

Abstract
An important medical problem with high mortality is shock, sepsis and multi-organ failure. They have currently no treatments other than alleviation of symptoms. Shock is accompanied by strong markers for inflammation and involves a cascade of events that leads to failure in organs even if they are not involved in the initial insult. Recent evidence indicates that pancreatic digestive enzymes carried in the small intestine after mixing with ingested food are a major cause for multi-organ failure. These concentrated and relatively non-specific enzymes are usually compartmentalized inside the intestinal lumen as requirement for normal digestion. But after breakdown of the mucosal barrier they leak into the wall of the intestine and start an autodigestion process that includes destruction of villi in the intestine. Digestive enzymes also generate cytotoxic mediators, which together are transported into the systemic circulation via the portal venous system, the intestinal lymphatics and via the peritoneum. They cause various degrees of cell and organ dysfunction that can reach the point of complete organ failure. Blockade of digestive enzymes in the lumen of the intestine in experimental forms of shock serves to reduce breakdown of the mucosal barrier and autodigestion of the intestine, organ dysfunctions and mortality.
Keywords: Pancreatic digestive enzymes, trypsin, mucosal barrier, mucin, epithelium, shock, sepsis, hemorrhage, mortality, inflammation, unbound free fatty acids
Introduction
Following almost two centuries of experimental and preclinical studies, the past two decades have witnessed a robust body of evidence to indicate that markers of inflammation accompany virtually all diseases. A rich assortment of hallmarks for inflammation has been documented, including leukocyte or platelet activation and adhesion to the endothelium, thrombosis, inflammatory gene expression. inflammatory molecules like reactive oxygen species, cytokines, lymphokines, complements or acute phase proteins (e.g. C-reactive proteins, fibrinogen) to name just a few (29, 45, 50). An increasing number of clinical trials confirm that markers for inflammation are detected in plasma of different patient groups irrespective of the particular organs exhibiting clinical symptoms in these groups (28, 34, 51). This is a remarkable body of evidence establishing an association between inflammatory markers and disease.
The inflammatory cascade fundamentally serves over a lifetime as a tissue repair mechanism after an injury to stop bleeding, prevent infections, remove injured tissue and replace it with new tissue and blood vessels (22, 57). It appears to be the only repair mechanism in adult tissues; embryonic tissues have alternative repair mechanisms (46). Key steps in tissue repair require the blood-clotting cascade, cell adhesion and cell-signaling molecules (e.g. chemokines, cytokines, acute phase proteins), degrading enzymes, growth factors, cell migration and phagocytosis, mitosis and stem cell differentiation. These same steps are observed in the inflammatory cascade. Thus biochemical markers in plasma, such as C-reactive protein levels, are a signature for a repair process in progress towards an eventual resolution of the injury.
If the inflammatory cascade serves as a repair mechanism, then what causes tissue injury in the first place? Identification of the primary event that leads to tissue injury is the “Holy Grail” for development of strategies to prevent disease and a requirement for optimization of treatment. While many injury mechanisms are recognized today (e.g. trauma, burns, infections, tumor growth, genetic mutations), we will highlight here a fundamental mechanism that has received little attention in the past, a process that is closely linked to the digestive system and which we designate as autodigestion (Fig. 1). It is evident for example in the case of Shock and Multi-organ Failure.
Figure 1. Food Digestion versus Autodigestion.
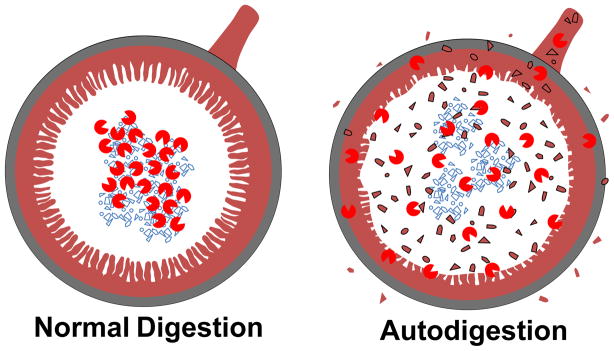
Small intestine cross section during normal digestion in which digestive enzymes (proteases, lipases, amylases, nucleases,
 ) are compartmentalized in the lumen to digest food particles and after escape of digestive enzymes into the wall of the intestine and autodigestion (e.g.
) are compartmentalized in the lumen to digest food particles and after escape of digestive enzymes into the wall of the intestine and autodigestion (e.g.
 ). Escape of digestive enzymes and inflammatory fragments they generate escape into the venular circulation and portal veins, into intestinal lymphatics and into the peritoneal space.
). Escape of digestive enzymes and inflammatory fragments they generate escape into the venular circulation and portal veins, into intestinal lymphatics and into the peritoneal space.
Shock and Multi-organ Failure
Physiological shock followed by multi-organ failure is the primary cause of death in younger people and patients in intensive care units. The number of deaths is of the order of hundreds of thousands each year in the US alone, and there is currently no treatment other than alleviation of symptoms (“early goal-directed therapy”) (48). Many clinical trials have been carried out to try to improve survival rates. The most recent treatment designed to interfere with coagulation and neutrophil rolling on the endothelium in shock was withdrawn from the US market in 2011 (36). The number of research reports listed in the US National Library of Medicine addressing “shock” and “sepsis” in early 2013 exceeds 290,000 and is especially rich in the trauma and critical care literature.
