

Abstract
Background
Genome-wide association studies (GWAS) have identified common genetic variants that predispose to atrial fibrillation (AF). It is unclear whether rare and low-frequency variants in genes implicated by such GWAS confer additional risk of AF.
Objective
To study the association of genetic variants with AF at GWAS top loci.
Methods
In the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Targeted Sequencing Study, we selected and sequenced 77 target gene regions from GWAS loci of complex diseases or traits, including 4 genes hypothesized to be related to AF (PRRX1, CAV1, CAV2, and ZFHX3). Sequencing was performed in participants with (n = 948) and without (n = 3330) AF from the Atherosclerosis Risk in Communities Study, the Cardiovascular Health Study, the Framingham Heart Study, and the Massachusetts General Hospital.
Results
One common variant (rs11265611; P = 1.70 × 10−6) intronic to IL6R (interleukin-6 receptor gene) was significantly associated with AF after Bonferroni correction (odds ratio 0.70; 95% confidence interval 0.58–0.85). The variant was not genotyped or imputed by prior GWAS, but it is in linkage disequilibrium (r2 = .69) with the single-nucleotide polymorphism, with the strongest association with AF so far at this locus (rs4845625). In the rare variant joint analysis, damaging variants within the PRRX1 region showed significant association with AF after Bonferroni correction (P = .01).
Conclusions
We identified 1 common single-nucleotide polymorphism and 1 gene region that were significantly associated with AF. Future sequencing efforts with larger sample sizes and more comprehensive genome coverage are anticipated to identify additional AF-related variants.
Keywords: Arrhythmia, Genetics, Atrial fibrillation, Epidemiology
Introduction
Atrial fibrillation (AF) is the most common arrhythmia and a major public health burden causing high morbidity,1–3 mortality,4 and societal cost.5 Both clinical6 and genetic7 factors predispose to AF. By using genome-wide association studies (GWAS), we and others have reported common genetic variants at 9 distinct chromosomal loci that are significantly related to AF.8–11 Although the heritability of AF has been estimated to be as high as 60%,12 the known AF-related genetic variants appear to explain only a fraction of the heritability.13 It has been hypothesized that the “missing heritability” of AF and other common diseases may be explained in part by rare variants with large effects.7
The Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE)14 Targeted Sequencing Study was initiated to identify genetic variants within genes implicated by GWAS. We hypothesized that GWAS loci might harbor functional variants as well as rare and low-frequency variants associated with AF. The analysis evaluated both individual disease-associated variants and joint analysis of low-frequency variants at each locus.
Methods
Study samples
The methods for the CHARGE Targeted Sequencing Study are summarized in the Online Supplement. For AF analyses, cases with AF were derived from both the cohort random sample and all the phenotype groups. In addition, we selected 200 patients from the Massachusetts General Hospital (MGH) with early-onset AF (≤66 years at AF onset and no structural heart disease at diagnosis). Study participants without AF from either the cohort random sample or the other phenotype groups were used as referents (Table 1). Participants with AF from the MGH were combined with participants with AF from the Framingham Heart Study (FHS) and were then compared with participants without AF from the FHS. This approach has been applied successfully in the past, since both participants from the MHS and participants from the FHS are derived from the same geographic region.10 Participants with AF from the Atherosclerosis Risk in Communities (ARIC) Study and the Cardiovascular Health Study (CHS) were compared with participants without AF from the ARIC Study and the CHS, respectively.9 All the participants in this study were of European ancestry. Institutional review boards at all participating centers approved the study, and participants gave informed consent.
