

Abstract
Helicobacter pylori UreF is involved in the insertion of Ni2+ in the urease active site. The recombinant protein in solution is a dimer characterized by an extensive α-helical structure and a well-folded tertiary structure. HpUreF binds two Ni2+ ions per dimer, with micromolar dissociation constant, as shown by calorimetry. X-ray absorption spectroscopic analysis indicates that the Ni2+ ions reside in a five-coordinate pyramidal geometry comprised exclusively of N/O-donor ligands derived from the protein, including one or two histidine imidazoles and carboxylate ligands. Binding of Ni2+ does not affect the solution properties of the protein. Mutation to alanine of His229 and/or Cys231, a pair of residues located on the protein surface that interacts with HpUreD, altered the affinity of the protein for Ni2+. This result, complemented by the XAS analysis, indicates that the Ni2+ binding site involves His229, and that Cys231 has an indirect structural role in metal binding. An in vivo assay of urease activation demonstrated that H229A-HpUreF, C231A-HpUreF and H229/C231-HpUreF are significantly less competent in this process, suggesting a role for a Ni2+ complex with UreF in urease maturation. This hypothesis was supported by calculations revealing the presence of a tunnel that joins the Cys-Pro-His metal binding site on UreG and an opening on the UreD surface, passing through UreF close to His229 and Cys231, in the structure of the H. pylori UreDFG complex. This tunnel could be used to transfer nickel into the urease active site during apo-to-holo enzyme activation.
INTRODUCTION
Helicobacter pylori (H. pylori, Hp) is a spiral gram-negative microaerophilic bacterium, representing the dominant species of the human gastric microbiome. H. pylori is the etiologic agent for chronic infection of the gastric mucosa, and an important risk factor for several diseases, such as gastric and duodenal ulcers and gastric adenocarcinoma [1]. This bacterium optimally grows at neutral pH, but tolerates the acidic conditions occurring in the gastric environment taking advantage of the activity of urease: urea degradation, catalyzed by this enzyme, ultimately produces ammonia and bicarbonate, and causes a local pH increase to values suitable for bacterial survival and growth, with physiological cytoplasmic pH in the 6.4 – 7.4 range [2]. This activity is thus required for bacterial gastric colonization [3], implying that understanding the structure, function, and activation of this enzyme is key to the development of specific drugs to eradicate H. pylori infections [4].
The structures of urease from three bacteria, Klebsiella aerogenes (Ka) [5], Bacillus pasteurii (Bp) [6], and H. pylori [7], and from the seeds of the plants Canavalia ensiformis (jack bean) [8] and Cajanus cajan (pigeon pea) [9], are available. In all these enzymes, the active site (Figure 1) features two essential Ni2+ ions, bridged by a conserved post-translationally carbamylated lysine residue, and coordinated by N/O-donor ligands from the side chains of histidine and aspartic acid residues, as well as by a hydroxide ion, which appears to act as the nucleophile in the catalytic mechanism [10]. The presence of Ni2+ ions is essential for the remarkable enhancement of the rate of the catalyzed hydrolysis reaction occurring with a kcat/kuncat of about 1015 [11].
Figure 1.

Scheme of the Helicobacter pylori urease activation process starting from the apo-enzyme and leading to holo-urease. The ribbon diagrams show the structure of Helicobacter pylori urease in its [(αβ)3]4 quaternary structure; each blue, gold and green highlighted chains represent one (αβ) heterodimer, and they all together reveal the similarity of the (αβ)3 moiety in this urease with the (αβγ)3 quaternary structure of other bacterial ureases such as that of Sporosarcina pasteurii and Klebsiella aerogenes. The details of the Ni2+ ions coordination environment in the active site are shown in the central inset. The crystal structures or models of the various protein complexes involved in the process are also shown as ribbon diagrams: HpUreD (light green), HpUreF (orange), HpUreG (red) and HpUreE (dark green).
