

Abstract
Fat1 is an atypical cadherin that controls vascular smooth muscle cell (VSMC) proliferation and migration. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (Nox1) is an important source of reactive oxygen species (ROS) in VSMCs. Angiotensin II (Ang II) induces the expression and/or activation of both Fat1 and Nox1 proteins. This study tested the hypothesis that Ang II-induced Fat1 activation and VSMC migration are mediated by Nox1-dependent ROS generation and redox signaling. Studies were performed in cultured VSMCs from Sprague-Dawley rats. Cells were treated with Ang II (1 μmol/L) for short (5 to 30 min) or long term stimulations (3 to 12 h) in the absence or presence of the antioxidant apocynin (10 μmol/L), extracellular-signal-regulated kinases 1/2 (Erk1/2) inhibitor PD98059 (1 μmol/L), or Ang II type 1 receptor (AT1R) valsartan (1 μmol/L). siRNA was used to knockdown Nox1 or Fat1. Cell migration was determined by Boyden chamber assay. Ang II increased Fat1 mRNA and protein levels and promoted Fat1 translocation to the cell membrane, responses that were inhibited by AT1R antagonist and antioxidant treatment. Downregulation of Nox1 inhibited the effects of Ang II on Fat1 protein expression. Nox1 protein induction, ROS generation, and p44/p42 MAPK phosphorylation in response to Ang II were prevented by valsartan and apocynin, and Nox1 siRNA inhibited Ang II-induced ROS generation. Knockdown of Fat1 did not affect Ang II-mediated increases in Nox1 expression or ROS. Inhibition of p44/p42 MAPK phosphorylation by PD98059 abrogated the Ang II-induced increase in Fat1 expression and membrane translocation. Knockdown of Fat1 inhibited Ang II-induced VSMC migration, which was also prevented by valsartan, apocynin, PD98059, and Nox1 siRNA. Our findings indicate that Ang II regulates Fat1 expression and activity and induces Fat1-dependent VSMC migration via activation of AT1R, ERK1/2, and Nox1-derived ROS, suggesting a role for Fat1 downstream of Ang II signalling that leads to vascular remodelling.
Keywords: Angiotensin II, Fat1, Nox1, ROS, ERK1/2 MAPK, migration, vascular smooth muscle cell
Introduction
Vascular remodeling is critical in the pathogenesis of clinically important cardiovascular disorders [1,2]. Angiotensin II (Ang II) activates signaling responses via the Ang II type 1 receptor (AT1R), thereby mediating the generation of reactive oxygen species (ROS), inflammation, proliferation, migration, hypertrophy, and fibrosis, all of which are important in vascular remodeling in cardiovascular disease [3–6].
Among the several sources of ROS in vascular smooth muscle cells (VSMCs), Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays a crucial role triggering the activation of many redox-sensitive pathways, such as mitogen-activated protein kinases (MAPKs), which are involved in proliferation events [7,8]. The NADPH oxidase, or Nox, family comprises seven isoforms of ROS-generating enzymes, Nox1-5 and DUOX (dual oxidase) 1 and 2. These enzymes differ not only in their ability to generate specific ROS, but also in their tissue distribution, subcellular regulation, requirement for cofactors, and oxidase subunits. Of the seven family members, four have been identified as important sources of ROS in the vasculature: Nox1, Nox2, Nox4, and Nox5. Nox1 is expressed in all layers of the vascular wall, and Ang II increases both its expression and activity, leading to augmented Nox1-derived ROS generation [3,9–11].
Cadherins are transmembrane adhesion proteins characterized as major mediators of cell homeostasis. Cadherins are involved in calcium (Ca2+)-dependent cell–cell adhesion, intracellular junction assembly, and tissue morphogenesis during development and vascular remodelling [12–14]. Previous studies from our laboratory showed that the vascular expression of Fat1 cadherin, an atypical cadherin, is significantly increased after arterial balloon injury and stimulation with growth factors or Ang II [14,15]. In addition, we found that Fat1 is involved in the regulation of key cellular functions for vascular remodeling, including migration and proliferation. Accordingly, the present study tested the hypothesis that Ang II leads to an increase in Fat1 protein expression and activity via redox-sensitive pathways, focusing on Nox1-derived ROS generation. We also sought to determine whether Fat1 activation plays a role in Ang II-induced redox-mediated VSMC migration.
Material and Methods
Animals
Housing conditions and experimental protocols were in accordance with the Ethical Animal Committees from the University of Sao Paulo and the Albert Einstein College of Medicine of Yeshiva University. Animals were housed under standard laboratory conditions with free access to food and water. Vascular smooth muscle cells were harvested from the aortas of male Sprague-Dawley rats (200 to 250 g, Charles River).