Table 1. Clinical characteristics of the study samples by cohort.
| No. of individuals | Clinical characteristic | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Cohort | Total | With AF | Without AF | Age (y)* | Women | Hypertension |
| ARIC Study | 1952 | 245 | – | 57.4 ± 5.6 | 87 (35.5%) | 116 (47.3%) |
| – | 1707 | 54.4 ± 5.6 | 868 (50.8%) | 488 (28.5%) | ||
| CHS | 1132 | 351 | – | 73.0 ± 5.8 | 185 (52.7%) | 221 (63.0%) |
| – | 781 | 72.3 ± 5.3 | 422 (54.0%) | 412 (52.8%) | ||
| FHS | 994 | 152 | – | 69.1 ± 9.4 | 63 (41.4%) | 63 (41.4%) |
| – | 842 | 60.8 ± 10.2 | 449 (53.3%) | 228 (27.1%) | ||
| MGH | 200 | 200 | – | 52.9 ± 11.1 | 37 (18.5%) | 50 (25.0%) |
Values are presented as mean ± SD and as n (%).
AF = atrial fibrillation; ARIC = Atherosclerosis Risk in Communities; CHS = Cardiovascular Health Study; FHS = Framingham Heart Study; MGH = Massachusetts General Hospital.
Age in the FHS was assessed at the time when DNA was drawn.
Sequencing, functional annotation, and data analysis
Sequencing methods, single-nucleotide polymorphism (SNP) functional annotation, and data analysis methods are described in detail in the Online Supplement. Briefly, approximately 2 Mb of target regions were captured by using a customized NimbleGen Capture array and sequenced by using the ABI SOLiD V4.0 platform. The raw short reads were aligned to the reference human genome by BFAST.15 SAMtools16 was used to pile up aligned reads and call variants with quality filters. We then performed quality control procedures on the resulting data. Variants were categorized as known or novel by comparison with the dbSNP database and the 1000 Genomes Project.17 The functional effect of identified variants on the encoded proteins was predicted by using the ANNOVAR software package.18 We selected and sequenced 77 targeted regions in this study. Thirty-three of these regions were previously implicated by GWAS to be associated with 1 of 14 investigated phenotypes. The remaining 44 targeted regions were pleiotropic loci that were associated with multiple phenotypes. In particular, 4 regions were specifically selected for AF7: ZFHX3, PRRX1, CAV1, and CAV2. By balancing gene size and space restrictions due to assay design, for AF we limited the analysis to the exonic regions of the 4 candidate genes. The location of capture probes is shown in Online Supplemental Figure 1.
For common variants (minor allele frequency [MAF] ≥ 1%), we performed both unweighted analyses and analyses weighted by the sampling probabilities to account for the study design. We report P values from unweighted analysis (which provides more power) and β estimates from the weighted analysis (which reflects population estimates of effect sizes).19 For rare variants (MAF <1%), we used a modified version of the sequence kernel association test to aggregate all the variants within each region and tested its association with AF.20 Our analyses were restricted to the exonic variants only. In our secondary analyses, we restricted the analyses to damaging variants only, which were defined as those that were nonsense variants or located in the splicing sites or missense variants but predicted to cause damaging effects to the encoded proteins by PolyPhen.21
Results
We identified a total of 52,736 variants across all sequenced individuals, including 4800 common variants and 47,936 rare variants. In particular, we identified 62 common variants and 818 rare variants within the 4 AF targeted genes.
Common variant results
No SNP within any of the 4 AF targeted regions was significantly associated with AF after Bonferroni correction for multiple testing (cutoff P value = 0.05/62 = 8.06 × 10−4). Only 4 SNPs reached nominal significance, including 2 SNPs located 46 kb upstream of PRRX1 (rs577676 and rs576736), 1 located in the 5′ untranslated region of CAV1 (rs45498702), and 1 missense SNP in ZFHX3 (rs2213978). The regional association plots are shown in Figure 1, which provides a detailed overview of the variation at each locus.
Figure 1.
