

Abstract
Corneocytes in mammalian stratum corneum are surrounded by a monolayer of covalently bound ω-OH-ceramides that form the corneocyte (-bound) lipid envelope (CLE). We review here the structure, composition, and possible functions of this structure, with insights provided by inherited and acquired disorders of lipid metabolism.
Keywords: acylceramides, corneocyte lipid envelope, cornified envelope, essential fatty acid deficiency, Gaucher disease, neutral lipid storage disease, Refsum disease
1. Structure and Composition of the CLE
Concurrent with epidermal terminal differentiation, the plasma membrane of granular (SG) cells disappears, paralleled by the formation of cornified envelope (CE) and its associated, external membrane monolayer, the corneocyte-bound lipid envelope (CLE) (Fig. 1). Both the origin and the function of this structure are still uncertain, but the recent identification of inherited and acquired disorders of lipid metabolism that impact this structure is providing new insights into the pathway leading to its formation, as well as to its putative functions.
Fig. 1.
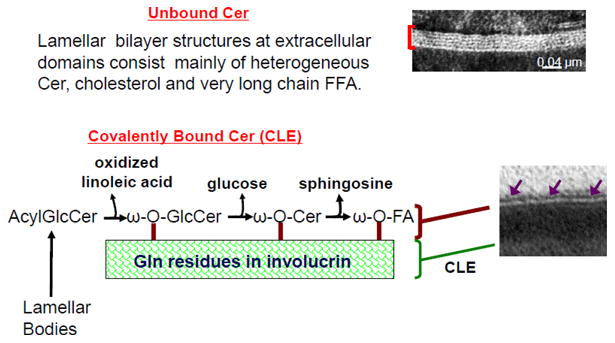
Model of Bound and Unbound Ceramides in Stratum Corneum Extracellular Domains.
The CLE is comprised of a monolayer of unusually long-chain, ω-acylated-hydroxy-ceramides (ω-OH-Cer; OS, or ω-OH-acyl-sphingosine), with lesser amounts of omega-hydroxy-fatty acids (ω-OH-FA), bound covalently primarily to glutamate residues in involucrin, located within the external portion of the cornified envelope (Fig. 1) [1-3]. Studies in Gaucher disease, type II, as well as in saposin-deficient transgenic mice show that the CLE initially is enriched in omega-hydroxy-glucosylceramides [ω-OH-(glucosyl)Cer], which subsequently are deglucosylated to ω-OH-Cer [4] (also see Sandhoff article in this issue) (Fig. 1). Of the five known isoforms of ceramidase, two (alkaline and acidic) are present in the stratum corneum (SC) [5-8]. While the alkaline form is present at low levels throughout the SC, the acidic isoform localizes in close proximity to the CLE [9]. Because of its localization, activity of the acidic isoform likely dominates the generation of ω-OH-FA. Yet, the low levels of free sphingoid bases in normal SC show that most of the covalently bound ω-OH-Cer remains intact even in the outer layers of normal SC (Table 1) (see also Loiseau, et al. paper, J Dermatol Sci In Press 2013).
Table 1.
Covalently Bound ω-OH Cer Content Declines in EFAD Mouse Epidermis
| CLE-ωOH Cer* (μg/mg dry epidermis) |
CLE Content** (% of normal mouse) |
|
|---|---|---|
| Normal | 1.88 + 0.12 | 100.0 + 13.0 |
| EFAD | 0.82 + 0.07*** | 49.7 + 10.5*** |
Legend:
Covalently bound lipids were isolated, and the resultant Cer fractions were quantitated by HPTLC-scanning densitometry, as described in [1].
Using randomly-obtained electron micrographs (n=10), CLE content was determined as the percent of the overall inter-desmosomal corneocyte-surface covered by CLE, as described by Behne, et al. [30].
P < 0.01 vs. normal control mice.