The use of Ni2+ ions for the activity of urease in H. pylori requires efficient systems for acquisition, intra-cellular trafficking, homeostasis and utilization of nickel [12, 13]. In vivo, urease is synthesized as an inactive apo-enzyme, and four accessory proteins, named UreD1, UreF, UreG and UreE are usually involved in a multistep process that produces the nickel-loaded active holo-enzyme (Figure 1). The key event of this process is the formation of a protein complex between the apo-enzyme and UreD, UreF, and UreG. In this complex, urease undergoes active site lysine carbamylation concomitant with GTP hydrolysis [14]. The latter process is catalyzed by UreG [15], a GTPase that is intrinsically disordered [16–18] but nevertheless able to function as an enzyme because of the substantial rigidity of the residues in the active site region [19]. Subsequently, Ni2+ ions productively enter into the active site of the enzyme, a process mediated by UreE, a Ni2+ metallo-chaperone [20].
Despite the significant amount of structural and biochemical information obtained so far on the proteins involved in urease maturation, the details of the protein interaction network that leads to Ni2+ incorporation into the urease active site are far from being fully understood, even though a hypothesis is generally accepted (Figure 1). UreD is the first protein to come into direct contact with urease, as revealed by chemical cross-linking experiments and mass spectrometry for KaUreD [21] as well as two-hybrid analyses and immuno-precipitation studies for Proteus mirabilis (Pm) UreD [22]. UreD mediates the interaction with other chaperones, likely acting as a protein platform [23, 24]. UreF binds the apo urease-UreD complex by directly interacting with UreD, as shown using chemical crosslinking and small-angle X-ray scattering (SAXS) experiments on K. aerogenes proteins [21, 25], and two-hybrid studies on P. mirabilis [22] and H. pylori [26] proteins. The same conclusion was drawn using in vitro light scattering experiments, pull-down assays and crystallography in the case of HpUreF [27]. The interaction of apo-urease with UreD and UreF appears to induce a conformational rearrangement of the urease enzyme, which promotes active site access to Ni2+ ions and CO2 needed for the carbamylation of the active site lysine [21]. Functional studies suggested that this event prevents Ni2+ ions from unproductively binding to the non-carbamylated active site [28]. On the basis of a structural model of BpUreF obtained by fold recognition algorithms [29], and following the observation that in the absence of KaUreF, KaUreG is unable to join the urease-KaUreD complex [15], UreF was proposed to have a role in modulating the GTPase activity of UreG through a direct protein-protein interaction [29]. Pull-down assays on H. pylori proteins indicated that this UreF-UreG interaction requires a pre-formed complex between UreF and UreD, suggesting that a conformational change on UreF, induced by UreD, is necessary in order to drive the formation of a UreF-UreG complex [27]. Considering that many GTPase activating proteins (GAP) are allosterically regulated by protein effectors [30], the role of UreD in determining the UreF-UreG interaction is consistent with the proposed role of HpUreF as an activator of the GTPase activity of UreG [29]. This hypothesis is also supported by weak but intriguing similarities between the structures of several GAP domains of different origins and the crystal structure of HpUreF [27, 31]. When expressed and purified alone, UreF from H. pylori strain 26695 features a truncation of the last 21 residues at the C-terminus, suggesting that this protein region is prone to hydrolytic cleavage [27, 31]. On the other hand, when the same protein is co-expressed and co-purified with HpUreD, a complex is formed (Figure 1) that directly involves the C-terminus of HpUreF at the interface with HpUreD, as revealed by the crystal structure of HpUreF-HpUreD complex [27]. The formation of this complex protects this region of UreF from degradation and indicates that it is essential for the stabilization of the UreF-UreD interaction. Site-directed mutagenesis of several conserved residues on the surface of KaUreF that are not located in the disordered and cleavable C-terminal region of the protein resulted in the identification of nine residues that appear to be important for the interaction of KaUreF with KaUreG based on in vitro pull-down assays, as well as for the full activation of urease based on in vivo experiments [32]. In the same study, UreF was proposed to act as a coupling factor between the GTPase activity of UreG and the process of metallocenter assembly, with UreF increasing the fidelity of activation [32].
The metal binding properties of HpUreG and HpUreE in solution and in the solid state have been extensively investigated [12, 18, 33, 34]. In particular, it has been established that these two proteins specifically bind both Ni2+ and Zn2+, with different affinities and coordination geometries, and that the HpUreE-HpUreG interaction depends on the metal ion bound state: Zn2+ binding promotes HpUreG dimerization [18] and stabilizes the formation of the (HpUreE)2-(HpUreG)2 hetero-tetramer [33] (Figure 1). The crosstalk between these metal ions thus significantly influences the urease activation process through modulation of the protein-protein interaction network [12].
