KNIRPS (KNI) and GIANT (GT).
KNI is expressed at high concentration in the presumptive fifth abdominal segment (A5) (Fig. 1) and can act as a short-range repressor when bound to an enhancer (Arnosti et al., 1996). The range of repression caused by bound KNI is thought to be restricted to distances between 50–100 bp from activator binding sites (Arnosti et al., 1996). knirps mutant embryos have a disrupted pattern of Abdominal-B (Abd-B) expression due to an expansion of the anterior boundary of expression (Casares and Sanchez-Herrero, 1995). The full length IAB7b enhancer in D. melanogaster contains three predicted KNI binding sites (proximal to abd-A relative to the signature motif) (Fig. 2a). All three putative KNI sites are in genomic regions which are highly conserved across Drosophila species. The closest predicted KNI site is 125 bp and 168 bp respectively from the two FTZ TF binding sites in the 2F2K signature motif. If KNI is acting as a repressor when bound to IAB7b, it may prevent the recruitment of the FTZ activator in A3 and A5, thus preventing transcriptional activation of the Abd-B target gene in these segments. To test this hypothesis, we created transgenic enhancers with all three predicted KNI sites in addition to the core signature motif (2F2K3N) (Fig. 2a). The inclusion of these additional KNI binding sites is sufficient to restrict reporter gene expression solely to A7 and A9 (Fig. 2c). However, it should be noted that the 2F2K3N construct also contains a number of additional predicted TF binding sites (Fig. 2a) which may contribute to repression at the IAB7b enhancer.
GT is expressed at high concentration in a broad band in the anterior of the embryo and in lower concentration in A5, A6 and A7 in the posterior of the embryo (Fig. 1). A single conserved GT binding site is predicted in the IAB7b signature motif (Fig. 2a). This GT binding site overlaps with the interior FTZ binding site (Fig. 2a). It is therefore possible that GT is also capable of contributing to the repression of the IAB7b CRM in the embryo, by directly competing with FTZ for binding in the signature motif. The loss of either FTZ binding site in the signature motif (either in the 2F2K SDM construct, which also results in a loss of the predicted GT site) or by deletion of the exterior FTZ site (1F2K) (Starr et al., 2011) results in no activation from the CRM (Fig. 2b). Given the presence of additional conserved predicted GT binding sites in the full length IAB7b enhancer it is also quite possible that GT is capable of mediating repression through different molecular mechanisms.
Thermodynamic-based model of enhancer activity
To fully understand the combinatorial TF inputs at the IAB7b cis-regulatory module and reconcile these with the in vivo transcriptional output in the embryo, we developed a quantitative mathematical model (Fig. 4a). The modeling approach we implemented is that of a thermodynamic-based model, which allowed us to consider the impacts of the type and number of binding sites present in the CRM, competitive binding events, short-range repression and cooperative interactions between FTZ activator molecules. TF binding affinities are represented in the model by the ‘K’ variables (Fig. 4a), with the underscore corresponding to the specific TF binding. All predictions resulted from a fixed set of parameter values for TF binding affinities (as described in Materials and Methods). TF concentrations, represented by square brackets (Fig. 4a), were taken from experimental data that is available through the BDTNP (full details of the data acquisition are in Materials and Methods). Competitive binding is incorporated into the model when determining the number of possible states of each enhancer segment and short-range repression is incorporated when determining which states are interpreted as ‘successful’, i.e. included in the numerator of model equations (Fig. 4a). The cooperative activity resulting from two FTZ binding sites bound simultaneously is represented by a cooperativity parameter, C (Fig. 4a). This value corresponds to the impact this state has on the transcriptional output, with a value of 1 corresponding to the output that would be observed if the proteins were activating independently (more details available in Materials and Methods).
Fig. 4. Mathematical model of FTZ cooperativity and repression at IAB7b.