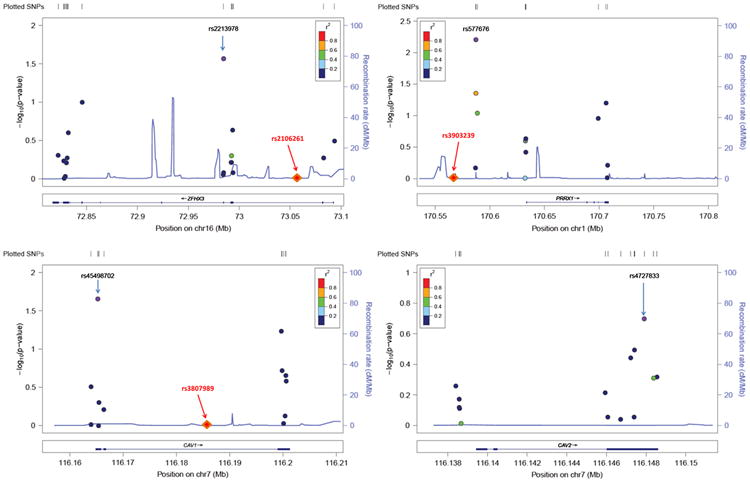
Regional association plots of atrial fibrillation (AF)–related genes. Common variants identified by sequencing in each of the 4 AF-related genes are plotted according to their chromosomal position on the x-axis (NCBI Build 37, 2009). The y-axis provided the −log10 P value of each variant's association with AF. Variants are clustered within the exons of these genes and colored on the basis of the linkage disequilibrium (r2) to the most significant single-nucleotide polymorphism (SNP; purple point) in each gene region. The diamond symbols indicate the location of the original genome-wide association study (GWAS) signal (the most significant SNP) relative to the sequence regions. Of note, the GWAS SNPs were located in introns or intergenic regions and thus not directly sequenced here. Also, CAV1 and CAV2 genes are adjacent, which is why a GWAS SNP is present only in the CAV1 plot.
We then expanded the analysis to variants in other targeted regions. Despite the complex design of our study, no obvious relevant population substructure was recognized (genomic control factor λ = 1.047; Online Supplemental Figure 2). Figure 2 shows the distribution of common variant associations across the 77 targeted regions in chromosomal order. Only 1 SNP, rs11265611 (P = 1.7 × 10−6), was significantly associated with AF after Bonferroni correction for multiple testing (cutoff P value = 0.05/4800 ≈ 1.0 × 10−5). The SNP was widely observed in patients from the FHS and MGH (MAF = 49%) and participants of the 1000 Genomes Project (MAF = 43%). However, it was filtered out in the ARIC Study and the CHS owing to the stringent quality control filters. The SNP is located in the first intron of IL6R (interleukin-6 receptor gene), which encodes the interleukin 6 receptor. It was not genotyped or imputed by prior GWAS, but it is in moderate linkage disequilibrium with rs4845625 (r2 = .69), an SNP that was previously reported to be associated with AF.22 We then performed conditional analysis by conditioning on rs4845625. The SNP, rs1 1265611, was still significantly associated with AF (P = 6.2 × 10−6) after adjusting the effect of rs4845625, suggesting that it might be an independent signal. The 10 SNPs with the smallest P values overall and the variants in AF-related target regions reaching nominal significance are presented in Table 2.
Figure 2.
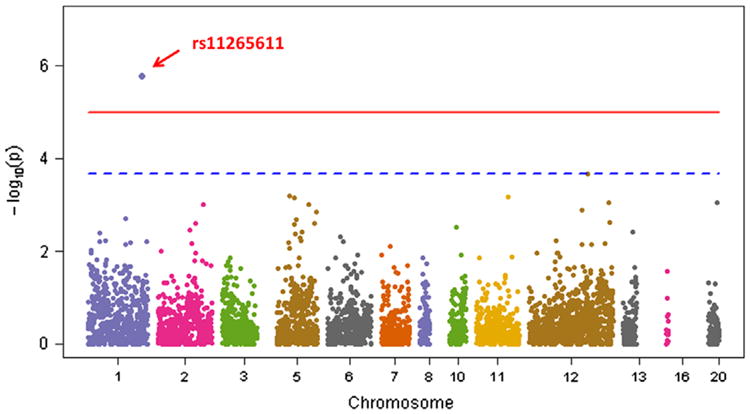
Manhattan plot of common variants. All common single-nucleotide polymorphisms (SNPs; n = 4800) were analyzed by using unweighted models. The −log10 P value of each SNP is plotted according to its chromosomal position. The red horizontal line indicates the Bonferroni corrected threshold of significance at P = 1.0 × 10−5, whereas the blue horizontal line indicates the 1 false-positive threshold at P = 1/4800 = 2.1 × 10−4.