Strong plasma markers for inflammation accompany physiological shock. The nature and level of such biochemical markers depends, however, on the specific insult that causes the shock. For example, experimental endotoxic shock (by infusion of bacterial endotoxins into the circulation) is associated with significant systemic leukocyte activation as well as cytokine release (e.g. TNFα), while in contrast experimental hemorrhagic shock (by temporary reduction of the blood volume and central arterial blood pressure) with similar mortality rate may show leukocyte activation while at the same time undetectable levels of TNFα (6). Septic shock patients have extensive cytokine release (16). These cytokines have been regarded in the past as possible targets for intervention, a hypothesis that was not confirmed in clinical trials (e.g. in the case of IL-1, TNFα). The evidence is consistent with the notion that cytokines are signaling molecules synthesized as part of the repair side of inflammation. Their inhibition in shock may be a questionable strategy. To develop a new therapeutic approach we need to bring to light the initial injury mechanisms in shock.
Trigger Mechanisms for Multi-organ Failure
Trauma, blood loss, infections, allergic reactions, or exposure to non-physiological temperatures (e.g. burns) or to cytotoxic chemicals are primary insults that can lead to physiological shock. If sufficiently severe, each of these insults causes loss of cell functions and multi-organ failure. Even though the trigger mechanisms are different for each case, there is an underlying mechanism that leads to cell dysfunctions with eventual multi-organ failure even in organs that are not victims of the initial insult.
We have obtained the first evidence for a tissue injury mechanism that is common to multiple shock forms. The mechanism was brought to light in the specific model of experimental hemorrhagic shock. Temporary reduction of the blood volume (e.g. to a mean arterial blood pressure of 35 mmHg for 1–2 hours) followed by return of blood volume leads to an initial blood pressure restoration but is followed within hours by a progressive reduction of blood pressure. The reduction of the blood pressure is accompanied by development of cell dysfunctions, loss of metabolic and immunological functions, abnormal enhancement of cell activation (e.g. leukocytes, endothelium), and development of microvascular and organ failure. Multi-organ failure after hemorrhagic shock can occur in healthy individuals at any age. Furthermore, the lethal outcome of hemorrhagic shock is also independent of bacteria as suggested by even early experiments with aseptic animals (59) as well as by the inability of even aggressive antibiotic treatment to prevent organ failure in many patients.
Inflammatory Mediators in Plasma during Shock
A remarkable feature is that during hemorrhagic shock inflammatory markers in plasma can be detected already within an hour after blood pressure reduction, and at this early stage is able to depress organ function or to provide strong activation, e.g. of naive donor leukocytes (5). One hour is too short for significant de-novo biosynthesis of mediators, like cytokines. The mediators in blood have the ability to suppress many cell function (e.g. reduction of cardiac contraction (31)), activate circulating leukocytes (27, 56) – in fact they affect most physiological functions.
Decades of analysis into the biochemistry of molecules that may cause pathophysiological activity in shock plasma has not led to a conclusive picture. Candidate mediators have been proposed (complement, endotoxins, lipid mediators, fibronectin fragments, and others), but it has not been possible to demonstrate that removal of these molecules reduces the activity of shock plasma, for example the ability to activate leukocytes. Thus in shock plasma an activity is present whose identification may require a different approach.
Pancreatic Digestive Enzymes
Analysis of a range of organs shows that during an ischemic state many of them have the ability to cause cell activation. But the pancreas stands out as an organ, which not only has the ability to generate pro-inflammatory but also cytotoxic mediators (26, 31, 56) (Fig. 2). Another organ that has the ability to produce cytotoxic mediators is the small intestine, but only in the presence of its luminal contents (43, 56). If the lumen is rinsed, the ability of the intestinal wall to generate inflammatory mediators is reduced, but it returns when digestive proteases are restored (56). The inflammatory mediators are independent of endotoxins. In light of the fact that the pancreas serves to synthesize and discharge digestive enzymes into the intestine as part of normal digestion, all the evidence points towards pancreatic digestive enzymes as a major source for cell dysfunction and cytotoxicity in shock.
Figure 2. Pancreatic Proteases as a Source for Inflammatory Mediators.
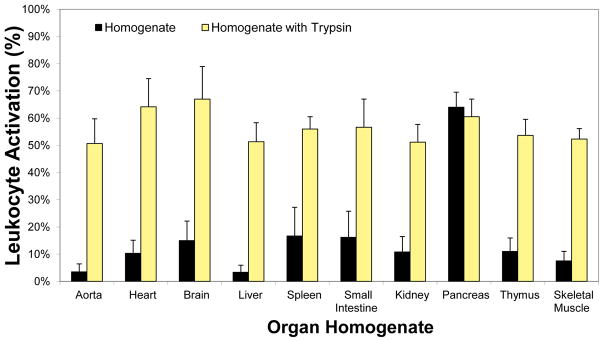
Activation of naive donor leukocytes (in form of pseudopod formation by freely suspended cells, (55)) generated by organ homogenates before (black bars) and after incubation (150 min) in pancreatic trypsin (yellow bars). The lumen of the intestine was rinsed before homogenization. The tissue homogenates contain insignificant endotoxin levels to stimulate significant leukocyte activation. All activation values with trypsin are significantly higher (p<0.05) except for the case of the pancreas.