All these data have recently been used to produce a structural model of the HpUreDFGE supercomplex (Figure 1) [35]. However, no information is yet available on the metal binding capability of UreD and UreF from any biological source, either in solution or in the solid state, with the available crystal structures of UreF and UreF-UreD complex from H. pylori strain 26695 not showing any metal ion bound [27, 31]. Several attempts carried out by us to purify the recombinant H. pylori UreF from the G27 strain, which features only a seven-residue difference in the protein sequence as compared to that of UreF from H. pylori strain 26695, consistently produced a protein that completely accumulated in the insoluble fraction of the cellular extract. The present study was undertaken using the recombinant HpUreF from H. pylori strain 26695, purified in a soluble form, to investigate the protein properties in solution. Our work revealed a novel Ni2+ binding activity of HpUreF in vitro, the role of Ni2+ in modulating the structural and functional properties of HpUreF, and its importance for urease activation in a cellular milieu.
MATERIALS AND METHODS
Materials
The pCR1.2 (3.9 kbp) sub-cloning vector was purchased from Eurofins; the pET15b (5.7 kbp) expression vector was from Novagen; NdeI and BamHI FastDigest® restriction enzymes were from Fermentas; plasmids were purified from E. coli XL10 – Gold Ultracompetent Cells (Stratagene) using Wizard Plus SV Minipreps DNA Purification Systems (Promega); DNA fragments were purified by electrophoresis on a 1% (w/v) Wizard SV agarose gel and PCR cleanup System (Promega); pGEM-T-easy sub-cloning vector and T4 DNA ligase were from Promega. Site directed mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis kit (Stratagene) on both pET15b::HpureF and pGEM::ureOP plasmids.
Gene cloning and mutagenesis
The ureF gene from H. pylori 26695 strain coding for sequence residues 1–254 was commercially synthesized and cloned into the pCR1.2 sub-cloning vector, introducing the recognition sites for NdeI and BamHI restriction enzyme at the 5′ and 3′ positions, respectively. The purified pCR1.2::HpureF construct was double digested with NdeI and BamHI. The purified DNA fragment, corresponding to the ureF gene, was ligated (T4 DNA ligase) into the pET15b expression vector previously digested with NdeI and BamHI endonucleases. This protocol introduced a His-tag at the N-terminus, followed by a GSH tri-peptide. The purified construct (pET15b::HpureF) was scrutinized by restriction analysis and sequenced at both strands. E. coli BL21(DE3) competent cells were transformed with the recombinant pET15b::HpureF construct.
According to the sequence of the urease operon from H. pylori G27 strain (NCBI code NC_011333.1), the two oligonucleotides 5′-AAAGGGGTATTAAACGCGCTTCTA-3′ and 5′-AGCGCTTTTAAATCTTTTGCGTGATGG-3′ were used to amplify the entire 6,144 bp urease operon by PCR from the genome of H. pylori G27 strain. The purified PCR product was ligated (T4 DNA ligase) into the pGEM-T-easy vector. The resulting construct (pGEM::ureOP) was purified, scrutinized by restriction analysis and sequenced at the 5′ and 3′ ends. The recombinant plasmid carries the urease operon in the same transcriptional direction of the T7 promoter, allowing induction of protein expression based on the T7 expression system. E. coli BL21(DE3) cells were transformed with pGEM::ureOP construct for the urease activity test.