Table 2. Common variant results.
| ARIC Study | CHS | FHS/MGH | Meta-analysis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||||||||
| SNP | Chr | Targeted region | β | SE | β | SE | β | SE | β | SE | OR | 95% CI | P |
| Ten most significant associations across all 77 target gene regions | |||||||||||||
| rs11265611 | 1 | IL6R | – | – | – | – | −0.35 | 0.10 | −0.35 | 0.10 | 0.70 | 0.58–0.85 | 1.7 × 10−6 |
| rs855222 | 12 | IGF1 | – | – | 0.38 | 0.13 | – | – | 0.38 | 0.13 | 1.45 | 1.15–1.88 | 2.1 × 10−4 |
| rs114779117 | 5 | MEF2C | 0.77 | 0.65 | 0.70 | 0.39 | 0.90 | 0.39 | 0.80 | 0.25 | 2.22 | 1.35–3.64 | 6.3 × 10−4 |
| rs10769256 | 11 | SPI1 | – | – | – | – | −0.29 | 0.10 | −0.29 | 0.10 | 0.75 | 0.61–0.91 | 6.5 × 10−4 |
| rs79435800 | 5 | NIPAL4 | 0.18 | 0.33 | 0.47 | 0.28 | 0.36 | 0.26 | 0.35 | 0.17 | 1.42 | 1.03–1.97 | 7.0 × 10−4 |
| rs72525420 | 20 | JAG1 | – | – | – | – | 0.40 | 0.17 | 0.40 | 0.17 | 1.49 | 1.06–2.09 | 8.9 × 10−4 |
| rs9805001 | 12 | C12orf51 | −1.59 | 0.87 | −0.81 | 0.61 | −0.98 | 0.76 | −1.04 | 0.42 | 0.35 | 0.16–0.80 | 9.0 × 10−4 |
| rs76416019 | 5 | ADAM19 | −0.00 | 0.31 | 0.40 | 0.27 | 0.30 | 0.26 | 0.25 | 0.16 | 1.29 | 0.94–1.77 | 9.7 × 10−4 |
| rs72965937 | 2 | IRS1 | 0.52 | 0.34 | 0.39 | 0.25 | 0.59 | 0.25 | 0.50 | 0.16 | 1.64 | 1.21–2.33 | 9.7 × 10−4 |
| rs1520220 | 12 | IGF1 | 0.36 | 0.16 | 0.19 | 0.14 | 0.22 | 0.13 | 0.25 | 0.08 | 1.28 | 1.09–1.51 | 1.3 × 10−3 |
| Nominally significant SNPs within AF-related target regions | |||||||||||||
| rs577676 | 1 | PRRX1 | −0.12 | 0.13 | 0.01 | 0.11 | −0.38 | 0.11 | −0.16 | 0.06 | 0.85 | 0.75–0.96 | 6.2 × 10−3 |
| rs45498702 | 7 | CAV1 | – | – | −0.13 | 0.26 | −0.72 | 0.26 | −0.43 | 0.19 | 0.65 | 0.45–0.94 | 2.2 × 10−2 |
| rs2213978 | 16 | ZFHX3 | −0.31 | 0.37 | −0.35 | 0.32 | −0.23 | 0.37 | −0.30 | 0.20 | 0.74 | 0.50–1.10 | 2.7 × 10−2 |
| rs576736 | 1 | PRRX1 | 0.12 | 0.13 | −0.01 | 0.11 | 0.29 | 0.12 | 0.13 | 0.07 | 1.14 | 1.00–1.31 | 4.4 × 10−2 |
AF = atrial fibrillation; ARIC = Atherosclerosis Risk in Communities; Chr = chromosome; CHS = Cardiovascular Health Study; CI = confidence interval; FHS = Framingham Heart Study; MGH = Massachusetts General Hospital; OR = odds ratio; SE = standard error; SNP = single-nucleotide polymorphism.