The Mucosal Barrier: Containment of Pancreatic Digestive Enzymes
Pancreatic digestive enzymes are the basic requirement for degradation of food in the small intestine. After synthesis and discharge from the pancreas into the duodenum, they are fully activated in the lumen of the intestine. They are relatively concentrated and non-specific in their ability to cleave biopolymers (e.g. proteins and peptides, polysaccharides, triglycerides, nucleic acid chains) to optimize digestion. It is possible to digest an intestine as part of a food source without digesting one’s own intestine. Protection against autodigestion is possible by compartmentalization of the digestive enzymes inside the lumen of the intestine, a mechanism facilitated by the mucosal barrier in the intestine (9). This barrier is made up of the mucosal epithelium and a mucin layer in addition to goblet cells, precursor stem cells and lymphocyte populations. The thickness of the mucin layer is up to several hundred micrometer. It consists of several isoforms, which in part are attached to the epithelium (e.g. mucin 13) and in part are secreted (e.g. mucin 2) from goblet cells into the lumen of the intestine and carried along with food by peristaltic transport. Powerful digestive enzymes, e.g. trypsin, need to be prevented from approaching the epithelial membrane in order to avoid cleavage of epithelial membrane receptors and interference with the normal transmembrane transport that is part of epithelial absorption during digestion.
Should the mucosal barrier be breached in the presence of digestive enzymes in the lumen of the intestine, these digestive enzymes escape into the intestinal wall and into the systemic circulation, leading to organ failure, blood pressure reduction and mortality within hours even in an otherwise normal circulation (25). Thus, the mucosal barrier in the intestine is a key requirement to protect against autodigestion, and it serves to minimize escape of other components from the lumen into the intestinal wall as well, such as bacteria and their products, partially digested food, peptide fragments and cytotoxic unbound free fatty acids.
Failure of the Mucosal Barrier in Shock
Pancreatic digestive enzymes are in direct contact with the mucosal barrier and the mucin layer. In spite of this exposure, the digestive enzymes in a normal intestine only minimally degrade the mucin layer. The glycosylated and hydrated superstructure of mucin serves to minimize proteolysis of mucin’s core protein structure (35) and passage of digestive enzymes across the mucin barrier is minimal during normal digestion (9, 25).
This evidence suggests that after a primary insult in shock, a set of mechanisms trigger elevation of the mucosal permeability to the digestive enzymes. These mechanisms likely depend on the specific primary events precipitating the shock. For example, ischemic conditions can lead to oxygen and ATP depletion in the mucosal epithelium and this depletion interferes with membrane transport, intercellular adhesion of the epithelium and paracellular permeability (37). Thus in hemorrhagic shock the ATP depletion during ischemia (and not the pancreatic digestive enzymes) causes the mucosal layer breakdown (8), so that supply of oxygen via oxygen carriers inside the lumen of the intestine serves to prevent entry of digestive enzymes into the wall of the intestine (7). The ischemia in hemorrhagic shock also causes activation of matrix metalloproteinases, which in turn have the ability to cleave the ectodomain of membrane receptors and thereby undermine epithelial barrier function (2). Alternatively, in the presence of inflammatory mediators stimulation of specific receptors may open the mucosal barrier (e.g. TLR4 in the presence of endotoxin) (21). Evidence for such a pathway comes from endotoxic and peritonitis shock models in which a similar entry of digestive enzymes into the wall of the intestine occurs (14, 19). The presence of endotoxin stimulates an enhancement of the mucosal permeability, facilitating leakage of the digestive enzymes out of the lumen of the small intestine. There may be other mechanisms by which the mucosal barrier becomes permeable to digestive enzymes, including trigger molecules derived from food components (42, 58).
Escape of Digestive Enzymes from the Lumen of the Intestine - Autodigestion
Zymographic tracing indicates that pancreatic digestive proteases enter the wall of the intestine in shock within minutes (Fig. 3). Their transport distances from the lumen into the wall of the intestine are short. In the rat digestive protease activity can be detected over the full thickness of the intestinal wall within a period of less than an hour (9).
Figure 3. Entry of Digestive Enzymes into the Wall of the Intestine.

Micrographs of pancreatic chymotrypsin labeled by immunohistochemistry on frozen sections of rat small intestine before (left panel, control), after 30 min of ischemia without (middle panel) and with a protease inhibitor (tranexamic acid, see (9))(right panel) in the lumen of the intestine. Food items in the lumen (L) and the spaces between villi (arrow) exhibit chymotrypsin labeling in controls with minimal labeling within the villi (V) and notable labeling in the muscularis (M). Chymotrypsin label is enhanced during ischemia and attenuated in the presence of enteral protease inhibition. Length of cross bar is 50 μm.
Entry of digestive enzymes into the intercellular spaces between epithelial cells leads to cleavage of the ectodomain of membrane receptors, including the inter-epithelial adhesion receptors (e.g. E-cadherin) (9). Further transport of digestive enzymes into the villi causes a dramatic destruction of their structures irrespective of the choice of the shock model (14, 38) (Fig. 4). The villi are degraded starting at their tips and over time reduce in length. Depending on the severity of shock, the villi and mucosal barrier may disappear altogether, fully exposing the underlying sub-mucosa and muscularis to the digestive enzymes. The tissue destruction includes cleavage of membrane signaling receptor (e.g. TLR4) undermining the ability of cells in the intestinal wall to signal physiological and immunological functions (8).