2. Formation of the CLE
It has been suggested that fusion of the limiting membrane of lamellar bodies generates the CLE, concurrent with exocytosis of these organelles at the apical plasma membrane of the outermost SG cells [10] (Fig. 1). By implication then, the limiting envelope of lamellar bodies would have to be enriched in ω-OH-(glucosyl)Cer, rather than the expected phospholipid-dominant composition of the limiting membrane of virtually all other cellular organelles. Support for this hypothesis comes from the recessively inherited ichthyosis, Harlequin ichthyosis, attributable to loss-of-function mutations in the transmembrane lipid transporter, ABCA12 [11, 12] (see article by Akiyama et al. in this issue). Although lack of this transporter results in a failure to deliver (glucosylCer) into nascent lamellar bodies [12], forme fruste organelles, with little or no internal lamellar contents, continue to be formed in large numbers, and presumably continues to be secreted in HI [10]. Despite a diminution of secreted lamellar membrane contents, a normal-appearing CLE appears external to the CE in this recessively inherited disorder (Fig. 2B vs. A), effectively ruling out lamellar body contents as the source of the CLE. But either fusion of these abortive lamellar bodies into the apical plasma membrane of the outermost cells of the SG, or insertion of other membrane fragments could account for the generation of the CLE [10]. Definitive proof of this mechanism will not be possible until biochemical analyses of both the bound lipids, as well as the composition of the limiting membrane of LBs are performed in HI.
Fig. 2.
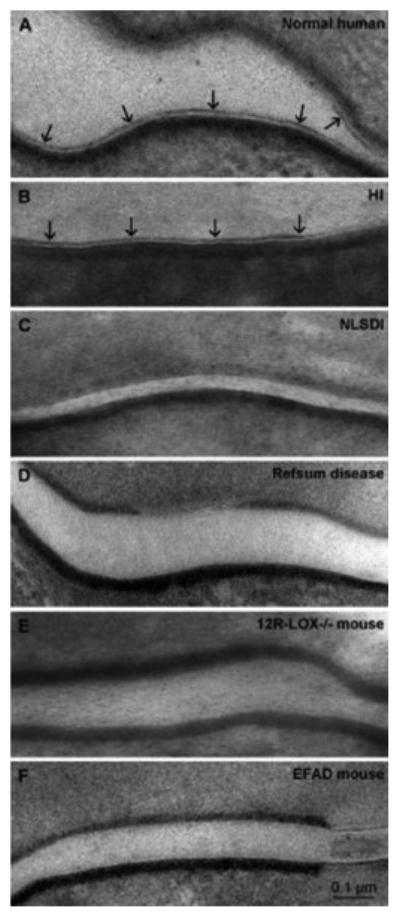
CLE in Normal and Diseased Skin (see text for abbreviations).
Nonetheless, it is still most widely accepted that the CLE is generated from a smaller pool of secreted extracellular acyl-(glucosyl)Cer. Hydrolysis of the omega-acyl linoleate moiety by an as-yet-uncharacterized, extracellular lipase would then generate a pool of ω-OH-(glucosyl)Cer that form the CLE (Fig. 1). With ultrastructural cytochemistry, lamellar bodies normally appear loaded with acidic lipase activity, which further appears to be secreted into the extracellular spaces of the SC (Fig. 3). This sequence parallels the secretion of other lamellar body-derived lipid and protein contents, including glucosylceramides. Since lamellar bodies contain little or no triglycerides [13], it has been unclear what the role of this abundant extracellular lipase activity could be. It now seems plausible that this secreted lipase activity within the SC extracellular domains could account for the hydrolysis of ω-esterified (oxidized) linoleic acid from acylCer, generating the ω-OH-Cer destined for covalent binding to the external face of the CE (Fig. 4).
Fig. 3.