Rare variant results
Table 3 displays the association results of rare variants with AF for the 4 targeted regions. None of them showed association with AF when all the exonic variants were included. However, 1 gene, PRRX1, was found to be associated with AF when only damaging variants were included (P = .01). The gene encodes a homeodomain transcription factor, and a common variant 46 kb upstream of this gene was found to be associated with AF in our prior GWAS.9 A total of 16 exonic rare variants were found within the PRRX1 region (Online Supplemental Table 1). These included 9 missense variants, 6 synonymous variants, and 1 nonsense variant. Three of these variants have already been reported in the dbSNP database. Six of 9 missense variants were predicted to cause damaging effects to the encoded protein.23 The nonsense variant (chr1: 170695522, G → A) resulted in a truncation of the last 25 amino acids of PRRX1.
Table 3. Rare variant results of AF targeted genes.
| Exonic variants | Damaging variants | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Targeted region | Chr | Start position* | Stop position* | P | CMAF | P | CMAF |
| PRRX1 | 1 | 170,586,793 | 170,708,641 | .64 | 0.02 | .01 | 0.0009 |
| ZFHX3 | 16 | 72,820,963 | 73,093,634 | 1.00 | 0.15 | .99 | 0.0102 |
| CAV2 | 7 | 116,138,344 | 116,148,695 | .67 | 0.01 | .58 | 0.0007 |
| CAV1 | 7 | 116,163,739 | 116,201,338 | .98 | 0.04 | .52 | 0.0002 |
AF = atrial fibrillation; Chr = chromosome; CMAF = cumulative minor allele frequency.
The chromosomal positions were based on the reference human genome (NCBI Build 37, 2009).
Discussion
By analyzing common genetic variants in the coding regions or in proximity to genes selected for targeted sequencing in almost 1000 individuals with AF and more than 3300 referents without AF, we could identify a single association that withstood correction for multiple comparisons, the SNP rs11265611. The association was still significant, although attenuated after adjusting the effect of a previously reported AF-associated SNP.22 The SNP rs11265611 was intronic to the gene IL6R, which encodes the IL6R. Interleukin-6 is an inflammatory cytokine that, through its receptor (IL6R), triggers the production of acute phase reactants.24,25 Inflammation is thought to play a central role in the initiation and maintenance of AF in many individuals.26–29 However, none of the common variants within the 4 AF targeted regions showed significant association with AF.
As in many other complex genetic disorders, the genetic variability of AF is not explained fully by the common variants identified to date.7 Several groups have advanced the hypothesis that rare variants might contribute to explaining genetic variability, both of AF and other complex traits.30,31 Sequencing genes implicated in GWAS revealed additional independent rare variants in the genes NOD, IL23R, CARD9, and at least 5 more genes.32 One hypothesis is that an individual rare genetic variant confers a stronger risk of disease than a common variant, for example, those identified by GWAS.32 In our study, we found that damaging rare variants within the PRRX1 region were associated with AF. The association, however, disappeared when all the exonic variants were included, suggesting that the rare variant test is susceptible to the inclusion of unrelated variants.
Nine genetic loci have been found to be significantly related to AF.8–11 However, 6 of these loci were located in the intronic gene regions whereas the remaining 3 loci were located in the intergenic regions. Given that these loci did not directly affect the encoded proteins, they are likely associated with AF through other mechanisms, which present a great challenge to identifying causal variants for AF. Additional efforts, including fine mapping of the discovered loci and deep resequencing of putative candidate genes, are thus necessary to uncover the genetic architecture underlying AF.