Figure 4. Morphological Destruction of the Intestinal Mucosal in Shock.

Micrograph of small intestine cross section (stained with toluidine blue) with villi in control rat (left panel), after hemorrhagic shock without (middle) and with enteral digestive protease inhibition (tranexamic acid, (14)) (right). The characteristic destruction of the villi in hemorrhagic shock (arrows) is attenuated after blockade of the pancreatic digestive proteases. Length of the crossbar is 50 μm.
Once digestive enzymes enter into the wall of the intestine, they also generate tissue degradation products among which unbound free fatty acids are the most cytotoxic ones. Unbound free fatty acids directly dissolve in membrane bi-lipid layers, cause bleb formation, membrane rupture and necrosis (44). While proteases may cleave membrane receptor, and thereby undermine cell function and cause cell detachment (1), they may not cause actual cell death. Cleaved membrane receptors may be absorbed into autophagosomes and are reconstituted into the membrane from intracellular pools, as is the case during cell culturing (e.g. by use of trypsin during cell passage from dish to dish). In contrast, the presence of even low concentrations of unbound free fatty acids is cytotoxic to the degree that the cells of the villi undergo apoptosis and necrotic destruction (44).
The entry of digestive enzymes and their degradation products into the wall of the intestine during shock is followed by further transport into the systemic circulation via the portal venous system, the intestinal lymphatics (47), and also via passage through the peritoneal cavity (23). Protease activity is detected in the plasma (2, 14), and consequently peripheral organs are exposed to the pathogenic combination of digestive enzymes and cytotoxic mediators. This mixture may mediate not only systemic cell death but also cleavage of membrane receptors and a compromise of their cell functions. For example, exposure of endothelial cells to digestive serine protease reduces the fluid shear stress response, and is accompanied by cleavage of the glycocalyx, the vascular endothelial growth factor receptor 2 as well as the insulin receptor (1). Unbound free fatty acids are cytotoxic to endothelial cells by the same mechanism as they are to epithelial cells, i.e. by bilipid membrane destruction.
Peripheral Organ Failure
Progressive loss of organ functions is a central problem in shock. The loss of function may occur not only in organs that are directly affected by the initial trauma, but over time includes innocent “bystander” organs. For example, local ischemia in the intestine by temporary interruption of its blood flow (without ischemia in other organs) leads to inflammation in the microcirculation of peripheral skeletal muscle, leukocyte activation and interaction with the endothelium and parenchymal cell death (19). The lung capillaries become permeable and fluid accumulates in the alveolar space while circulating neutrophils become entrapped in the pulmonary capillary sheet (38), early complications that lead to acute respiratory failure (13). Specific cell functions, like glucose transport, may be compromised (33). Cell and organ failure go hand in hand. The more organs fail the lower the probability for survival; when two or three organs have failed the probability of survival is reduced to less than 50%.
The question arises, is release of digestive enzymes and cytotoxic mediators from the intestine a central part of multi-organ failure? The evidence we present above supports this hypothesis. But to investigate further we need an approach to block the pancreatic digestive enzymes and determine whether it reduces signatures for auto-digestion. Thus, we are not only testing a potentially important hypothesis, but we may at the same time develop a potential intervention.
A New Opportunity: Inhibition of Pancreatic Enzymes in the Lumen of the Intestine
The “first line of defense” against autodigestion is preservation of the mucosal barrier. But the clinical reality in many shock situations with injured and permeable intestines at the time of arrival in a hospital requires the development of a “second line of defense”. The therapeutic strategy in this case is to minimize autodigestion of the intestinal wall and maximize preservation of the mucosal barrier, even though digestive enzymes may already have escaped from the lumen of the intestine. We have developed an approach that is designed to block digestive enzymes directly inside the lumen of the intestine (“enteral blockade”). Enteral treatment allows application of digestive enzyme inhibitors at concentrations that match the high concentrations of digestive enzymes inside the lumen of the intestine. The enteral administration of an enzyme inhibitor can be achieved by injection into an exposed intestine after an abdominal incision (14), or via infusion by a catheter (with an inflatable balloon) placed at the entry of the small intestine (24), or slow infusion into the intestine via a nasogastric tube in the lower part of the stomach (30).
These experiments show that pharmacological blockade of pancreatic digestive enzymes in the lumen of the intestine reduces morphological damage to the intestine (Fig. 2, 3) and attenuates inflammation in multiple shock models (including splanchnic artery occlusion, hemorrhagic and endotoxic shock) (2, 12, 18–20, 38–40) and can be demonstrated in multiple species (17, 24). Enteral blockade of digestive enzymes also significantly reduces the level of inflammatory markers in the intestine, liver, heart, lung and in the systemic circulation (14, 38) and attenuates plasma cytokine levels (19, 38). The characteristic lesion formation at the level of the microcirculation in form of acute red blood cell leakage from microvessels into the tissue, e.g. in the lung and heart muscle, is reduced after blockade of digestive enzymes in the intestine. Enteral blockade of digestive enzymes also reduces cytotoxicity in the microcirculation of peripheral organs, indicating that the ischemic intestine serves as a source of inflammatory mediators and contributor to peripheral organ failure, i.e. a source for the systemic inflammatory response syndrome (SIRS) and mechanism for multi-organ failure (19).