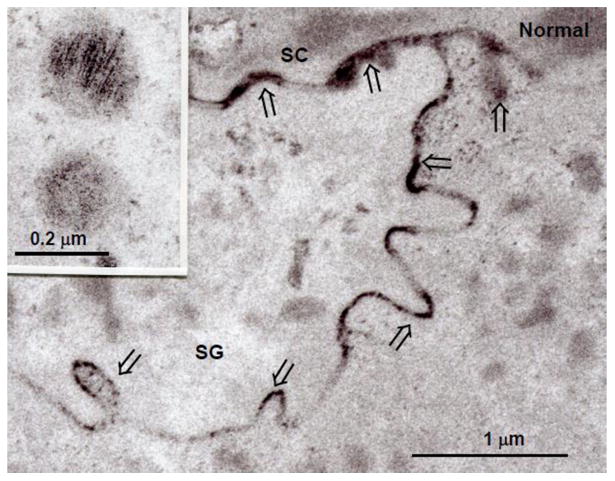
Normal Human Epidermis: Lipase activity is restricted to lamellar bodies (inset), and after secretion, within the extracellular spaces (open arrows). Methods for assessing upon localization on an ultrastructural level are cited in [24].
Fig. 4.
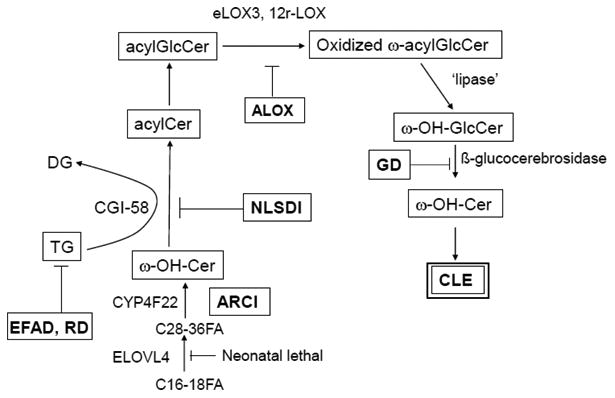
Pathways Leading To CLE Formation: Key insights from inherited and acquired lipid metabolic disorders are highlighted (see text for further details)
3. Pathways Leading To the Formation of the CLE: Insights from Inherited and Acquired Metabolic Disorders
While the pathway that leads to ceramide and acylCer formation is now well characterized (see articles by R. Sandhoff and K. Sandhoff in this issue), the biochemical sequence leading to acylCer and ω-OH-Cer formation remained uncertain until relatively recently. Issues such as: i) How are the unusually long-chained ω-acylated FA generated? ii) How does a subset of acylCer acquire a specific class of FFA; i.e., linoleic acid, for purposes of ω-esterification? iii) How does the putative lipase or transferase that generates ω-OH-Cer target the oxidized linoleate moiety in acylCer? Recent studies in a group of recessively inherited and acquired disorders are helping to provide answers questions such as these.
Two key steps are involved in ω-OH-Cer formation. An epidermis-unique isoform of the ELOV family, ELOVL4, generates the very-long chain N-acyl groups [14], while a cytochrome P450 isoform, CYP4F22, generates the ω-OH-Cer [15, 16]. While loss of ELOVL4 appears to be neonatal lethal, deficiency in CYP4F22 leads to a form of non-syndromic autosomal recessive ichthyosis [17] We have shown that the CLE is absent in the recessive disorder, neutral lipid storage disease with ichthyosis (NLSDI or Chanarin-Dorfman syndrome) [14] (Fig. 2C). The cause of NLSDI is loss of function mutations in CGI-58, a co-factor that is required for the activation of a subset of acidic lipases which recognize linoleic acid-enriched triglycerides (Fig. 4). CGI-58 is also a co-factor for ATGL in adipose tissue, but ATGL ko mice lack skin features, while loss of CGI-58 results in a syndromic form of ichthyosis [18, 19] (see article by R. Zechner, et al. in this issue). Together, these studies suggest that a pool of linoleate-enriched triglycerides transfers this EFA to previously synthesized ω-OH-(glucosyl)Cer, generating ω-acyl-(glucosyl) ceramides (Figs. 1 & 4).