Site directed mutagenesis was performed on both pET15b::HpureF and pGEM::ureOP plasmids. The primers 5′-GATTGAATTGAAAAAGGCTTTAAGGGCTTATCTTTATGCGCAAACTTCTAAC-3′ and 5′-GTTAGAAGTTTGCGCATAAAGATAAGCCCTTAAAGCCTTTTTCAATTCAATC-3′ were used for the preparation of the mutant H180A-HpUreF in pET15b::HpureF, while the primers 5′-CCTAGAACTAGACGAAAGCGCCCTGGCCACGGCAAGCGTTCAAAAC-3′ and 5′-GTTTTGAACGCTTGCCGTGGCCAGGGCGCTTTCGTCTAGTTCTAGG-3′ (mutated bases underlined) were used to obtain the double mutant H229A/C231A-HpUreF in pET15b::HpureF and pGEM::ureOP. To obtain HpUreF single mutants, the following primers were used: 5′-AGAACTAGACGAAAGCGCCCTGTGCACGGCAAGC-3′ and 5′-GCTTGCCGTGCACAGGGCGCTTTCGTCTAGTTCT-3′ for H229A-HpUreF in pET15b::HpureF and pGEM::ureOP, 5′-GACGAAAGCCACCTGGCCACGGCAAGCGTTCA-3′ and 5′-TGAACGCTTGCCGTGGCCAGGTGGCTTTCGTC-3′ for C231A-HpUreF in pET15b::HpureF and pGEM::ureO (mutated bases underlined).
Protein heterologous expression and purification
Recombinant wild type UreF from H. pylori 26695 strain was produced using a protocol adapted from a previous study [31]. Expression of His-tagged HpUreF and its mutants was obtained by growing the transformed cells in 2 L of Luria-Bertani (LB) medium at 37 °C until the OD600 reached 0.5 – 0.6. The expression was induced with 0.5 mM isopropyl β -thiogalactopyranoside (IPTG), the temperature was reduced to 16 °C, and expression was carried out for 21 h. 15N-labeled protein was obtained with the same protocol, using 15N-enriched minimal M9 medium (6 g L−1 of Na2HPO4, 3 g L−1 of KH2PO4, 0.5 g L−1 NaCl, 1.25 g L−1 (15NH4)2SO4, 0.246 g L−1 of MgSO4, 5 g L−1 of glycerol and 4 g L−1 of glucose). The cells were harvested by centrifugation at 8,000 × g for 20 min at 4 °C. The cellular pellet was re-suspended in 15 – 20 mL of buffer containing 50 mM HEPES pH 7, 0.5 M NaCl, 0.2 mM tris(2-carboxyethyl)phosphine (TCEP), 5 % w/v glycerol, 0.5 % w/v CHAPS, 10 mM MgCl2 and 20 μg mL−1 of DNase I. After incubation at 37 °C for 15 min, the cells were disrupted by two passages through a French Pressure cell (SLM, Aminco) at 20,000 pounds/square inch. The cell pellet was separated from the supernatant by centrifugation at 20,000 × g for 20 min at 4 °C. The soluble fraction, containing the recombinant protein, was loaded onto a column containing 5 mL of Ni-NTA affinity resin pre-equilibrated with 5 volumes of 50 mM HEPES pH 7, containing 0.5 M NaCl, 0.2 mM TCEP, 5 % w/v glycerol, 0.5 % w/v CHAPS, and 50 mM imidazole. The column was washed with five volumes of the same buffer not containing CHAPS, and the His-tagged HpUreF was eluted with 0.5 M imidazole. After elution, the protein solution was dialyzed twice for 3 hours at 4 °C against 3 L of 10 mM TrisHCl at pH 8.3, containing 0.15 M NaCl, 0.1 mM TCEP and 10 % w/v glycerol, in order to remove imidazole and thus obtain the optimal buffer conditions for the subsequent enzymatic reaction. The His-tag was removed by incubating the protein with three units of thrombin protease (Sigma) per mg of protein at 20 °C for 4 h. The sequence of thrombin-cleaved HpUreF is expected to retain the additional GSH sequence at the N-terminus. The uncleaved protein was removed by incubating the protein samples at room temperature for 20 min in the presence of 100 μL Ni-NTA affinity resin and then centrifuged at 4,000 × g for 15 min. In order to remove thrombin protease, the supernatant was collected and incubated at room temperature for 20 min with benzamidine agarose resin (Sigma). The resulting slurry was centrifuged at 4,000 × g for 15 min, the supernatant was collected, and 10 mM EDTA was added. The protein was concentrated using 10 kDa molecular weight cut-off Amicon ultra-filtration units (Millipore UFC801024) until reaching a final volume of 2 mL. The sample was then eluted using a size-exclusion chromatography using a Superdex 75 XK 16/60 column (GE Healthcare) in 10 mM TrisHCl buffer at pH 8.0, containing 0.15 M NaCl and 0.2 mM TCEP. This final purified protein (ca. 15 mg per liter of culture) was concentrated to 1 mg mL−1 using Amicon Ultrafiltration units (Millipore), and stored at −80 °C. The purification of the protein mutants followed exactly the same protocol. The yield of protein expression, the size exclusion elution profiles, and the protein stability over time did not change comparing the wild type and the mutated proteins. The thermal stability of the wild type and double mutant was determined using differential scanning fluorimetry experiments in triplicate (Thermofluor, [36] see