(A) The thermodynamic-based modeling equations used for the mathematical modeling of the IAB7b enhancer. The equations, from top to bottom, correspond to the different assumptions: (1) Enhancer is activated by two non-overlapping FTZ binding sites capable of cooperative activation; (2) Enhancer is activated by two non-overlapping FTZ binding sites capable of cooperative activation and repressed by two non-overlapping KR binding sites; (3) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two non-overlapping KR binding sites and the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site); and (4) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two non-overlapping KR binding sites, the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site), and three non-overlapping KNI binding sites. In each equation, K’s represent binding affinities and square brackets represent protein concentrations (F =FTZ, R = KR, G = GT, N = KNI). (B) Each of the four model predictions is shown for different values of the cooperativity parameter, C. In the first row, a schematic of the CRM assumed to be driving expression is shown. In the second row, the 1D TF concentrations along the A–P axis used as model input are shown. The third through ninth rows illustrate, on a virtual embryo, the model predictions when different cooperativity values for FTZ interactions were used.
Some general trends were observed with this model, regardless of the cooperativity value for FTZ. When just the two FTZ binding sites are included in the model, the most extensive pattern of gene expression is predicted (Fig. 4b). As the two KR, one GT, and three KNI binding sites are added to the model there is a predicted loss of expression in some stripes present in the anterior regions of the embryo (Fig. 4b).
The model predictions that most precisely reproduce the experimental data are those obtained using a relatively weak FTZ cooperativity value of C = 1.1 (Fig. 2c and 4b). When C = 1.1, starting only with the two FTZ binding sites in the signature motif, seven stripes of gene expression output are predicted, corresponding to the nuclei along the A–P axis in the embryo with a localized high concentration of FTZ (Fig. 4b, C = 1.1). The addition of the two KR binding sites in the signature motif results in a loss of expression in stripes 2 (T2) and 3 (A1) (Fig. 4b, C = 1.1), presumably mediated by short-range repression by KR due to the proximity to the FTZ sites (Fig. 3). If we then add GT to the model, we see additional loss of stripe 1 (C3) and, in combination with KR, stripe 4 (A3). This repression is most likely due to direct competition for binding at the interior FTZ site in the signature motif, but may also be mediated in part by short-range repression of the exterior FTZ site (Fig. 4b, C = 1.1). Significantly, with these inputs, the model predicts that expression persists in stripe 5 (A5) due to the fact that there is a lower concentration of GT in the posterior region of the embryo when compared to the anterior (Fig. 4b, C = 1.1). Finally, the addition of KNI to the model is sufficient to inhibit expression in stripe 5 (A5), but not 6 (A7) and 7 (A9) (Fig. 4b, C = 1.1). Given the distance between the KNI sites and FTZ sites this effect must represent a longer-range repression (or quenching) than had previously been considered possible for KNI (Arnosti et al., 1996). When compared to the other predictions, one should note that the C = 1.1 value for cooperativity is the only value that gives predictions exhibiting the same qualitative behavior for the enhancer as those observed experimentally (Fig. 2c and 4b). Even small deviations in the cooperativity value result in predicted expression patterns that appear to be missing stripes of expression (C ≤ 0.9) or contain extraneous stripes of expression (C ≥ 1.3). As the C value deviates further from C = 1.1 the model predictions become increasing inaccurate. This supports the notion of a distance-dependent cooperativity function (Dresch and Drewell, 2012) and the hypothesis that the spacing between the two FTZ sites is crucial for the proper cooperative activation and gene regulation by IAB7b (Starr et al., 2011).
Our mathematical model is also able to further test the potential for FTZ cooperativity directly as it can take in any TF concentration inputs. For example, if one wanted to use the model to predict expression output in mutant backgrounds, this could be easily done, as long as one knows how the mutant background impacts the concentration of the TFs feeding into the model. An example is provided by a FTZ heterozygote mutant if we make the simplifying assumption that in the mutant there is 50% reduction in the level of FTZ protein when compared to wild type, but the cooperativity value remains unchanged at C=1.1. In this mutant background the model predicts seven stripes of gene expression when only the two FTZ binding sites are considered (as is the case for the wild type situation), but only the most posterior stripe 7 of expression when either KR and GT or KR, GT and KNI are added to the model. This deviates markedly from the model predictions in the FTZ wild-type background (see above and Fig. 4b) and supports the hypothesis that FTZ may well be acting in a concentration dependent and cooperative manner at IAB7b.