Similarly, the CLE and bound ceramides are almost entirely absent in another recessively inherited subgroup of ichthyoses, attributable to loss-of-function mutations in either the 12R lipoxygenase (12R-LOX) or epidermal lipoxygenase 3 (eLOX3) (Fig 2E) [20, 21] (see also article by P. Krieg in this issue). While mice with a transgenic knock-out of 12R-LOX display a prominent barrier abnormality, whether a CLE is present or absent in these mice was not noted. Subsequently, Alan Brash's laboratory at Vanderbilt University showed that these two ALOX enzymes, which localize solely to keratinizing epithelia, sequentially oxidize the linoleate moiety of acylCer [22, 23] Oxidized linoleate then becomes the substrate for the putative extracellular lipase that generates ω-OH-(glucosyl)Cer that becomes covalently bound to the CE (Fig. 4) (further details of this biochemical sequence can be found in an article from the Brash group in this issue). Accordingly, the CLE and bound Cer are largely absent in 12R-Lox-/- transgenic mice (Fig. 2E) [22]. Together, these studies clearly demonstrate that oxidation of the omega-esterified linoleic acid moiety is a prerequisite for CLE formation. Finally, the release of ω-OH-Cer is paralleled by the liberation of linoleate products, such as hepoxilin and trioxilin that have putative pro-barrier signaling functions in the epidermis [24, 25] (see also A. Brash article in this issue).
Ultrastructural studies show that the CLE is either absent, partially formed, or loosely attached to the CE in the exceedingly rare, neurocutaneous disorder, Refsum disease (RD) [26] (Fig. 2D), which is due to loss of function mutations in phytanic acid oxidase, a peroxisomal enzyme that metabolizes unsaturated, branched-chain fatty acids normally found in plant-derived glycerolipids into species that can be more readily eliminated. In the absence of this oxidase, these abnormal free fatty acids likely replace some of the usual acylated fatty acids found in membrane lipids, producing often severe, multisystem abnormalities [27]. While biochemical data are still lacking, the ultrastructural abnormalities suggest that unmetabolized phytanic acid incorporates into the triglyceride pool, competing with the linoleic acid that normally incorporates into the triglyceride pool that contributes to CLE formation (Fig. 4). It seems reasonable to assume that these plant-derived fatty acids would not serve as an appropriate substrate for either the ALOXs or the putative lipase/transferase that generates ω-OH-(glucosyl)Cer, explaining the reduced CLE in this disorder.
Finally, in still unpublished work, we have shown that dietary essential fatty acid deficiency (EFAD) leads to reduced CLE formation (Fig. 2F), which is paralleled by a marked reduction in bound ω-OH-Cer (Table 1; see also [28]). In EFAD, omega esterified linoleate is replaced by oleic acid [29], which cannot serve as a substrate for the ALOXs. As in RD, it is doubtful that the oleate moiety in ω-OH-(glucosyl)Cer would be an appropriate substrate for the lipase/transferase catalytic step that leads to the formation of the CLE (Fig. 4). While it has been suggested that substitution of oleate for linoleate in acylCer comprises the molecular defect that accounts for the barrier abnormality in EFAD (Ibid.), it is possible that failure to generate a CLE instead provokes the barrier abnormality.
Together, these inherited disorders have provided important insights into the pathway that leads to formation of the CLE. Needless to say, each of these disorders is accompanied by profound functional abnormalities, but to what extent dysfunction can be attributed to loss of the CLE vs. parallel abnormalities in extracellular lamellar bilayers is uncertain, because structures other than the CLE are impacted in each of these disorders.
4. What Is the Function of the CLE?
While the functional implications of the above metabolic sequence is still emerging, it seems likely that ω-OH-(glucosyl)Cer resist ceramidases, allowing the persistence of this monolayer, unchanged at least during the initial stages of corneocyte maturation [4] (Fig. 1). This would allow the CLE to function as a scaffold for the organization of the extracellular lamellar bilayers, as needed for barrier function [28, 30].