Cell culture
Primary culture of rat aortic VSMCs was prepared as previously described [16] and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen Life Technologies) containing 10% fetal bovine serum (FBS, HyClone), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 mmol/L Hepes (pH 7.4, Sigma-Aldrich). VSMCs were used between passages 4 and 8. Cells were made quiescent by incubation in medium containing 0.4% horse serum for 24 h before stimulation. Total cellular protein extracts were prepared at designated time points.
Protocols for cell stimulation
Growth-arrested VSMCs were treated with Ang II (1 μmol/L, Sigma-Aldrich) for short (5 to 30 min) or long (3 to 12 h) term stimulations. In some experiments, cells were exposed for 30 minutes to apocynin (10 μmol/L, Santa Cruz), valsartan (10 μmol/L, Sigma-Aldrich) or PD98059 (1 μmol/L, Tocris) prior to Ang II addition. Concentrations of Ang II and inhibitors were chosen based on preliminary experiments and previous assays.
Lucigenin assay
NADPH oxidase-derived superoxide anion generation was measured in VSMC homogenates by lucigenin chemiluminescence [17]. Stimulated VSMCs were washed in ice-cold phosphate-buffered saline (PBS) and scraped off in assay buffer (50 mmol/L KH2PO4, 1 mmol/L EGTA, 150 mmol/L sucrose, pH 7.4). The reaction (final volume 250 μL) wasstarted by the addition of NADPH (0.1 mmol/L) assay buffer containing sample homogenate (50 μL) and lucigenin (5 μmol/L). Luminescence was measured every 1.8 seconds (30 cycles) in a Victor II multilabel luminometer (Wallac). The lucigenin-derived chemiluminescence measured without the samples was subtracted from each test reading to yield net of ROS generation, which was expressed as relative luminescence units (RLU).
ROS detection in intact cells with dihydroethidium
ROS generation in intact VSMCs was measured by using dihydroethidium (DHE, Santa Cruz) fluorescent dye [11]. VSMCs were incubated in Hank’s balanced salt solution (HBSS) containing 1.3 mmol/L CaCl2 and 5.5 mmol/L glucose supplemented with 2 mmol/L DHE in a light-protected chamber at 37°C for 20 min. Cells were washed with HBSS and images were obtained before and after stimulation with Ang II in presence or absence of inhibitors. Images were obtained using an Axio Observer .Z1 microscope (Zeiss, Thornwood, NY).
Real Time PCR
Total RNA was extracted from VSMCs by homogenization in TRIzol (Invitrogen), treated with DNase I (1 U/μl, Promega), and used for first-strand cDNA synthesis. The mRNA levels were quantified in triplicate by qPCR in the Mx3000P Real-Time PCR System with the Brilliant SYBR Green qPCR kit (Stratagene). Specific primers for qPCR were rat Fat1, 5′-CCCCTTCCAACTCTCCCTCA-3′ (forward) and 5′-CAGGCTCTCCCGGGCACTGT-3′ (reverse); rat Nox1, 5′-CTTCCTCAGTGGCTGGGATA-3′ (forward) and 5′-GCAAGCATTTGCGCAGGCT-3′ (reverse); rat NOXA1, 5′-CATCCCTGACCACAACTCG-3′ (forward) and 5′-CTCCTCGGGCTTTGTAGCTGAA-3′ (reverse); rat p47phox, 5′-GCTGCCCACACCTCTTGAACTT-3′ (forward) and 5′-TCTTCAGGCAAAACCACCA-3′ (reverse). PCR cycling conditions included 10 min at 95° C for 1 cycle followed by 45 cycles at 95° C for 30 s, 60° C for 30 s, and 72° C for 60 s. Dissociation curve analysis confirmed that signals corresponded to unique amplicons. Specific mRNA expression levels were normalized relative to hypoxanthine phosphoribosyltransferase (HPRT) mRNA levels using the comparative ΔΔCt method 18.
Western Blot analysis
For Fat1 analysis, total or fractionated proteins from VSMCs cells were extracted in radio-immunoprecipitation assay (RIPA) buffer with protease inhibitors. Whole cell lysate (40 μg) was separated by electrophoresis through 3–8% Novex Tris-acetate or 4–12% Bis-Tris polyacrylamide gels (Invitrogen) and transferred to Immobilon-P PVDF membrane (Millipore). For ERK1/2 MAPK and Nox1 analysis, proteins from VSMCs were separated by electrophoresis on a 10% polyacrylamide gel and transferred onto a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline solution with Tween for 1 hour at 24°C. Membranes were then incubated with specific antibodies overnight at 4°C. Antibodies were as follows: anti-ERK1/2 MAPK [Thr202/Tyr204] (1:1000, Millipore), and anti-Nox1 (1:1000, Sigma-Aldrich). The anti-Fat1 antibody has been previously described14. After incubation with secondary antibodies, signals were revealed with chemiluminescence, visualized by autoradiography and quantified densitometrically. To assess equivalence of protein loading, the same membranes were re-probed with loading control antibodies: anti-ERK1/2 MAPK (1:1000, Millipore) was used as loading control and was carried out on the same membranes for phosphorylated proteins, and antibody to β-actin (1:3000, Abcam) was also used as an internal housekeeping control.