The enteral protease blockade is also associated with significantly improved post-shock animal activities and blood pressure, reduced peripheral organ damage, as well as relatively rapid return to normal food consumption, weight gain and intestinal motility (14, 38, 39). Recovery of the intestine is a sine-qua-non for survival. Without exception non-survivors after shock have lesions in the intestine.
Enteral blockade of the digestive enzymes is accompanied by reduction of long-term mortality (≥3 months) (14). We demonstrated this protection in hemorrhagic, peritonitis and endotoxic shock models and with three different digestive protease inhibitors administered one hour after induction of shock. These results indicate that autodigestion by the powerful digestive enzymes - in both ischemic and septic shock models - is a common denominator for multi-organ failure and mortality. Furthermore, we obtained the first human evidence in a septic shock patient who was treated upon consent by nasogastric tube administration of pancreatic digestive protease inhibitor that enteral blockade of digestive enzymes reduced shock indicators and allowed full recovery and hospital discharge (30).
Enteral versus Intravenous Blockade of Digestive Enzymes
In the experiments described above, the digestive enzyme inhibitors were administered directly into the lumen of the intestine as the most direct route to reach the digestive enzymes in the intestine, even if they may already have leaked into the wall of the intestine. In contrast to this enteral treatment, intravenous administration of digestive enzyme inhibitors is less effective to block digestive enzymes in the intestine (12, 38). There are multiple reasons. Transport into the lumen of the intestine via the intestinal circulation is ineffective if intestinal microvessels are underperfused during autodigestion. High concentrations of inhibitors are required to block the concentrated digestive enzymes, which if directly administered into the circulation may compromise thrombosis and other vascular functions. Preliminary evidence in septic patients suggests that intravenous administration of digestive enzymes inhibitors may be ineffective (30).
Protease Activity and Receptor Cleavage in Chronic Inflammation
Many diseases are accompanied by a gastrointestinal co-morbidity. It remains to be determined whether the intestine with its unique pool of powerful digestive enzymes is a victim and secondary participant in the inflammation that accompanies such diseases, or whether the intestine is in fact a driving mechanism for disease. This requires a systems analysis in which the intestine needs to be looked at in the context of its digestive activity and the choice of macronutrients that are selected as food source. There is evidence to suggest that unchecked proteolytic activity may not only occur in extreme situations, like shock, but also in chronic diseases, e.g. in hypertension and diabetes, but to a milder degree and below the level of the tissue destruction seen in shock.
In the spontaneously hypertensive rat (a genetic model of hypertension) matrix metalloproteinase activity can be detected in the plasma and in endothelial cells (53, 54). The activity is lower than in shock, but sufficient to produce cell dysfunctions, many of which are symptomatic for patients with hypertension and diabetes. Ectodomain proteolytic cleavage of the insulin receptor in the spontaneously hypertensive rat leads to hyperglycemia and a loss of the ability to transport glucose into cells, i.e. insulin resistance (15). Proteolytic destruction of the beta-2 adrenergic receptor causes constriction of arterioles and consequently elevation of central blood pressure, i.e. hypertension (49), and cleavage of the ectodomain of the vascular endothelial growth factor 2 causes apoptosis of endothelial cells, which at the level of capillaries leads to actual loss of these vessels, i.e. capillary rarefaction (53), a common vascular complication in hypertension. Furthermore, proteolytic cleavage of adhesion receptors leads to deficient rolling and adhesion of leukocytes to the endothelium in the spontaneously hypertensive rat (3, 11, 52), and ectodomain cleavage of chemotactic receptor (formal peptide receptor) leads to deficient pseudopod projection and mechanotransduction response (10). The characteristically low sleep quality of the spontaneously hypertensive rat is associated with ectodomain cleavage of the serotonin receptor in the brain (55). Each of these diverse cell dysfunctions can be restored by chronic blockade of protease inhibitors, suggesting that the underlying mechanism for this chronic disease may involve unchecked activity of multiple families of degrading proteases in the systemic circulation. There is a need for further exploration of ectodomain destruction of membrane receptors by extracellular proteases in chronic human hypertension and diabetes. The role of digestive enzymes in this process needs identification, be it by direct involvement or by conversion of pro- into active degrading enzymes, like the matrix metalloproteinases,
Summary
The term ”autodigestion” has been coined in the past for the case of pancreatitis in which the pancreas is digested by its own enzymes (4, 41). In the context of physiological shock and sepsis, “autodigestion” refers to a broader phenomenon that includes, besides the pancreas, the small intestine with its active digestive enzymes (Fig. 4). Containment of the digestive process in the lumen of the small intestine requires an intact mucosal barrier. Compromise of this barrier after ischemia or other injuries to the intestine allows leakage of digestive enzymes into the lamina propria of the villi, the mucosa and muscularis of the intestinal wall. Exposure of the wall of the intestine to its own digestive enzymes leads to autodigestion with catastrophic consequences since the intestinal tissue and the circulation have inadequate concentrations of protease inhibitors to block the concentrated digestive enzymes. The digestive enzymes generate cytotoxic fragments, e.g. unbound free fatty acids, and the mixture of digestive enzymes and cytotoxic fragments is transported into the systemic circulation where they reach other organs and generate multiple cell dysfunctions and eventual organ failure. The price to pay for a lifelong digestive process may be autodigestion. At the end of life the digestive system appears to lose its ability to digest food only and instead it also digests autologous tissue.