Alternatively, it has been suggested that the CLE contributes to the cohesion of the SC [3]. The basis for this assertion lies in the observation that exhaustive lipid solvent extraction, while removing all of the lamellar bilayers, induces the collapse and firm attachment of corneocytes to one another, rather than separating them [1, 3]. Yet, whether the CLE serves this function under normal conditions, when the lamellar bilayers are present, remains uncertain.
Finally, the CLE could also function as a semi-permeable membrane that could allow the free transmembrane passage of water, while restricting the loss of larger hygroscopic molecules, such as filaggrin breakdown products, out of the corneocyte [9]. Then, after deglucosylation of ω-OH-(glucosyl)Cer, the CLE becomes vulnerable to attack by one or both of the two ceramidase isoforms, known to be present in SC [5, 8]. We demonstrated a large amount of acidic ceramidase activity in close proximity to the CLE, as well as the extracellular lamellar bilayers within the mid- to outer SC by in situ zymography [9]. The subsequent replacement of some ω-OH-(glucosyl)Cer by ω-OH-FFA would render the CLE less effective as a semi-permeable membrane, eventually allowing the parallel loss of water and the hygroscopic contents out of corneocytes in the SC. Should this feature become more prominent, it could have clinical consequences. Indeed, accelerated degradation of the CLE, indicated by a higher ratio of omega-hydroxy-fatty acids to omega-hydroxy-ceramides, is an overlooked, but potentially important feature of atopic dermatitis [31] (see related article by Elias et al, in this issue). Indeed, atopic dermatitis is often complicated by colonization with bacteria that secrete ceramidase [32], and/or activation of endogenous ceramidase activity [33]. Either mechanism could contribute to the xerosis that is such a prominent feature of atopic dermatitis. Specifically, the dry skin of aopics can be attributed not only to the reduced generation of hygroscopic breakdown products of filaggrin, but also to an accelerated leakage of these materials out of corneocytes.
5. Conclusion
While the function(s) of the CLE remain uncertain, its structure, composition, and pathways leading to its formation are in the process of being clarified through insights provided by both inherited and acquired disorders of lipid metabolism.
Acknowledgments
This work was supported by NIH grant AR019098, the Rene Touraine Foundation, and by the Medical Research Service, Department of Veterans Affairs. These contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAMS or NIH.
Abbreviations
- acylCer
acylceramides
- CE
cornified envelope
- CLE
corneocyte-bound lipid envelope
- EFAD
essential fatty acid deficiency
- (F)FFA
(free) fatty acids
- HI
harlequin ichthyosis
- NLSDI
neutral lipid storage disease with ichthyosis
- ω-OH-Cer
omega-hydroxy-ceramides
- ω-OH-FA
omega-hydroxy-fatty acids
- ω-OH-(glucosyl)Cer
omega-hydroxy-glucosylceramides
- RD
Refsum disease
- SC
stratum corneum
- SG
stratum granulosum
Footnotes
This article is part of a Special Issue entitled The Important Role of Lipids in the Epidermis and their Role in the Formation and Maintenance of the Cutaneous Barrier.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wertz PW, Downing DT. Covalently bound omega-hydroxyacylsphingosine in the stratum corneum. Biochim Biophys Acta. 1987;917:108–111. doi: 10.1016/0005-2760(87)90290-6. [DOI] [PubMed] [Google Scholar]
- 2.Marekov LN, Steinert PM. Ceramides are bound to structural proteins of the human foreskin epidermal cornified cell envelope. J Biol Chem. 1998;273:17763–17770. doi: 10.1074/jbc.273.28.17763. [DOI] [PubMed] [Google Scholar]