Assessment of Fat1 and p47phox membrane translocation
Activation of Fat1 and p47phox was assessed by evaluating their translocation from the cytosol to the membrane. VSMCs were homogenized in lysis buffer (50 mmol/L Tris/HCl [pH 7.4], 5 mmol/L EGTA and 2 mmol/L EDTA, 0.1 mmol/L phenylmethylsulfonyl fluoride [PMSF], 0.2 mmol/L sodium orthovanadate [Na3VO4], 1 μmol/L pepstatin A, 1 μmol/L leupeptin, and 1 μmol/L aprotinin) and fractionated to obtain cytosol- and membrane-rich fractions. Homogenates were centrifuged at 100,000 g for 1 hour at 4°C, thereby isolating cytosolic fraction in the supernatant. The particulate fraction was resuspended in lysis buffer containing 1% Triton X-100. Protein analysis was performed by western blotting as described above using anti-Fat1 and anti-p47phox (1:500, Santa Cruz) antibodies.
Immunofluorescent imaging
VSMCs were cultured on glass coverslips in DMEM, fixed and permeabilized for 10 min at room temperature with PBS containing 2% paraformaldehyde and 0.3% Triton X-100, and then blocked with PBS/2% BSA. Anti-Fat1 antibody (1:500) incubation was performed for 1 h at room temperature in PBS/2% BSA followed by incubation with Alexa Fluor 488 anti-rabbit secondary antibody (Invitrogen) plus Texas Red-X phalloidin (1:100; Invitrogen) for cytoskeleton staining. Coverslips were mounted with Prolong Gold anti-fade mounting medium containing diamidino-2-phenylindole (DAPI, Invitrogen). Images were acquired with a Leica TCS SP2 SE inverted microscope using a 40X, 1.25 numerical aperture oil immersion objective (Leica HCX PL APO).
RNA Interference and Cell Transfection
siRNA for Fat1 and Nox1 were purchased from Ambion, as described previously [14] and transfected with X-treme GENE Reagent (Roche) according to the manufacturer’s recommendations. Fat1 and Nox1 knockdown efficiency was assessed by western blot analysis.
Cell migration assays
VSMC migration was assessed by modified Boyden chamber assay using Transwell 24-well cell culture inserts with 8 μm pores (Costar). VSMCs were harvested and added to the insert (5 × 104 cells/well), and culture medium with or without Ang II was added to the chamber. In some experiments, AT1R antagonist, antioxidant agents, or ERK1/2 inhibitor were added 30 min prior to Ang II stimulation. Migration was also assessed in cells transfected with siRNA for Nox1 or Fat1. After 12 h, non-migrating cells were removed from upper filter surfaces and the filter was washed, fixed, and stained. Six randomly selected 200× fields were photographed and the number of cells that migrated was determined.
Statistical analysis
Data are presented as mean ± standard error deviation of mean (SEM). Comparisons between two groups were analyzed by t test, and comparisons among three or more groups were assessed by analysis of variance (ANOVA) with a Bonferroni post hoc test. Significance was accepted for values of P < 0.05.
Results
Ang II induces Fat1 and Nox1 mRNA and protein expression increase
The effect of Ang II on Fat1 expression is demonstrated in the Figure 1. Ang II increased Fat1 mRNA (Figures 1A) and protein levels (Figures 1B) in VSMCs in a time-dependent manner. Increased protein expression of Fat1 was inhibited by the AT1R antagonist valsartan and the antioxidant agent, apocynin (Figure 1C). To further evaluate the contribution of ROS to Ang II induced Fat1 expression increase, VSMCs were subjected to Nox1 gene knockdown. Downregulation of Nox1 inhibited the effects of Ang II on Fat1 protein expression (Figure 1D).
Figure 1. Ang II increases Fat1 mRNA and protein expression.

Time-course (3 to 12 h) effect of 10−6 mol/L Ang II on Fat1 gene (A) and protein (B) expression in VSMCs. (C) Effect of Ang II (12 h) on Fat1 protein expression in the absence and in the presence of apocynin or valsartan. (D) Effect of Ang II (12 h) on Fat1 protein expression in non-transfected and Nox1 siRNA-transfected VSMCs. Representative immunoblots: Fat1, Nox1, and β-actin. In this and subsequent figures, immunoblots probed initially for Fat1 or Nox1 were reprobed with a reference protein, as indicated, to assess loading. Results are mean ± SEM of 4 experiments. *P<0.05, vs vehicle.