Supplementary Information). In each purification step, the purity of wild type and mutant HpUreF proteins, as well as its molecular mass under denaturing conditions, was assessed by electrophoresis on NuPAGE Novex Pre-Cast Gel System (Invitrogen) with NuPAGE 4 – 12 % Bis-Tris gels. The identity of the purified HpUreF, as well as its molecular mass, were demonstrated by ESI-QTOF mass spectrometry. The absence of metal ions bound to the purified proteins was confirmed by inductively coupled plasma emission spectroscopy (ICP-ES), using a procedure previously described [37]. Protein concentration was estimated using the theoretical extinction coefficient (ε280= 16,390 M−1 cm−1) calculated from the amino acid sequence using the ProtParam web site (http://web.expasy.org/protparam/). The concentration of the protein is expressed as protein dimer throughout the manuscript.
Isothermal titration calorimetry
Ni2+ binding titrations of wild type and mutant HpUreF proteins were performed at 25 °C using a high-sensitivity VP-ITC micro-calorimeter (MicroCal LLC, Northampton, MA, U.S.A.). Immediately before each ITC measurement, the protein sample was eluted through a Superdex S75 10/300 GL (GE Healthcare) size-exclusion chromatography column using a 10 mM TrisHCl pH 8.0 buffer containing 0.15 M NaCl and 0.2 mM TCEP as eluent. The same buffer was used to dissolve the nickel salt. The reference cell was filled with deionized water. Before each experiment the baseline stability was verified. An interval of 300 – 400 s was applied between the injections to allow the system to reach thermal equilibrium. Control experiments were carried out by titration of the metal ion solution into the buffer alone, under identical conditions, and the heat of dilution was negligible. The solution containing HpUreF (25 μM) or its mutants (15 – 20 μM), loaded into the sample cell (1.4093 mL), was titrated with 55 × 5 μL injections of a solution containing 500 – 750 •M NiSO4. The data were processed with the Origin software package (MicroCal), and fitted using a non-linear least-squares minimization algorithm to theoretical titration curves that involved different binding models. The reduced chi-square parameter χ2ν (χ2ν = χ2/N, where N is the degrees of freedom, N =Nidp − Npar; Nidp = number of points, Npar = number of parameters floating in the fit) was used to establish the best fit among the tested models. Values for the enthalpy change of the reaction (ΔH), the binding affinity constant (Kd), and the number of sites (n) were parameters of the fit. The reaction entropy was calculated using ΔG = − RT lnKd (R = 1.9872 cal mol−1 K−1, T = 298 K) and ΔG = ΔH − TΔS. The values given for ΔH and ΔS are apparent, and include contributions not only from metal binding but also from associated events such as protonation/deprotonation of the amino acid residues involved in the binding and consequent change in the buffer ionization state. Indeed, the present study does not have the goal of determining the absolute thermodynamic parameters for Ni2+ binding to HpUreF, but rather to determine the equilibrium constant and stoichiometry in comparison with binding events involving HpUreG and HpUreE, determined by some of us in previous studies [18, 33] using the same conditions and protocols.
Light scattering measurements
The oligomerization state and the hydrodynamic radius of HpUreF in the absence and presence of Ni2+ were determined using a combination of size-exclusion chromatography (SEC), multiple-angle light scattering (MALS) and quasi-elastic light scattering (QELS). HpUreF (500 μL, 35 μM, in the absence and in the presence of 70 μM NiSO4 in the incubation and elution buffer) was diluted in 10 mM TrisHCl pH 8.0, containing 0.15 mM NaCl and 0.2 mM TCEP. The samples were loaded onto a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with the same buffer and eluted at the flow rate of 0.6 mL/min. The column was connected downstream to a multiple-angle laser light (690.0 nm) scattering DAWN EOS photometer (Wyatt Technology) and to a quasi-elastic light scattering apparatus (WyattQELS). The specific refractive index increment (dn/dc) for the protein was taken as 0.185 mL/g [38]. The value of 1.331 for the solvent refractive index and the concentration of the eluted protein were determined using a refractive index detector (Optilab DSP, Wyatt). Molecular weights were determined from a Zimm plot. Data were processed using the Astra version 5.1.7 (Wyatt Technology), following the manufacturer’s instructions.