- 3.Wertz PW, Swartzendruber DC, Kitko DJ, Madison KC, Downing DT. The role of the corneocyte lipid envelope in cohesion of the stratum corneum. J Invest Dermatol. 1989;93:169–172. doi: 10.1111/1523-1747.ep12277394. [DOI] [PubMed] [Google Scholar]
- 4.Doering T, Proia RL, Sandhoff K. Accumulation of protein-bound epidermal glucosylceramides in beta-glucocerebrosidase deficient type 2 Gaucher mice. FEBS Lett. 1999;447:167–170. doi: 10.1016/s0014-5793(99)00274-4. [DOI] [PubMed] [Google Scholar]
- 5.Houben E, Holleran WM, Yaginuma T, Mao C, Obeid LM, Rogiers V, Takagi Y, Elias PM, Uchida Y. Differentiation-associated expression of ceramidase isoforms in cultured keratinocytes and epidermis. J Lipid Res. 2006;47:1063–1070. doi: 10.1194/jlr.M600001-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Downing DT. Lipid and protein structures in the permeability barrier of mammalian epidermis. J Lipid Res. 1992;33:301–313. [PubMed] [Google Scholar]
- 7.Wertz PW, Downing DT. Ceramidase activity in porcine epidermis. Febs Letters. 1990;268:110–112. doi: 10.1016/0014-5793(90)80985-r. [DOI] [PubMed] [Google Scholar]
- 8.Houben E, Hachem JP, De Paepe K, Rogiers V. Epidermal ceramidase activity regulates epidermal desquamation via stratum corneum acidification. Skin Pharmacol Physiol. 2008;21:111–118. doi: 10.1159/000114872. [DOI] [PubMed] [Google Scholar]
- 9.Lin TK, Crumrine D, Ackerman LD, Santiago JL, Roelandt T, Uchida Y, Hupe M, Fabrias G, Abad JL, Rice RH, Elias PM. Cellular changes that accompany shedding of human corneocytes. J Invest Dermatol. 2012;132:2430–2439. doi: 10.1038/jid.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elias PM, Fartasch M, Crumrine D, Behne M, Uchida Y, Holleran WM. Origin of the corneocyte lipid envelope (CLE): observations in harlequin ichthyosis and cultured human keratinocytes. J Invest Dermatol. 2000;115:765–769. doi: 10.1046/j.1523-1747.2000.00124-5.x. [DOI] [PubMed] [Google Scholar]
- 11.Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, Stephens KG, Sybert VP, Mallory SB, North BV, Witt DR, Sprecher E, Taylor AE, Ilchyshyn A, Kennedy CT, Goodyear H, Moss C, Paige D, Harper JI, Young BD, Leigh IM, Eady RA, O'Toole EA. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794–803. doi: 10.1086/429844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, Sawamura D, Shimizu H. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777–1784. doi: 10.1172/JCI24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grayson S, Johnson-Winegar AG, Wintroub BU, Isseroff RR, Epstein EH, Jr, Elias PM. Lamellar body-enriched fractions from neonatal mice: preparative techniques and partial characterization. J Invest Dermatol. 1985;85:289–294. doi: 10.1111/1523-1747.ep12276826. [DOI] [PubMed] [Google Scholar]
- 14.Uchida Y, Cho Y, Moradian S, Kim J, Nakajima K, Crumrine D, Park K, Ujihara M, Akiyama M, Shimizu H, Holleran WM, Sano S, Elias PM. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J Invest Dermatol. 2010;130:2497–2499. doi: 10.1038/jid.2010.145. [DOI] [PubMed] [Google Scholar]
- 15.Vasireddy V, Uchida Y, Salem N, Jr, Kim SY, Mandal MN, Reddy GB, Bodepudi R, Alderson NL, Brown JC, Hama H, Dlugosz A, Elias PM, Holleran WM, Ayyagari R. Loss of functional ELOVL4 depletes very long-chain fatty acids (> or =C28) and the unique omega-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida Y, Hama H, Alderson NL, Douangpanya S, Wang Y, Crumrine DA, Elias PM, Holleran WM. Fatty acid 2-hydroxylase, encoded by FA2H, accounts for differentiation-associated increase in 2-OH ceramides during keratinocyte differentiation. J Biol Chem. 2007;282:13211–13219. doi: 10.1074/jbc.M611562200. [DOI] [PubMed] [Google Scholar]