Ang II induces Fat1 translocation to membrane
We next examined whether Ang II, in addition to stimulating Fat1 expression, induces trafficking of this cadherin from the cytosol to cell membrane. Immunofluorescence image analysis demonstrated that Fat1 is localized and further dynamically targeted to the plasma membrane upon Ang II stimulation (Figure 2A). This finding is consistent with the increased Fat1 protein content in membrane enriched fractions observed in VSMCs stimulated with Ang II (Figure 2B). This effect of Ang II on Fat1 membrane recruitment required both AT1R activity and ROS generation, as evidenced by the inhibitory effects of valsartan and apocynin.
Figure 2. Fat1 subcellular localization and Fat1 content in membrane fractions.

Top panels, representative images of anti-Fat1 staining in VSMCs stimulated with 10−6 mol/L Ang II or vehicle. VSMCs were double-labeled with antibody to Fat1 (Alexa Fluor 488, anti-rabbit secondary antibody, green fluorescence) and Texas red-X phalloidin. DAPI was used for fluorescent staining of DNA. Arrows indicate Fat1 staining at cellular free edges (B) VSMCs were subjected to membrane and cytosol fractionation after stimulation with 10−6 mol/L Ang II (5 and 30 min). Membrane to cytosol ratio of Fat1 protein content is expressed as % of vehicle. Representative immunoblots: Fat1and β-actin. Results are mean ± SEM of 5 experiments. *P<0.05, vs vehicle.
Ang II induces ROS generation and Nox1 mRNA and protein expression increase
As shown in Figure 3, Ang II increased Nox1 mRNA (Figure 3A) and protein expression (Figure 3B) in VSMCs in a time-dependent manner. Increased protein expression of Nox1 was inhibited by valsartan and apocynin (Figure 3C). The knockdown of Fat1 with siRNA did not affect the modulatory effect of Ang II on Nox1 expression (Figure 3D). Ang II-induced ROS generation was determined by lucigenin chemiluminescence and assay DHE imaging. Our data demonstrate that Ang II induces ROS generation in short (Figure 4A) and long (Figure 4B) term stimulations of VSMCs. These effects were inhibited by valsartan and apocynin. The knockdown of Nox1, but not Fat1, reduced Ang II induced long term ROS generation (Figure 4C). Thus Fat1 expression is induced by ROS, but does not appear to feed back to affect ROS generation. Activation of NADPH oxidase was assessed by evaluating the translocation from the cytosol to the membrane of its regulatory subunit, p47phox, in short term stimulation (Figure 4D). Ang II induced the increase of p47phox content in VSMC membrane fractions, indicating its recruitment to the plasma membrane.
Figure 3. Ang II increases Nox1 mRNA and protein expression.

Time-course (3 to 12 h) effect of 10−6 mol/L Ang II on Nox1 mRNA (A) and protein (B) expression in VSMCs. (C) Effect of Ang II (12 h) on Nox1 protein expression in the absence or presence of apocynin and valsartan. (D) Effect of Ang II (12 h) on Nox1 protein expression in VSMCs, non-transfected or transfected with Fat1 siRNA. Representative immunoblots: Nox1, Fat1, and β-actin. Results are mean ± SEM of 3 experiments. *P<0.05, vs vehicle.
Figure 4. Ang II-induced ROS generation in VSMCs is inhibited by AT1R antagonist, antioxidants, and Nox1 siRNA.

ROS generation was evaluated by lucigenin chemiluminescence and DHE fluorescence. VSMCs were stimulated with 1 μmol/L Ang II for short (A) and long (B) term in the absence or in the presence of apocynin (10 μmol/L). (C) VSMCs non-transfected and transfected and with Fat1 or Nox1 siRNA. (D) p47phox translocation was evaluated in VSMCs subjected to membrane and cytosol fractionation after stimulation with 10−6 mol/L Ang II. Membrane to cytosol ratio of p47phox protein content is expressed as % of vehicle. Top panels, representative images of DHE fluorescence. Representative immunoblot: p47phox. Results are mean ± SEM of 6 experiments. *P<0.05, vs vehicle.
Ang II-induced ROS-dependent Fat1 expression increase and activation is associated with ERK1/2 MAPK phosphorylation
ROS are likely to transduce rapid activation signals that lead to longer-term genomic events. Redox-sensitive signalling via the ERK1/2 MAPK pathway is associated with proliferative and growth-related mechanisms mediated by Ang II [18]. We assessed the potential involvement of ERK1/2 activity in Ang II-induced Fat1 expression increase and activation. As shown in the Figure 5, Ang II induced p44/p42 MAPK phosphorylation, an effect inhibited by valsartan and apocynin (Figures 5A, 5B. The ERK1/2 inhibitor, PD98059, prevented both Ang II-induced Fat1 expression increase (Figure 5C) and recruitment to the plasma membrane in VSMCs (Figure 5D).