Outlook
Early blockade of digestive enzymes in the lumen of the intestine may serve as an intervention to stop the destruction of cells and tissue structures in the intestine and in the systemic circulation. It may be applied to patients at risk for (e.g. in cases of elective surgery) or already in shock after an insult (14). To optimize such an approach, there is a need for detailed analysis of the transport and activity of pancreatic digestive enzymes in-vivo during normal digestion and analysis of the mucosal barrier properties in the digestive track. This information will serve as the basis for understanding the mechanisms that lead to the escape of digestive enzymes from the lumen of the intestine in pathophysiological conditions (9). More sensitive and specific diagnostic tools to detect degrading enzyme activity in patients are required (32). To minimize autodigestion, more specific digestive enzyme inhibitors and optimal delivery methods need to be developed while at the same time the nutritional needs of the patients may have to be met by alternative pathways, for example by intravenous delivery.
Acknowledgments
We thank Erik Kistler, Alex Penn, Angelina Altshuler, Frank A. DeLano for many discussions regarding the autodigestion hypothesis. Supported by NIH Grants HL 67825 and GM 85072 and by an unrestricted gift from Leading Biosciences Inc.. The authors thank Mr. Frank A. DeLano for preparation of Figure 4.
The studies described in this review were funded in part by an unrestricted gift from Leading Biosciences Inc..
Footnotes
Conflict of Interest: G.W.S.-S. is scientific advisor to Leading Biosciences Inc. and owns equity in InflammaGen, a company by Leading Bioscience Inc., which develops therapy for shock patients.
References
- 1.Altshuler AE, Morgan MJ, Chien S, Schmid-Schönbein GW. Proteolytic activity attenuates the response of endothelial cells to fluid shear stress. Cell Mol Bioeng. 2012;5:82–91. doi: 10.1007/s12195-011-0207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshuler AE, Penn AH, Yang JA, Kim GR, Schmid-Schönbein GW. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS One. 2012;7:e32672. doi: 10.1371/journal.pone.0032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arndt H, Smith CW, Granger DN. Leukocyte-endothelial cell adhesion in spontaneously hypertensive and normotensive rats. Hypertension. 1993;21:667–73. doi: 10.1161/01.hyp.21.5.667. [DOI] [PubMed] [Google Scholar]
- 4.Bacsy E, Nagy Z, Papp M. Intra-acinar lipolysis, an early sign of intravital pancreatic autodigestion. Acta Med Acad Sci Hung. 1970;27:331–5. [PubMed] [Google Scholar]
- 5.Barroso-Aranda J, Schmid-Schönbein GW. Transformation of neutrophils as indicator of irreversibility in hemorrhagic shock. Am J Physiol. 1989;257:H846–H852. doi: 10.1152/ajpheart.1989.257.3.H846. [DOI] [PubMed] [Google Scholar]
- 6.Barroso-Aranda J, Zweifach BW, Mathison JC, Schmid-Schönbein GW. Neutrophil activation, tumor necrosis factor, and survival after endotoxic and hemorrhagic shock. J Cardiovasc Pharmacol. 1995;25(Suppl 2):S23–9. doi: 10.1097/00005344-199500252-00006. [DOI] [PubMed] [Google Scholar]
- 7.Chang M. Mucin Disruption and Transport of Digestive Enzymes in Early Stages of Intestinal Ischemia. University of California; San Diego: 2012. Ph.D. [Google Scholar]
- 8.Chang M, Alsaigh T, Kistler EB, Schmid-Schönbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen AY, DeLano FA, Valdez SR, Ha JN, Shin HY, Schmid-Schönbein GW. Receptor cleavage reduces the fluid shear response in neutrophils of the spontaneously hypertensive rat. Am J Physiol Cell Physiol. 2010;299:C1441–9. doi: 10.1152/ajpcell.00157.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen AY, Ha JN, Delano FA, Schmid-Schönbein GW. Receptor cleavage and P-selectin-dependent reduction of leukocyte adhesion in the spontaneously hypertensive rat. J Leukoc Biol. 2012;299:C1441–C1449. doi: 10.1189/jlb.0112010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deitch EA, Shi HP, Lu Q, Feketeova E, Xu DZ. Serine proteases are involved in the pathogenesis of trauma-hemorrhagic shock-induced gut and lung injury. Shock. 2003;19:452–6. doi: 10.1097/01.shk.0000048899.46342.f6. [DOI] [PubMed] [Google Scholar]
- 13.Del Sorbo L, Slutsky AS. Acute respiratory distress syndrome and multiple organ failure. Curr Opin Crit Care. 2011;17:1–6. doi: 10.1097/MCC.0b013e3283427295. [DOI] [PubMed] [Google Scholar]
- 14.Delano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. 2013;5:169ra11. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage: mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52:415–23. doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112:321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- 17.Doucet JJ, Hoyt DB, Coimbra R, Schmid-Schönbein GW, Junger WG, Paul LW, Loomis WH, Hugli TE. Inhibition of enteral enzymes by enteroclysis with nafamostat mesilate reduces neutrophil activation and transfusion requirements after hemorrhagic shock. J Trauma. 2004;56:501–10. doi: 10.1097/01.ta.0000114536.98447.f7. discussion 510–1. [DOI] [PubMed] [Google Scholar]