Circular dichroism spectroscopy
The secondary structure content of HpUreF was estimated by circular dichroism (CD). Apo-HpUreF (2.5 μM) was diluted in i) 10 mM TrisHCl pH 8.0 containing 0.15 M NaCl and 0.2 mM TCEP, or ii) 20 mM phosphate buffer pH 8.0, in absence and in the presence of two equivalents of NiSO4 per protein dimer. Only in the case of phosphate buffer could the low wavelength range be used for quantitative estimation of secondary structure content. The CD spectra were recorded at room temperature using a JASCO 810 spectropolarimeter flushed with N2 and a cuvette with 0.1 cm path-length. The spectra were registered from 185 to 250 nm (in phosphate buffer) or from 200 to 250 nm (in TrisHCl buffer) at 0.2 nm intervals. The spectra obtained were corrected for the spectrum of the buffer by subtraction. The secondary structure composition of HpUreF was evaluated using the CSSDTR algorithm available at the Dichroweb server (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml) and the best t determined through the NRMSD (normalized root mean square deviation) parameter.
NMR spectroscopy experiments
Uniformly 15N – labeled HpUreF (250 – 500 •M in 10 mM TrisHCl pH 8.0, containing 0.15 M NaCl and 0.2 mM TCEP) was used to carry out NMR experiments on the protein in its apo form. NMR spectra were acquired at 298 K with a SGU-based Bruker Avance 900 MHz spectrometer operating at the proton nominal frequency of 899.07 MHz and equipped with a 5-mm TCI cryo-probe with a shielded z-axis pulse field gradient. The Bruker pulse sequence trosyetf3gpsi2 [39, 40] was used to obtain phase-sensitive gradient-enhanced sensitivity-improved 1H-15N TROSY-HSQC spectra, with gradient selection using the f3-channel. In this pulse scheme, water suppression is achieved via coherence pathway rejection. The spectra were acquired with a relaxation delay of 2.0 s and an acquisition time of 390 ms. A typical acquisition consisted of 144 scans per each FID for a matrix of 2048×256 complex points. Spectral widths of 11.6 ppm, with the carrier at 4.77 ppm for the 1H dimension, and of 40.0 ppm, with the carrier at 118.58 ppm for the 15N dimension, were used. Data were processed using the iNMR software (www.inmr.net). Linear prediction was used in the 15N dimension to improve digital resolution. Data were zero-filled to 2048 × 512 points and weighted using a squared sine wave shifted by 60°.
X-ray absorption spectroscopy
Two protein samples containing HpUreF (0.39 mM in 10 mM TrisHCl pH 8, 150 mM NaBr, 0.2 mM TCEP) were prepared by adding 0.9 equivalents of NiSO4 per protein dimer. After metal addition, the sample was incubated for five minutes, loaded into sample cells, frozen in liquid nitrogen and stored at 77 K until data collection.
XAS data were collected at SSRL (Stanford Synchrotron Radiation Lightsource, 3 GeV ring) on beam line 7-3 equipped with a Si(220) (phi = 0°) double crystal monochromator, a liquid helium cryostat for the sample chamber and a 30-element Ge X-ray fluorescence detector array. Scattering was reduced by placing a Söller slit with a 3 μm Z-1 element filter between the sample and the detector. Internal energy calibration was performed by collecting spectra simultaneously in transmission mode on a nickel foil.
The SixPack software was used to average the data and perform calibrations [41]. Energy calibration was achieved by setting the first inflection point from the XANES spectral region to 8,331.6 eV for nickel foil. The Athena software, using the AUTOBK algorithm, was employed for data reduction and normalization [42]. A linear pre-edge function and a quadratic polynomial for the post-edge data were used for background subtraction, followed by normalization of the edge-jump.