Figure 5. Ang II-induced Fat1 expression/activation via ERK1/2 phosphorylation.

ERK1/2 MAPK phosphorylation was evaluated in VSMCs stimulated with 1 μmol/L Ang II for short (A) and long (B) term in the absence or in the presence of apocynin (10 μmol/L). The effects of Ang II on Fat1 expression (C) and translocation to the cell membrane (D) were evaluated in the absence or in the presence of PD98059 (1 μmol/L). Representative immunoblots: ERK1/2 MAPK, ERK1/2 MAPK [Thr202/Tyr204], Fat1, β-actin. Results are mean ± SEM of 6 experiments. *P<0.05, vs vehicle.
Ang II induces VSMCs migration via Fat1-dependent mechanism
To identify the functional significance of Fat1 in Ang II signaling, we evaluated this pathway in VSMC migration. As observed in the Figure 6, Ang II induced VSMC migration, which was inhibited by valsartan, apocynin, and PD98059. Ang II typically promotes VSMC hypertrophy, rather than hyperplasia, and the 12 h duration of this modified Boyden chamber assay is substantially less than VSMC doubling time, which limits any potential contribution of concurrent cell division. Of importance, Ang II-induced cell migration was not observed in VSMCs transfected with Nox1 or Fat1 siRNA. These data suggest that Ang II-mediated VSMC migration requires Fat1 expression and an intact AT1R/Nox1/Erk1/2 signaling axis.
Figure 6. Ang II induces VSMC migration through redox signaling and Fat1-dependent mechanisms.
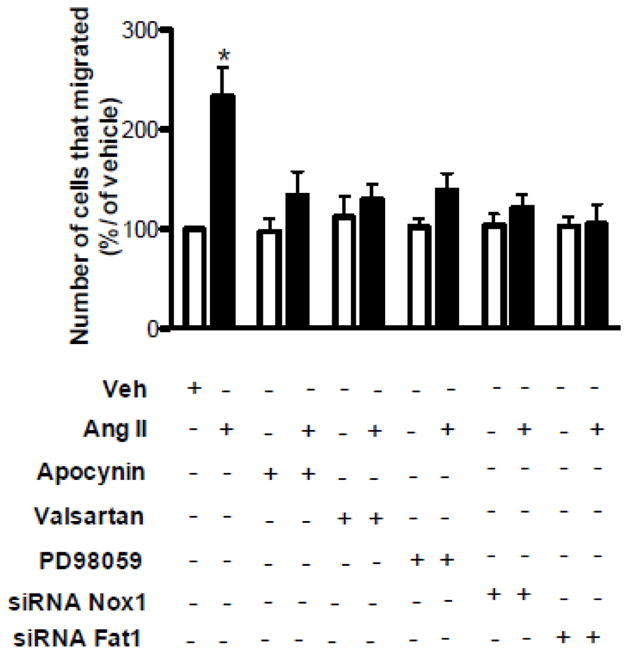
VSMC migration in response to Ang II stimulation (1 μmol/L, 12 h) was determined using a Boyden chamber assay. Ang II-induced VSMC migration was determined in the absence or in the presence of apocynin (10 μmol/L), valsartan (10 μmol/L), PD98059 (1 μmol/L), and in cells transfected with siRNA for Nox1 or Fat1. Results are mean ± SEM of 4 experiments. *P<0.05, vs vehicle.
Discussion
Major findings of the present study demonstrate that: 1) Ang II through AT1R-dependent redox mechanisms increases the expression and activity of Fat1, 2) Nox1 and Erk1/2 are upstream signaling components involved in Ang II-induced short and long term Fat1 regulation, and 3) Fat1 expression is necessary for Ang II-mediated VSMC migration. The cadherin superfamily of cell surface proteins includes classical, proto-, atypical, and other cadherin families that share characteristic extracellular Ca2+-binding domains known as cadherin repeats [19]. In epithelial cells, classical cadherins mediate cell-cell adhesion and regulate cell growth, migration, and polarity [20]. The function of cadherins in VSMC biology and vascular disease is still poorly understood. The levels of most cadherins expressed in VSMCs [21] decrease in response to growth factors or vascular injury [22]. In contrast, the atypical cadherin Fat1 is highly expressed during vascular remodeling and activated by growth factors associated with VSMC proliferation and migration [14]. These findings suggest a unique role for Fat1 among other cadherins in the vasculature.