In our current approach, we balanced dense coverage of targeted regions vs limited array capacities. As with GWAS for more common alleles, it can be expected that hypothesis-free, large-scale approaches will lead to the detection of novel variants, some of them rare. Such approaches will include exome-wide sequencing in sufficient numbers of AF cases and controls, genotyping by newly designed arrays that primarily contain rare alleles, such as the Illumina Exome Chip,33 or ultimately the sequencing of the entire genome in large samples.
Conclusions
We identified 1 common SNP and 1 genomic region that were significantly associated with AF. On the basis of our experience with increasing sample sizes in GWAS, we anticipate that robust novel rare and low-frequency variants for AF will be detected by broader efforts including exome-wide sequencing and exome chip genotyping in studies involving greater numbers of individuals with AF.
Supplementary Material
Acknowledgments
We thank the staff and participants of the ARIC Study for their important contributions.
Funding support for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE Consortium” was provided by the National Institutes of Health (NIH) through the American Recovery and Reinvestment Act of 2009 (5RC2HL102419). Data for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE Consortium” were provided by Eric Boerwinkle on behalf of the Atherosclerosis Risk in Communities Study; L. Adrienne Cupples, principal investigator for the Framingham Heart Study; and Bruce Psaty, principal investigator for the Cardiovascular Health Study. Sequencing was carried out at the Baylor Genome Center (U54 HG003273). The ARIC Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute (NHLBI) contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN2682011000010C, HHSN2682011000011C, and HHSN2682011000012C). The Framingham Heart Study is conducted and supported by the NHLBI in collaboration with the Boston University (contract no. N01-HC-25195) and its contract with Affymetrix, Inc, for genome-wide genotyping services (contract no. N02-HL-6-4278) and for quality control by Framingham Heart Study investigators using genotypes in the SNP Health Association Resource project. A portion of this research was conducted by using the Linux Cluster for Genetic Analysis computing resources at Boston University Medical Campus. This CHS research was supported by NHLBI contracts N01-HC-85239, N01-HC-85079, N01-HC-85080, N01-HC-85081, N01-HC-85082, N01-HC-85083, N01-HC-85084, N01-HC-85085, N01-HC-85086; N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, and HHSN268201200036C as well as by NHLBI grants HL080295, HL087652, HL105756, with additional contribution from NINDS. Additional support was provided through National Institute on Aging of the National Institutes of Public Health grants AG-023629, AG-15928, AG-20098, and AG-027058. See also http://www.chs-nhlbi.org/pi.htm. This project was also supported by NIH grants N01-HC 25195 and 6R01-NS 17950 (to Dr Benjamin); 1RO1HL092577, 5R21DA027021, 1RO1HL104156, 1K24HL105780, 1R01 HL102214, and 1RC1HL101056 (to Dr Ellinor); 1RC1HL099452 and American Heart Association grant 09SDG2280087 (to Dr Alonso), and the Netherlands Organisation for Scientific Research grant (Veni grant 016.136.055; to Dr Rienstra). This work was partially supported by the Evans Center for Interdisciplinary Biomedical Research ARC on Atrial Fibrillation at Boston University (http://www.bumc.bu.edu/evanscenteribr/). Dr Psaty serves on the DSMB of a clinical trial of a device funded by Zoll LifeCor and on the Steering Committee of the Yale Open Data Access Project funded by Medtronic.