- 17.Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, Coudiere P, DiGiovanna JJ, Elias P, Fischer J, Fleckman P, Gina M, Harper J, Hashimoto T, Hausser I, Hennies HC, Hohl D, Hovnanian A, Ishida-Yamamoto A, Jacyk WK, Leachman S, Leigh I, Mazereeuw-Hautier J, Milstone L, Morice-Picard F, Paller AS, Richard G, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taieb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297:E289–296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem. 2004;279:30490–30497. doi: 10.1074/jbc.M403920200. [DOI] [PubMed] [Google Scholar]
- 20.Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177:173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krieg P, Rosenberger S, de Juanes S, Latzko S, Hou J, Dick A, Kloz U, van der Hoeven F, Hausser I, Esposito I, Rauh M, Schneider H. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol. 2013;133:172–180. doi: 10.1038/jid.2012.250. [DOI] [PubMed] [Google Scholar]
- 22.Zheng Y, Yin H, Boeglin WE, Elias PM, Crumrine D, Beier DR, Brash AR. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: a proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem. 2011;286:24046–24056. doi: 10.1074/jbc.M111.251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 24.Jobard F, Lefevre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Ozguc M, Lathrop M, Prud'homme JF, Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11:107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 25.Brash AR, Yu Z, Boeglin WE, Schneider C. The hepoxilin connection in the epidermis. Febs J. 2007;274:3494–3502. doi: 10.1111/j.1742-4658.2007.05909.x. [DOI] [PubMed] [Google Scholar]
- 26.Menon GKO, Schmitz EG, Crumrine D, Elias PM. Abnormalities in ultrastructure of epidermal lamellar bodies and corneocyte lipid envelope in Refsum disease. JID Annual Meeting; Phoenix, AZ; 2011. p. S52. [Google Scholar]
- 27.Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meguro S, Arai Y, Masukawa Y, Uie K, Tokimitsu I. Relationship between covalently bound ceramides and transepidermal water loss (TEWL) Arch Dermatol Res. 2000;292:463–468. doi: 10.1007/s004030000160. [DOI] [PubMed] [Google Scholar]
- 29.Melton JL, Wertz PW, Swartzendruber DC, Downing DT. Effects of essential fatty acid deficiency on epidermal O-acylsphingolipids and transepidermal water loss in young pigs. Biochim Biophys Acta. 1987;921:191–197. doi: 10.1016/0005-2760(87)90018-x. [DOI] [PubMed] [Google Scholar]
- 30.Behne M, Uchida Y, Seki T, de Montellano PO, Elias PM, Holleran WM. Omega-hydroxyceramides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function. J Invest Dermatol. 2000;114:185–192. doi: 10.1046/j.1523-1747.2000.00846.x. [DOI] [PubMed] [Google Scholar]
- 31.Macheleidt O, Kaiser HW, Sandhoff K. Deficiency of epidermal protein-bound omega-hydroxyceramides in atopic dermatitis. J Invest Dermatol. 2002;119:166–173. doi: 10.1046/j.1523-1747.2002.01833.x. [DOI] [PubMed] [Google Scholar]
- 32.Ohnishi Y, Okino N, Ito M, Imayama S. Ceramidase activity in bacterial skin flora as a possible cause of ceramide deficiency in atopic dermatitis. Clinical and diagnostic laboratory immunology. 1999;6:101–104. doi: 10.1128/cdli.6.1.101-104.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kita K, Sueyoshi N, Okino N, Inagaki M, Ishida H, Kiso M, Imayama S, Nakamura T, Ito M. Activation of bacterial ceramidase by anionic glycerophospholipids: possible involvement in ceramide hydrolysis on atopic skin by Pseudomonas ceramidase. Biochem J. 2002;362:619–626. doi: 10.1042/0264-6021:3620619. [DOI] [PMC free article] [PubMed] [Google Scholar]