The Ni2+ XANES spectrum of Ni-HpUreF was fit over the 8280–8355 eV range, using the PeakFit v4.12 software package [43]. Pseudo-Voigt area functions were used for the absorption features, and a cumulative Gaussian function was used for the edge jump.
The Ni2+-HpUreF EXAFS data was extracted using an Rbkg of 0.8, a spline from k = 2 to 12.5 Å−1 with a rigid clamp at high k-values. The k3-weighted data were fit in the r-space over the k = 2 – 12.5 Å−1 range, with E0 for Ni set to 8340 eV. The data set was processed using a Kaiser-Bessel window with a dk = 2 (window sill).
The Artemis software, which uses the FEFF6 and IFEFFIT algorithms, was used to generate and fit scattering paths to the data [42, 44, 45]. Fits utilizing both single-scattering and multiple-scattering pathways were performed as previously described [34, 46] and illustrated in subsequent sections, using the general EXAFS equation below [47]:
Histidine ligands were fit as rigid imidazole rings, constructed using average values and bond lengths obtained from crystallographic data [48]. The distance of the imidazole ring from the metal center was fit in terms of the metal-to-ligand bond distance (Reff) and a rotation angle α, as previously described [49, 50]. To assess the goodness-of-fit from different fitting models, the parameters χ2, reduced χ2 (χ2ν) and R-factor were minimized. Increasing the number of adjustable parameters is generally expected to improve the R-factor; however χ2ν may go through a minimum then increase indicating that the model is over-fitting the data. These parameters are defined as follows:
and
Nidp is the number of independent data points defined as:
Δr is the fitting range in r-space••Δk is the fitting range in k-space, Npts is the number of points in the fitting range, Nvar is the number of variables floating during the fit••ε is the measurement of uncertainty, Re() is the real part of the EXAFS Fourier-transformed data and theory functions, Im() is the imaginary part of the EXAFS Fourier-transformed data and theory functions••χ(Ri) is the Fourier-transformed data or theory function, and
Urease activation assay in the cellular milieu
E. coli BL21(DE3) cells were freshly transformed with the wild type HpUreF, H229A, C231A and H229A/C231A mutant pGEM::ureOP plasmids. For each clone, cultures were performed in duplicate from two different colonies of the same bacterial transformation. The bacterial cells were cultured at 37 °C in 40 mL of LB, supplemented with 100 μg/mL ampicillin and 0.5 mM NiSO4 and 0.5% glucose (w/v). Protein expression was induced by adding 0.5 mM IPTG when OD600 reached 0.5. Cells were harvested by centrifugation after 16 hours from induction and re-suspended in 2 mL of 20 mM TrisHCl pH 7.5, containing 100 mM NaCl, 10 mM MgCl2 and 20 μg/mL DNase. Cellular lysis was performed using 25 mg/mL CelLytic Express (Sigma-Aldrich). The cell pellet was separated from the soluble fraction by centrifugation at 20,000 rpm for 20 min at 4 °C. The total protein concentration in the soluble fraction was determined using the Bradford reagent [51] and used to normalize the measured urease activity. Specific urease activity was quantitatively determined at 25 °C using the pH-stat method, as previously reported [52, 53]. Measurements were performed twice on the same cellular extract and the results were averaged. One unit of urease activity is defined as the amount (mg) of enzyme capable of hydrolyzing 1 μmol of urea per minute. The reaction mixture (10 mL) contained 1 – 2 mL of cellular extract and 200 mM urea. A negative control, performed using a cellular extract of non-recombinant BL21 (DE3) cells, showed the absence of detectable activity.
Analysis of metal transport pathways in the UreG2-UreF2-UreD2 complex from H. pylori
The CAVER 3.0 [54] program was used for calculating all pathways departing from HpUreG Cys-Pro-His motif region within the structure of the UreG2-UreF2-UreD2 complex from H. pylori strain 26695 (PDB: 4HI0) [55]. All water molecules were removed before the tunnel calculation. The starting point of the tunnel search was calculated as the average position between the HpUreG Cys66 Sγ atoms from each HpUreG monomer. The tunnel search was performed using a probe of 0.9 Å radius. The identified tunnels were clustered by hierarchical average link clustering based on the pairwise distances of the tunnels computed. The results were analyzed using the UCSF CHIMERA package [56].