The renin-angiotensin system (RAS) is a central component of the physiological and pathological responses in the cardiovascular system. This is supported by extensive experimental and clinical data demonstrating that inhibition of the RAS prevents development of atherosclerosis, hypertension, congestive heart failure, and coronary artery disease. Besides being a potent vasoactive effector peptide of the RAS, Ang II, via the AT1R, promotes proliferative and migratory events by inducing the expression of adhesion molecules, integrins, cytokines and growth and pro-fibrotic mediators. Our study demonstrated Fat1 activation as novel non-genomic signalling pathway for Ang II via AT1R in VSMCs. Of importance, Ang II also plays a versatile role in Fat1 regulation by modulating its expression at gene and protein levels and affecting its distribution within cells
ROS generation mediated by Ang II through AT1R activation and NADPH oxidase has been extensively explored [23–26]. In VSMCs, Ang II increases ROS production by enhancing Nox1 activity and expression [27]. The overall activity of Nox1 in ROS production in VSMCs is thought to depend on the assembly of a functional multimeric protein complex at the cell surface. When the cytosolic subunit p47phox is phosphorylated, it is recruited to the cell membrane, and together with NOXA1 and Rac1/2, forms a complex with Nox1 and p22phox. This event triggers oxygen reduction leading to superoxide anion formation [28,29]. ROS activates short term signaling regulatory events that in turn affect gene protein expression. Our data and those accumulating in the literature demonstrate that Ang II induces ROS generation in both short and long term stimulations of VSMCs. The translocation of p47phox from the cytosol to the cell membrane induced by Ang II supports the activation of NADPH oxidase. We employed two strategies to examine the role of Nox1 and ROS in Ang II-mediated Fat1 activity and expression regulation. Firstly we used siRNA to knockdown Nox1 in VSMCs, and secondly we studied the effect of antioxidants in the responses to Ang II. Our observations suggest that Nox1 and associated ROS generation are involved in Ang II-mediated Fat1 activation and increased expression. Interestingly, Fat1 knockdown did not affect Ang II-induced Nox1 expression or ROS generation. While Nox1 is an upstream regulator of Fat1, the latter does not appear to be involved in feedback regulation of this pathway.
ROS can activate many signalling pathways involved in inflammatory, proliferative, and migratory responses, which contribute to vascular remodelling [3]. The MAPKs, a family of serine/threonine-specific protein kinases that include p38, JNK, and ERK1/2, are sensitive to the cellular redox status [30–32]. ERK1/2 MAPK activity, which is increased upon Ang II stimulation of VSMCs, is strongly linked to proliferative and migratory responses. In addition, ERK1/2 expression is modulated by ROS-dependent mechanisms [33,34]. In the present study, we found that Ang II-induced ERK1/2 activation was dependent on ROS generation, since apocynin inhibited the MAPK phosphorylation. The importance of redox signalling in Ang II-induce Fat1 activation and expression increase is underscored by our finding that inhibition of p44/p42 MAPK phosphorylation prevented these effects.
The ability of VSMCs to migrate is critical during the remodelling process. We previously found that Fat1 promotes VSMCs migration [14,15]. In order to confirm the functional importance of Fat1 in Ang II-mediate remodelling responses, we evaluated VSMC migration and the contribution of signalling pathways upstream of Fat1 activation. We found that Ang II stimulated VSMC migration, and that this effect was inhibited by Fat 1 or Nox1 knockdown, or by antioxidant treatment. Our data with PD98059 demonstrate that p44/p42 MAPK phosphorylation is also involved in Ang II-induced migration event.
In summary, our findings demonstrate a new pathway implicating Nox1-derived ROS generation and ERK1/2 MAPK in Fat1 activation and VSMC migration in response to Ang II (Figure 7). Important clinical conditions that confer a heightened risk of cardiovascular disease, such as diabetes and hypertension, are associated with high circulating levels of Ang II [35–38]; at least part of this elevated risk may be a consequence of increased vascular remodelling. Ang II-activated signalling pathways are well recognized as potential targets for pharmacologic intervention to limit the incidence and progression of cardiovascular disease. The present studies extend this knowledge to include the effects of Ang II on the Fat1 cadherin, and suggest new strategies to interrupt Fat1 cadherin-mediated VSMC migration that may in turn limit vascular remodelling.
Figure 7. Signalling scheme linking Ang II and ROS generation to Fat1 and VSMC migration.