To further investigate the model’s predictive power, the model was run with the same fixed parameter values as those used to generate the predictions in Fig. 4b with C = 1.1, including the bioinformatically predicted HB binding site (Fig. S1a). Our resulting model output is similar to that shown in Fig 4b. (Fig. S1b), implying that HB plays only a modest role in the regulation of the IAB7b enhancer. This is most likely due to the very low levels of HB in the anterior portion of the embryo at this time point in development (approximately 2 hours into development). The model was also run on the 2F2K SDM and 1F2K (Starr et al., 2011) constructs (Fig. S2a). Both of these constructs represent CRMs with only a single FTZ binding site, therefore removing the possibility of any cooperative activation from occurring. The model results predict no gene expression driven by either enhancer (Fig. S2b) in agreement with the in vivo experimental results (Starr et al., 2011), even though the model does not a priori require there to be more than one FTZ binding site to drive gene expression.
Our final model therefore confirms that the IAB7b enhancer is a CRM acting as a logic processor of combinatorial TF inputs to generate a regulatory transcriptional output (Fig. 5). Indeed, only two activator (FTZ) and six repressor (one GT, two KR and three KNI) binding sites are required to produce the final output (Fig. 5). The potential for FTZ to bind cooperatively at the IAB7b enhancer could mediate large changes in Abd-B expression in response to relatively small changes in FTZ concentration. The ability of transcription factors to bind cooperatively is known to play an important role in Drosophila development. For example, Bcd mutants unable to bind cooperatively fail to create the sharp hb expression boundaries necessary and result in embryonic lethality (Burz and Hanes, 2001; Lebrecht et al., 2005). Given the possibility that each of the three TF repressors are functioning through distinct mechanisms (GT direct competition, KR short-range repression and KNI longer-range quenching) the spatial organization of the binding sites for these factors is likely to be under tight evolutionary selective pressure. This is evidenced by the high level of conservation observed for these key binding sites in IAB7b orthologs across diverse Drosophila species (Fig. 2a). Taken together with the requirement that there are two FTZ activator binding sites in IAB7b spaced exactly 43 bp apart, this suggests that the sequence architecture at this enhancer is critical to maintain robust regulatory function.
Fig. 5. Combinatorial transcriptional inputs control the regulatory output of IAB7b.
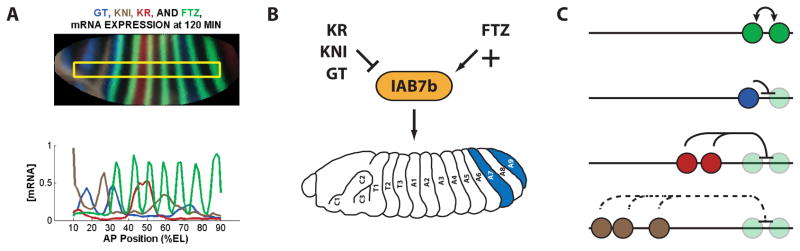
(A) The mRNA expression profiles 120 minutes into development for the gap repressor TFs (GT, KNI and KR) and the pair-rule activator TF (FTZ) are shown on a virtual embryo obtained via projection of the BDTNP data set along the anterior-posterior (AP) axis. Positions indicate the percentage of embryo length (%EL). (B) The combinatorial input of the three repressors (GT, KNI and KR) and single activator (FTZ) result in the in vivo confirmed regulatory output of the IAB7b CRM. (C) Distinct molecular mechanisms of transcriptional repression are at play in IAB7b. The FTZ activator (green) TF cooperatively binds (arrows) at two sites in the IAB7b CRM. GT (blue) directly competes for binding at the internal FTZ site, but may also prevent recruitment of FTZ (faded green) at the neighboring binding site by short-range repression. The two KR binding sites are in close proximity to the FTZ binding sites and therefore recruitment of the KR (red) TF permits short-range repression. In contrast, given the greater distance between the three KNI sites and FTZ sites, repression by KNI (brown) must be mediated by a longer-range quenching.