- 18.Fitzal F, Delano FA, Young C, Rosario HS, Junger WG, Schmid-Schönbein GW. Pancreatic enzymes sustain systemic inflammation after an initial endotoxin challenge. Surgery. 2003;134:446–56. doi: 10.1067/s0039-6060(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 19.Fitzal F, DeLano FA, Young C, Rosario HS, Schmid-Schönbein GW. Pancreatic protease inhibition during shock attenuates cell activation and peripheral inflammation. J Vasc Res. 2002;39:320–9. doi: 10.1159/000065544. [DOI] [PubMed] [Google Scholar]
- 20.Fitzal F, DeLano FA, Young C, Schmid-Schönbein GW. Improvement in early symptoms of shock by delayed intestinal protease inhibition. Arch Surg. 2004;139:1008–16. doi: 10.1001/archsurg.139.9.1008. [DOI] [PubMed] [Google Scholar]
- 21.Hanson PJ, Moran AP, Butler K. Paracellular permeability is increased by basal lipopolysaccharide in a primary culture of colonic epithelial cells; an effect prevented by an activator of Toll-like receptor-2. Innate Immun. 2011;17:269–82. doi: 10.1177/1753425910367813. [DOI] [PubMed] [Google Scholar]
- 22.Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol. 2011;2:65. doi: 10.3389/fimmu.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishimaru K, Mitsuoka H, Unno N, Inuzuka K, Nakamura S, Schmid-Schönbein GW. Pancreatic proteases and inflammatory mediators in peritoneal fluid during splanchnic arterial occlusion and reperfusion. Shock. 2004;22:467–71. doi: 10.1097/01.shk.0000142253.31006.8c. [DOI] [PubMed] [Google Scholar]
- 24.Kim HD, Malinoski DJ, Borazjani B, Patel MS, Chen J, Slone J, Nguyen XM, Steward E, Schmid-Schönbein GW, Hoyt DB. Inhibition of intraluminal pancreatic enzymes with nafamostat mesilate improves clinical outcomes after hemorrhagic shock in swine. J Trauma. 2010;68:1078–83. doi: 10.1097/TA.0b013e3181da78b1. [DOI] [PubMed] [Google Scholar]
- 25.Kistler EB, Alsaigh T, Chang M, Schmid-Schönbein GW. Impaired small-bowel barrier integrity in the presence of lumenal pancreatic digestive enzymes leads to circulatory shock. Shock. 2012;38:262–7. doi: 10.1097/SHK.0b013e31825b1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kistler EB, Hugli TE, Schmid-Schönbein GW. The pancreas as a source of cardiovascular cell activating factors. Microcirculation. 2000;7:183–92. [PubMed] [Google Scholar]
- 27.Kistler EB, Lefer AM, Hugli TE, Schmid-Schönbein GW. Plasma activation during splanchnic arterial occlusion shock. Shock. 2000;14:30–4. doi: 10.1097/00024382-200014010-00006. [DOI] [PubMed] [Google Scholar]
- 28.Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, Hutchinson WL, Pepys MB. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99:237–42. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 29.Kollias G, Douni E, Kassiotis G, Kontoyiannis D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann Rheum Dis. 1999;58(Suppl 1):I32–9. doi: 10.1136/ard.58.2008.i32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YT, Wei J, Chuang YC, Chang CY, Chen IC, Weng CF, Schmid-Schönbein GW. Successful treatment with continuous enteral protease inhibitor in a patient with severe septic shock. Transplant Proc. 2012;44:817–9. doi: 10.1016/j.transproceed.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 31.Lefer AM, Barenholz Y. Pancreatic hydrolases and the formation of a myocardial depressant factor in shock. Am J Physiol. 1972;223:1103–1109. doi: 10.1152/ajplegacy.1972.223.5.1103. [DOI] [PubMed] [Google Scholar]
- 32.Lefkowitz RB, Schmid-Schönbein GW, Heller MJ. Whole blood assay for elastase, chymotrypsin, matrix metalloproteinase-2, and matrix metalloproteinase-9 activity. Anal Chem. 2010;82:8251–8. doi: 10.1021/ac101462c. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Messina JL. Acute insulin resistance following injury. Trends Endocrinol Metab. 2009;20:429–35. doi: 10.1016/j.tem.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65:S140–6. doi: 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 35.Lichtenberger LM. The hydrophobic barrier properties of gastrointestinal mucus. Annu Rev Physiol. 1995;57:565–83. doi: 10.1146/annurev.ph.57.030195.003025. [DOI] [PubMed] [Google Scholar]
- 36.Marti-Carvajal A, Salanti G, Cardona AF. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst Rev. 2008:CD004388. doi: 10.1002/14651858.CD004388.pub3. [DOI] [PubMed] [Google Scholar]
- 37.Matthews JB, Smith JA, Tally KJ, Menconi MJ, Nguyen H, Fink MP. Chemical hypoxia increases junctional permeability and activates electrogenic ion transport in human intestinal epithelial monolayers. Surgery. 1994;116:150–7. discussion 157–8. [PubMed] [Google Scholar]