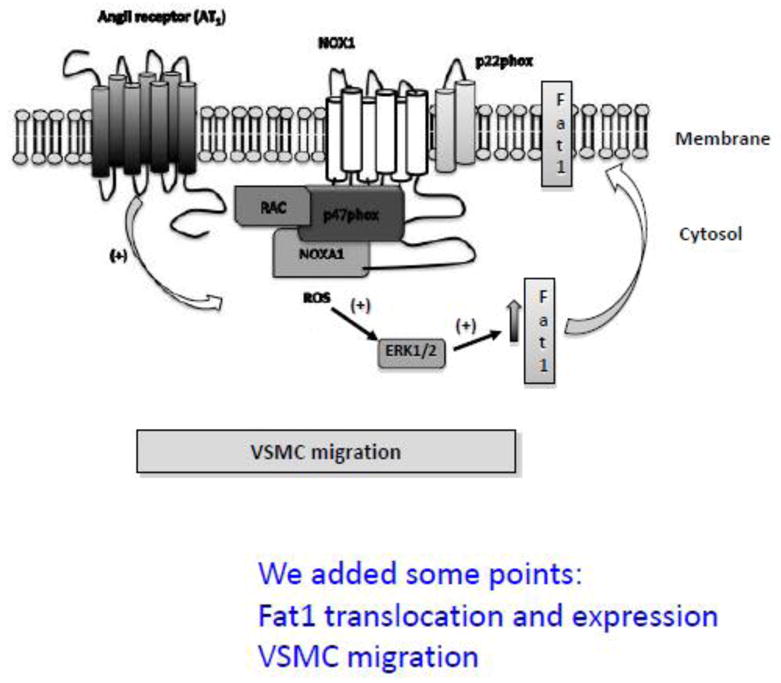
In VSMCs, Ang II induces activation of the AT1R, which triggers Nox1 activation and ROS generation; ROS activates ERK1/2 MAPK, leading to increased Fat1 cadherin activity and expression, which is required for Ang II-induced cellular migration.
Highlights.
Ang II regulates Fat1 through AT1R-dependent redox mechanisms
Nox1 and Erk1/2 are involved in short- and long-term regulation of Fat1 by Ang II
Fat1 expression is necessary for Ang II-mediated VSMC migration
Acknowledgments
Sources of funding
This study was funded by FAPESP (Foundation for Research Support of the State of Sao Paulo 2011/01785-6; 2011/22035-5) to RT and by National Institutes of Health Grants HL088104 and HL104518 to NS.
Abbreviations
- Ang II
Angiotensin II
- AT1R
Ang II type 1 receptor
- NADPH
Nicotinamide adenine dinucleotide phosphate
- Erk1/2
extracellular-signal-regulated kinases 1/2
- ROS
Reactive Oxygen Species
- VSMC
Vascular Smooth Muscle Cell
- MAPKs
Mitogen-activated protein kinases
- DUOX
Dual oxidase
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
Fetal bovine serum
- PBS
Phosphate-buffered saline
- HBSS
Hank’s balanced salt solution
- DHE
dihydroethidium
- HPRT
hypoxanthine hosphoribosyltransferase
- ANOVA
analysis of variance
Footnotes
Conflicts of interest
No potential conflicts of interest are reported.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yet SF, Layne MD, Liu X, Chen YH, Ith B, Sibinga NE, et al. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17(12):1759–61. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid Redox Signal 2012; Antioxid Redox Signal. 2013;1;19(10):1110–20. doi: 10.1089/ars.2012.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemarie CA, Schiffrin EL. The angiotensin II type 2 receptor in cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2010;11:19–31. doi: 10.1177/1470320309347785. [DOI] [PubMed] [Google Scholar]
- 5.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 6.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–72. [PubMed] [Google Scholar]
- 7.Touyz RM, El Mabrouk M, He G, Wu XH, Schiffrin EL. Mitogen-activated protein/extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ Res. 1999;19;84(5):505–15. doi: 10.1161/01.res.84.5.505. [DOI] [PubMed] [Google Scholar]
- 8.Knock GA, Ward JP. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal. 2011;15:1531–47. doi: 10.1089/ars.2010.3614. [DOI] [PubMed] [Google Scholar]
- 9.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;29;302(2):148–58. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky, et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;1:51;7, 1271–88. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, et al. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens. 2011;5(3):137–53. doi: 10.1016/j.jash.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997;13:119–46. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 13.Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. 2001;114:629–41. doi: 10.1242/jcs.114.4.629. [DOI] [PubMed] [Google Scholar]
- 14.Hou R, Liu L, Anees S, Hiroyasu S, Sibinga NE. The Fat1 cadherin integrates vascular smooth muscle cell growth and migration signals. J Cell Biol. 2006;8;173(3):417–29. doi: 10.1083/jcb.200508121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou R, Sibinga NE. Atrophin proteins interact with the Fat1 cadherin and regulate migration and orientation in vascular smooth muscle cells. J Biol Chem. 2009;13;284(11):6955–65. doi: 10.1074/jbc.M809333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sibinga NE, Foster LC, Hsieh CM, Perrella MA, Lee WS, Endege WO, et al. Collagen VIII is expressed by vascular smooth muscle cells in response to vascular injury. Circ Res. 1997;80(4):532–41. doi: 10.1161/01.res.80.4.532. [DOI] [PubMed] [Google Scholar]
- 17.Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension. 2005;45(4):773–9. doi: 10.1161/01.HYP.0000154365.30593.d3. [DOI] [PubMed] [Google Scholar]
- 18.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovascular Research. 2006;71:216–25. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiedon A, Tölle M, Bastine J, Schuchardt M, Huang T, Jankowski V, et al. Uridine adenosine tetraphosphate (Up4A) is a strong inductor of smooth muscle cell migration via activation of the P2Y2 receptor and cross-communication to the PDGF receptor. Biochem Biophys Res Commun. 2012;20;417(3):1035–40. doi: 10.1016/j.bbrc.2011.12.088. [DOI] [PubMed] [Google Scholar]