Abbreviations
- AF
atrial fibrillation
- ARIC
Atherosclerosis Risk in Communities
- CHARGE
Cohorts for Heart and Aging Research in Genomic Epidemiology
- CHS
Cardiovascular Health Study
- FHS
Framingham Heart Study
- GWAS
genome-wide association studies
- IL6R
interleukin-6 receptor
- MAF
minor allele frequency
- MGH
Massachusetts General Hospital
- SNP
single-nucleotide polymorphism
Footnotes
Appendix Supplementary data: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.hrthm.2013.11.012
References
- 1.Ott A, Breteler MM, de Bruyne MC, van Harskamp F, Grobbee DE, Hofman A. Atrial fibrillation and dementia in a population-based stud:. the Rotterdam study. Stroke. 1997;28:316–321. doi: 10.1161/01.str.28.2.316. [DOI] [PubMed] [Google Scholar]
- 2.Wang TJ, Larson MG, Levy D, et al. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation. 2003;107:2920–2925. doi: 10.1161/01.CIR.0000072767.89944.6E. [DOI] [PubMed] [Google Scholar]
- 3.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–952. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 5.Kim MH, Johnston SS, Chu BC, Dalal MR, Schulman KL. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovas Qual Outcomes. 2011;4:313–320. doi: 10.1161/CIRCOUTCOMES.110.958165. [DOI] [PubMed] [Google Scholar]
- 6.Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort: the Framingham Heart Study. JAMA. 1994;271:840–844. [PubMed] [Google Scholar]
- 7.Sinner MF, Ellinor PT, Meitinger T, Benjamin EJ, Kaab S. Genome-wide association studies of atrial fibrillation: past, present, and future. Cardiovasc Res. 2011;89:701–709. doi: 10.1093/cvr/cvr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudbjartsson DF, Arnar DO, Helgadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 9.Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellinor PT, Lunetta KL, Glazer NL, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–244. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin EJ, Rice KM, Arking DE, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–881. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christophersen IE, Ravn LS, Budtz-Joergensen E, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–383. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lubitz SA, Yin X, Fontes JD, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. 2010;304:2263–2269. doi: 10.1001/jama.2010.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Psaty BM, O'Donnell CJ, Gudnason V, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.1000 Genomes Project Consortium. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lumley T, Dupuis J, Rice KM, et al. http://stattech.wordpress.fos.auckland.ac.nz/files/2012/05/design-paper.pdf.
- 20.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnabel RB, Kerr KF, Lubitz SA, et al. Large-scale candidate gene analysis in whites and African Americans identifies IL6R polymorphism in relation to atrial fibrillation: the National Heart, Lung, and Blood Institute's Candidate Gene Association Resource (CARe) project. Circ Cardiovasc Genet. 2011;4:557–564. doi: 10.1161/CIRCGENETICS.110.959197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Women's Health Initiative Study Group. Design of the Women's Health Initiative clinical trial and observational study. Control Clin Trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 24.Rafiq S, Frayling TM, Murray A, et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8:552–559. doi: 10.1038/sj.gene.6364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reich D, Patterson N, Ramesh V, et al. Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aviles RJ, Martin DO, Apperson-Hansen C, et al. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–3010. doi: 10.1161/01.CIR.0000103131.70301.4F. [DOI] [PubMed] [Google Scholar]
- 27.Issac TT, Dokainish H, Lakkis NM. Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J Am Coll Cardiol. 2007;50:2021–2028. doi: 10.1016/j.jacc.2007.06.054. [DOI] [PubMed] [Google Scholar]
- 28.Marcus GM, Smith LM, Glidden DV, et al. Markers of inflammation before and after curative ablation of atrial flutter. Heart Rhythm. 2008;5:215–221. doi: 10.1016/j.hrthm.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcus GM, Smith LM, Ordovas K, et al. Intracardiac and extracardiac markers of inflammation during atrial fibrillation. Heart Rhythm. 2010;7:149–154. doi: 10.1016/j.hrthm.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birch PJ, Dekker LV, James IF, Southan A, Cronk D. Strategies to identify ion channel modulators: current and novel approaches to target neuropathic pain. Drug Discov Today. 2004;9:410–418. doi: 10.1016/S1359-6446(04)03043-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.