- 38.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1772–7. doi: 10.1073/pnas.97.4.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Protease inhibition in the intestinal lumen: attenuation of systemic inflammation and early indicators of multiple organ failure in shock. Shock. 2002;17:205–9. doi: 10.1097/00024382-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 40.Mitsuoka H, Schmid-Schönbein GW. Mechanisms for blockade of in vivo activator production in the ischemic intestine and multi-organ failure. Shock. 2000;14:522–7. doi: 10.1097/00024382-200014050-00005. [DOI] [PubMed] [Google Scholar]
- 41.Nagai H, Henrich H, Wunsch PH, Fischbach W, Mossner J. Role of pancreatic enzymes and their substrates in autodigestion of the pancreas. In vitro studies with isolated rat pancreatic acini. Gastroenterology. 1989;96:838–47. [PubMed] [Google Scholar]
- 42.Penn AH, Altshuler AE, Small JW, Taylor SF, Dobkins KR, Schmid-Schönbein GW. Digested formula but not digested fresh human milk causes death of intestinal cells in vitro: implications for necrotizing enterocolitis. Pediatr Res. 2012;72:560–7. doi: 10.1038/pr.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. 2007;27:296–304. doi: 10.1097/01.shk.0000235139.20775.7f. [DOI] [PubMed] [Google Scholar]
- 44.Penn AH, Schmid-Schönbein GW. The intestine as source of cytotoxic mediators in shock: free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol. 2008;294:H1779–92. doi: 10.1152/ajpheart.00902.2007. [DOI] [PubMed] [Google Scholar]
- 45.Raman K, Chong M, Akhtar-Danesh GG, D’Mello M, Hasso R, Ross S, Xu F, Pare G. Genetic markers of inflammation and their role in cardiovascular disease. Can J Cardiol. 2013;29:67–74. doi: 10.1016/j.cjca.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 46.Redd MJ, Cooper L, Wood W, Stramer B, Martin P. Wound healing and inflammation: embryos reveal the way to perfect repair. Philos Trans R Soc Lond B Biol Sci. 2004;359:777–84. doi: 10.1098/rstb.2004.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richter MD. MS Thesis. UC San Diego: 2012. Protease activity in mesenteric lymph following splanchnic arterial occlusion. [Google Scholar]
- 48.Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–77. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 49.Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schönbein GW. Matrix metalloproteinases cleave the beta2-adrenergic receptor in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;299:H25–35. doi: 10.1152/ajpheart.00620.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid-Schönbein GW. Analysis of inflammation. Annu Rev Biomed Eng. 2006;8:93–131. doi: 10.1146/annurev.bioeng.8.061505.095708. [DOI] [PubMed] [Google Scholar]
- 51.Tonelli M, Sacks F, Pfeffer M, Jhangri GS, Curhan G. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005;68:237–45. doi: 10.1111/j.1523-1755.2005.00398.x. [DOI] [PubMed] [Google Scholar]
- 52.Tong S, Neboori HJ, Tran ED, Schmid-Schönbein GW. Constitutive expression and enzymatic cleavage of ICAM-1 in the spontaneously hypertensive rat. J Vasc Res. 2011;48:386–96. doi: 10.1159/000323474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tran ED, DeLano FA, Schmid-Schönbein GW. Enhanced matrix metalloproteinase activity in the spontaneously hypertensive rat: VEGFR-2 cleavage, endothelial apoptosis, and capillary rarefaction. J Vasc Res. 2010;47:423–31. doi: 10.1159/000281582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tran ED, Yang M, Chen A, Delano FA, Murfee WL, Schmid-Schönbein GW. Matrix metalloproteinase activity causes VEGFR-2 cleavage and microvascular rarefaction in rat mesentery. Microcirculation. 2011;18:228–37. doi: 10.1111/j.1549-8719.2011.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valdez SR. Serotonin 5HT-1A Receptor Density In The Brain Of The Spontaneously Hypertensive Rats. Univ. Calif; San Diego, M.S: 2010. [Google Scholar]
- 56.Waldo SW, Rosario HS, Penn AH, Schmid-Schönbein GW. Pancreatic digestive enzymes are potent generators of mediators for leukocyte activation and mortality. Shock. 2003;20:138–43. doi: 10.1097/01.shk.0000073866.47824.ae. [DOI] [PubMed] [Google Scholar]
- 57.Weidenbusch M, Anders HJ. Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis. J Innate Immun. 2012;4:463–77. doi: 10.1159/000336717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westergaard H, Dietschy JM. Delineation of the dimensions and permeability characteristics of the two major diffusion barriers to passive mucosal uptake in the rabbit intestine. J Clin Invest. 1974;54:718–32. doi: 10.1172/JCI107810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zweifach BW. Hemorrhagic Shock in Germ-free Rats. Ann N Y Acad Sci. 1959;78:315–319. doi: 10.1111/j.1749-6632.1959.tb53116.x. [DOI] [PubMed] [Google Scholar]