Supplementary Material
(A) The thermodynamic-based modeling equations, with the predicted HB binding site included, used for the mathematical modeling of the IAB7b enhancer. The equations, from top to bottom, correspond to two different assumptions: (1) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, and the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site); and (2) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site), and three non-overlapping KNI binding sites. In each equation, K’s represent binding affinities and square brackets represent protein concentrations (F = FTZ, R = KR, G = GT, N = KNI, H = HB). (B) Each of the two model predictions is shown for the FTZ cooperativity value C = 1.1. In the first row, a schematic of the CRM assumed to be driving expression is shown. In the second row, the 1D TF concentrations along the A–P axis used as model input are shown. The third row illustrates, on a virtual embryo, the model predictions.
(A) The thermodynamic-based modeling equations, with all predicted binding sites included, used for the mathematical modeling of the IAB7b SDM and 1F2K constructs (Starr et al., 2011). The equations, from top to bottom, correspond to two different assumptions: (1) Enhancer is activated by one non-overlapping FTZ binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression; and (2) Enhancer is activated by one FTZ binding site which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, and the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site). In each equation, K’s represent binding affinities and square brackets represent protein concentrations (F = FTZ, R = KR, G = GT, H = HB). (B) Each of the two model predictions is shown for the FTZ cooperativity value C = 1.1, although since each equation has only a single FTZ site, this cooperativity value does not affect the model output at all. In the first row, a schematic of the CRM assumed to be driving expression is shown. In the second row, the 1D TF concentrations along the A–P axis used as model input are shown. The third row illustrates, on a virtual embryo, the model predictions.
Highlights.
We investigate the sequence architecture at the IAB7b enhancer
An evolutionarily conserved signature motif is identified
FUSHI-TARAZU is the activator transcription factor for the enhancer
Three transcriptional repressors regulate the enhancer
The combinatorial input of the activator and repressors are critical
Acknowledgments
The research in this paper was supported by funding to R.A.D. from the National Institutes of Health (HD54977 and GM090167), the National Science Foundation (IOS-0845103) and Howard Hughes Medical Institute Undergraduate Science Education Program grants (520051213 and 52006301) to the Biology department at Harvey Mudd College. J.M.D. was funded as a Teaching and Research Postdoctoral Fellow at Harvey Mudd College, supported in part by NSF Grant DMS-0839966.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbari OS, Bousum A, Bae E, Drewell RA. Unraveling cis-regulatory mechanisms at the abdominal-A and Abdominal-B genes in the Drosophila bithorax complex. Developmental Biology. 2006;293:294. doi: 10.1016/j.ydbio.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Arnone MI, Davidson EH. The hardwiring of development: Organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- Arnosti DN, Gray S, Barolo S, Zhou J, Levine M. The gap protein knirps mediates both quenching and direct repression in the Drosophila embryo. The EMBO Journal. 1996;15:3659–3666. [PMC free article] [PubMed] [Google Scholar]