- 20.Shang Y, Sibinga NES. Fat and Dachsous cadherins in mammalian development and disease. In: Yoshida K, editor. Molecular and Functional Diversities of Cadherin and Protocadherin. Kerala, India: Research Signpost; 2010. pp. 253–74. [Google Scholar]
- 21.Wedlich D. The polarising role of cell adhesion molecules in early development. Curr Opin Cell Biol. 2002;14(5):563–8. doi: 10.1016/s0955-0674(02)00374-5. [DOI] [PubMed] [Google Scholar]
- 22.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52(3):372–86. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 23.Tanoue T, Takeichi M. Mammalian Fat1 cadherin regulates actin dynamics and cell-cell contact. J Cell Biol. 2004;165:517–28. doi: 10.1083/jcb.200403006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griendling KK, Ushio-Fukai M, Lassègue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29(1Pt2):366–73. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 25.Hitomi H, Fukui T, Moriwaki K, Matsubara K, Sun GP, Rahman M, et al. Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens. 2006;24(6):1089–95. doi: 10.1097/01.hjh.0000226199.51805.88. [DOI] [PubMed] [Google Scholar]
- 26.Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun. 2009;26;384(2):149–54. doi: 10.1016/j.bbrc.2009.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skultetyova D, Filipova S, Riecansky I, Skultety J. The role of angiotensin type 1 receptor in inflammation and endothelial dysfunction. Recent Pat Cardiovasc Drug Discov. 2007;2(1):23–7. doi: 10.2174/157489007779606130. [DOI] [PubMed] [Google Scholar]
- 28.Wingler K, Wünsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med. 2001;1;31(11):1456–64. doi: 10.1016/s0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
- 29.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–71. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montezano AC, Touyz RM. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol. 2012;110(1):87–94. doi: 10.1111/j.1742-7843.2011.00785.x. [DOI] [PubMed] [Google Scholar]
- 31.Hartney T, Birari R, Venkataraman S, Villegas L, Martinez M, Black SM, et al. Xanthine oxidase-derived ROS upregulate Egr-1 via ERK1/2 in PA smooth muscle cells; model to test impact of extracellular ROS in chronic hypoxia. PLoS One. 2011;6(11):e27531. doi: 10.1371/journal.pone.0027531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC, Miller FJ., Jr Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc Res. 2012;1;93(3):406–13. doi: 10.1093/cvr/cvr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yogi A, Callera GE, Mecawi AS, Batalhão ME, Carnio EC, Antunes-Rodrigues J, et al. Acute ethanol intake induces superoxide anion generation and mitogen-activated protein kinase phosphorylation in rat aorta: A role for angiotensin type 1 receptor. Toxicol Appl Pharmacol. 2012;1;264(3):470–8. doi: 10.1016/j.taap.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 34.Luo X, Xiao Y, Song F, Yang Y, Xia M, Ling W. Increased plasma S-adenosyl-homocysteine levels induce the proliferation and migration of VSMCs through an oxidative stress-ERK1/2 pathway in apoE(−/−) mice. Cardiovasc Res. 2012;15;95(2):241–50. doi: 10.1093/cvr/cvs130. [DOI] [PubMed] [Google Scholar]
- 35.Braun GS, Kretzler M, Heider T, Floege J, Holzman LB, Kriz W, et al. Differentially spliced isoforms of FAT1 are asymmetrically distributed within migrating cells. J Biol Chem. 2007;3;282(31):22823–33. doi: 10.1074/jbc.M701758200. [DOI] [PubMed] [Google Scholar]
- 36.Diet F, Pratt RE, Berry GJ, Momose N, Gibbons GH, Dzau VJ. Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation. 1996;94:2756–67. doi: 10.1161/01.cir.94.11.2756. [DOI] [PubMed] [Google Scholar]
- 37.Schieffer B, Schieffer E, Hilfiker-Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M, et al. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation. 2000;101:1372–8. doi: 10.1161/01.cir.101.12.1372. [DOI] [PubMed] [Google Scholar]
- 38.Nosadini R, Tonolo G. Cardiovascular and renal protection in type 2 diabetes mellitus: the role of calcium channel blockers. J Am Soc Nephrol. 2002;13(3):S216–23. doi: 10.1097/01.asn.0000034687.62568.9b. [DOI] [PubMed] [Google Scholar]