- Bae E, Calhoun VC, Levine M, Lewis EB, Drewell RA. Characterization of the intergenic RNA profile at abdominal-A and Abdominal-B in the Drosophila bithorax complex. PNAS. 2002;99:16847–16852. doi: 10.1073/pnas.222671299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachy PA, Varkey J, Young KE, von Kessler DP, Sun BI, Ekker SC. Cooperative binding of an Ultrabithorax homeodomain protein to nearby and distant DNA sites. Molecular and Cellular Biology. 1993;13:6941–6956. doi: 10.1128/mcb.13.11.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Carlson JW, Celniker SE. Drosophila DNase I footprint database: a systematic genome annotation of transcription factor binding sites in the fruitfly, Drosophila melanogaster. Bioinformatics. 2005;21:1747–1749. doi: 10.1093/bioinformatics/bti173. [DOI] [PubMed] [Google Scholar]
- Berleth T, Burri M, Thoma G, Bopp D, Richstein S, Frigerio G, Noll M, Nüsslein-Volhard C. The role of localization of bicoid RNA in organizing the anterior pattern of the Drosophila embryo. EMBO J. 1988;7:1749–1756. doi: 10.1002/j.1460-2075.1988.tb03004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. PNAS. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borok MJ, Tran DA, Ho MC, Drewell RA. Dissecting the regulatory switches of development: lessons from enhancer evolution in Drosophila. Development. 2010;137:5–13. doi: 10.1242/dev.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burz DS, Hanes SD. Isolation of Mutations that Disrupt Cooperative DNA Binding by the Drosophila Bicoid Protein. Journal of Molecular Biology. 2001;305:219–230. doi: 10.1006/jmbi.2000.4287. [DOI] [PubMed] [Google Scholar]
- Busturia A, Bienz M. Silencers in Abdominal-B, a homeotic Drosophila gene. The EMBO Journal. 1993;12:1415–1425. doi: 10.1002/j.1460-2075.1993.tb05785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB, Scott MP. Zygotically active genes that affect the spatial expression of the fushi tarazu segmentation gene during early Drosophila embryogenesis. Cell. 1986;45:113–126. doi: 10.1016/0092-8674(86)90543-x. [DOI] [PubMed] [Google Scholar]
- Casares F, Sanchez-Herrero E. Regulation of the infraabdominal regions of the bithorax complex of Drosophila by gap genes. Development. 1995;121:1855–1866. doi: 10.1242/dev.121.6.1855. [DOI] [PubMed] [Google Scholar]
- Dresch JM, Drewell RA. Sequence and Genome Analysis III: Emerging Methods, Techniques and Applications. iConcepts Press; 2012. Decoding the cis-regulatory grammar behind enhancer architecture. [Google Scholar]
- Driever W, Nüsslein-Volhard C. A gradient of bicoid protein in Drosophila embryos. Cell. 1988;54:83–93. doi: 10.1016/0092-8674(88)90182-1. [DOI] [PubMed] [Google Scholar]
- Florence B, Handrow R, Laughon A. DNA-binding specificity of the fushi tarazu homeodomain. Mol Cell Biol. 1991;11:3613–3623. doi: 10.1128/mcb.11.7.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowlkes CC, Hendriks CL, Keränen SV, Weber GH, Rübel O, Huang MY, Chatoor S, DePace AH, Simirenko L, Henriquez C, Beaton A, Weiszmann R, Celniker S, Hamann B, Knowles DW, Biggin MD, Eisen MB, Malik J. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm. Cell. 2008;133:364–374. doi: 10.1016/j.cell.2008.01.053. [DOI] [PubMed] [Google Scholar]
- Gray S, Szymanski P, Levine M. Short-range repression permits multiple enhancers to function autonomously within a complex promoter. Genes & Devel. 1994;8 doi: 10.1101/gad.8.15.1829. [DOI] [PubMed] [Google Scholar]
- Han K, Levine MS, Manley JL. Synergistic activation and repression of transcription by Drosophila homeobox proteins. Cell. 1989;56:573–583. doi: 10.1016/0092-8674(89)90580-1. [DOI] [PubMed] [Google Scholar]
- Harding K, Levine M. Gap genes define the limits of Antennapedia and Bithorax gene expression during early development in Drosophila. The EMBO Journal. 1988;7:205–214. doi: 10.1002/j.1460-2075.1988.tb02801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Ho MC, Johnsen H, Goetz SE, Schiller BJ, Bae E, Tran DA, Shur ASA, JM, Rau C, Bender W, Fisher WW, Celniker SE, Drewell RA. Functional evolution of cis-regulatory modules at a homeotic gene in Drosophila. PLoS Genetics. 2009;5:e1000709. doi: 10.1371/journal.pgen.1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch M, Jackle H. Transcriptional regulation and spatial patterning in Drosophila. Curr Opin Gen Dev. 1993;3:566–573. doi: 10.1016/0959-437x(93)90092-4. [DOI] [PubMed] [Google Scholar]
- Ilsley GR, Fisher J, Apweiler R, Depace AH, Luscombe NM. Cellular resolution models for even skipped regulation in the entire Drosophila embryo. Elife. 2013;2:e00522. doi: 10.7554/eLife.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga JT. Regulation of RNA Polymerase II Transcription by Sequence-Specific DNA Binding Factors. Cell. 2004;116:247–257. doi: 10.1016/s0092-8674(03)01078-x. [DOI] [PubMed] [Google Scholar]
- Lebrecht D, Foehr M, Smith E, Lopes FJP, Vanario-Alonso CE, Reinitz J, Burz DS, Hanes SD. Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc Natl Acad Sci USA. 2005;102:13176–13181. doi: 10.1073/pnas.0506462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Manley JL. Transcriptional Repression of Eukaryotic Promoters. Cell. 1989;59:405–408. doi: 10.1016/0092-8674(89)90024-x. [DOI] [PubMed] [Google Scholar]
- Lewis E. Gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- Lewis EB, Knafels JD, Mathog DR, Celniker SE. Sequence analysis of the cis-regulatory regions of the bithorax complex of Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8403–7. doi: 10.1073/pnas.92.18.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda RK, Karch F. The ABC of the BX-C: the bithorax complex explained. Development. 2006;133:1413–1422. doi: 10.1242/dev.02323. [DOI] [PubMed] [Google Scholar]
- Martin CH, Mayeda CA, Davis CA, Ericsson CL, Knafels JD, Mathog DR, Celniker SE, Lewis EB, Palazzolo MJ. Complete sequence of the bithorax complex of Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8398–402. doi: 10.1073/pnas.92.18.8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaly J, Barges S, Sipos L, Maeda R, Cleard F, Hogga I, Bender W, Gyurkovics H, Karch F. Dissecting the regulatory landscape of the Abd-B gene of the bithorax complex. Development. 2006;133:2983–2993. doi: 10.1242/dev.02451. [DOI] [PubMed] [Google Scholar]
- Papadopoulos DK, Skouloudaki K, Adachi Y, Samakovlis C, Gehring WJ. Dimer formation via the homeodomain is required for function and specificity of Sex combs reduced in Drosophila. Developmental Biology. 2012;367:78–89. doi: 10.1016/j.ydbio.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Papatsenko D, Levine MS. Dual regulation by the Hunchback gradient in the Drosophila embryo. Proc Natl Acad Sci USA. 2008;105:2901–2906. doi: 10.1073/pnas.0711941105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. A Genetic Switch. 3. Cold Spring Harbor Laboratory Press; 2004. Phage Lambda Revisited. [Google Scholar]
- Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- Qian S, Capovilla M, Pirrotta V. The bx region enhancer, a distant cis-control element of the Drosophila Ubx gene and its regulation by hunchback and other segmentation genes. EMBO J. 1991;10:1415–1425. doi: 10.1002/j.1460-2075.1991.tb07662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez_Herrero E. Control of the expression of the bithorax complex genes abdominal-A and abdominal-B by cis-regulatory regions in Drosophila embryos. Development (Cambridge, England) 1991;111:437–49. doi: 10.1242/dev.111.2.437. [DOI] [PubMed] [Google Scholar]
- Sauer F, Rivera-Pomar R, Hoch M, Jäckle H. Gene regulation in the Drosophila embryo. Philos Trans R Soc Lond B Biol Sci. 1996;351:579–587. doi: 10.1098/rstb.1996.0057. [DOI] [PubMed] [Google Scholar]
- Sauer F, Tjian R. Mechanisms of transcriptional activation: differences and similarities between yeast, Drosophila, and man. Curr Opin Genet Dev. 1997;7:176–181. doi: 10.1016/s0959-437x(97)80126-8. [DOI] [PubMed] [Google Scholar]
- Small S, Blair A, Levine M. Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J. 1991a;11:4047–4057. doi: 10.1002/j.1460-2075.1992.tb05498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S, Blair A, Levine M. Regulation of two pair-rule stripes by a single enhancer in the Drosophila embryo. Dev Biol. 1996;175:314–324. doi: 10.1006/dbio.1996.0117. [DOI] [PubMed] [Google Scholar]
- Small S, Kraut R, Hoey T, Warrior R, Levine M. Transcriptional regulation of a pair-rule stripe in Drosophila. Genes Dev. 1991b;5:827–839. doi: 10.1101/gad.5.5.827. [DOI] [PubMed] [Google Scholar]
- Starr MO, Ho MC, Gunther EJM, Tu YK, Shur AS, Goetz SE, Borok MJ, Kang V, Drewell RA. Molecular dissection of cis-regulatory modules at the Drosophila bithorax complex reveals critical transcription factor signature motifs. Developmental Biology. 2011;359:290–302. doi: 10.1016/j.ydbio.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Struhl K, Macdonald PM. The gradient morphogen bicoid is a concentration-dependent transcriptional activator. Cell. 1989;57:1259–1273. doi: 10.1016/0092-8674(89)90062-7. [DOI] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol. 2004;21:36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Ueda H, Sonoda S, Brown JL, Scott MP, Wu C. A sequence-specific DNA-binding protein that activates fushi tarazu segmentation gene expression. Genes & Devel. 1990;4:624–635. doi: 10.1101/gad.4.4.624. [DOI] [PubMed] [Google Scholar]
- Yoo J, Ko S, Kim H, Sampson H, Yun JH, Choe KM, Chang I, Arrowsmith CH, Krause HM, Cho HS, Lee W. Crystal structure of fushi tarazu factor 1 ligand binding domain/fushi tarazu peptide complex identifies new class of nuclear receptors. J Biol Chem. 2011;286:31225–31231. doi: 10.1074/jbc.M111.252916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ashe H, Burks C, Levine M. Characterization of the transvection mediating region of the abdominal-B locus in Drosophila. Development. 1999;126:3057–65. doi: 10.1242/dev.126.14.3057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) The thermodynamic-based modeling equations, with the predicted HB binding site included, used for the mathematical modeling of the IAB7b enhancer. The equations, from top to bottom, correspond to two different assumptions: (1) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, and the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site); and (2) Enhancer is activated by two FTZ binding sites capable of cooperative activation, one of which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site), and three non-overlapping KNI binding sites. In each equation, K’s represent binding affinities and square brackets represent protein concentrations (F = FTZ, R = KR, G = GT, N = KNI, H = HB). (B) Each of the two model predictions is shown for the FTZ cooperativity value C = 1.1. In the first row, a schematic of the CRM assumed to be driving expression is shown. In the second row, the 1D TF concentrations along the A–P axis used as model input are shown. The third row illustrates, on a virtual embryo, the model predictions.
(A) The thermodynamic-based modeling equations, with all predicted binding sites included, used for the mathematical modeling of the IAB7b SDM and 1F2K constructs (Starr et al., 2011). The equations, from top to bottom, correspond to two different assumptions: (1) Enhancer is activated by one non-overlapping FTZ binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression; and (2) Enhancer is activated by one FTZ binding site which is overlapping with a GT binding site, and repressed by two KR binding sites, one of which is overlapping with a HB binding site also capable of repression, and the overlapping GT binding site (i.e.: this includes competitive binding between FTZ and GT at the overlapping site). In each equation, K’s represent binding affinities and square brackets represent protein concentrations (F = FTZ, R = KR, G = GT, H = HB). (B) Each of the two model predictions is shown for the FTZ cooperativity value C = 1.1, although since each equation has only a single FTZ site, this cooperativity value does not affect the model output at all. In the first row, a schematic of the CRM assumed to be driving expression is shown. In the second row, the 1D TF concentrations along the A–P axis used as model input are shown. The third row illustrates, on a virtual embryo, the model predictions.