RESULTS AND DISCUSSION
Purification of HpUreF
The purified HpUreF was analyzed by mass spectrometry, which typically showed the presence of a major species in solution that corresponds to the sequence of HpUreF with the initial GSH fragment, but lacking the C-terminal 21 residues. The same C-term truncation was previously reported in two independent studies [27, 31], indicating an intrinsic propensity of this region of HpUreF to be lost during expression and purification. Consistently, according to ProtParam (http://web.expasy.org/protparam/) the C-terminal truncated form of the protein is stable, as opposed to the full protein (with, or without, the GSH fragment), or to the truncated form without the GSH, all of which are predicted to be unstable due to hydrolysis. During initial attempts of protein purification, the mass spectrum of the protein showed an additional peak interpreted as due to the presence of a Ni2+ ion bound to the truncated form of HpUreF, possibly derived from the first purification step of the His-tagged HpUreF that involved affinity chromatography on a Ni2+-loaded resin. This observation suggested that in solution HpUreF is able to bind Ni2+, and that this property is maintained after His-tag removal. Before any subsequent analyses, the wild type protein obtained following the His-tag digestion, as well as its single and double mutants, were extensively treated with EDTA to remove any bound metal ions. EDTA was removed in the last size-exclusion purification step, giving the fully metal-voided proteins, as confirmed using ICP-ES analysis.
HpUreF nickel binding properties
The capability of HpUreF to tightly bind Ni2+ was confirmed by the presence of exothermic peaks following each injection in isothermal titration calorimetry (ITC) experiments (Figure 2A). Fits of the integrated heat data (Figure 2B), carried out using a simple model entailing a single binding event, yielded a stoichiometry of two Ni2+ bound per HpUreF dimer, with a dissociation constant Kd(Ni) = 6.4 ± 0.4 μM, a negative enthalpy ΔH(Ni) = −5.4 ± 0.1 kcal mol−1 and a positive entropy ΔS(Ni) = +5.5 cal mol−1 K−1.
Figure 2.

Representative plots of titration data showing the thermal effect of 55 × 5 μL injections of Ni2+ onto a solution of HpUreF. In A, raw heat data are shown for wild type HpUreF. In B, integrated heat data of wild type HpUreF (black filled circles), H229A HpUreF (blue filled circles) mutant, C231A HpUreF mutant (red filled circles) and H229A/C231A HpUreF double mutant (hollow circles) are indicated together with the best fits (solid lines).
The observed binding affinity for Ni2+ by HpUreF is similar to that reported for H. pylori HypA (Kd = 3.7 μM at pH 8.3 from ITC data [57]), HypB (Kd = 0.15 μM at pH 7.6 from competition experiments [58]), and SlyD (Kd = 2.7 μM at pH 7.6 from equilibrium dialysis [59]), metallochaperones important in Ni delivery to [Ni,Fe]-hydrogenase and urease, as well as H. pylori UreE (Kd = 0.15 μM at pH 7.0 [33]) and UreG (Kd = 10.0 μM at pH 8.0 [18]), proteins involved in urease maturation, both measured by ITC. This observation supports the idea that Ni2+ is shuffled among a battery of protein transporters that obtain the metal ion from exchangeable cellular pools in which metal concentration is regulated by related metallosensors [60, 61].
Solution structural properties of HpUreF and nickel binding
Structural information on HpUreF is available only from solid-state crystallographic data [27, 31]. We investigated the fold of HpUreF in solution, and examined whether Ni2+ binding influences protein secondary, tertiary and quaternary structure, using circular dichroism, NMR spectroscopy, and light scattering.
The CD spectrum of apo-HpUreF (Figure 3A) shows the prevalence of α-helices, with negative deflections around 222 nm and 208 nm and a positive peak at 190 nm. The quantitative analysis of the spectrum (NRMSD = 0.009) estimated a secondary structure composition of 69% α-helix, 8% β-strand, 7% turns, and 17% random coil. This composition is similar (62% α-helix, 8% turns, and 30% coil) to that calculated by STRIDE [62] using the crystallographic structure of isolated apo-HpUreF [31]. The CD spectrum of the protein is not significantly affected by the addition of two equivalents of Ni2+ per protein dimer, indicating that nickel binding does not change the secondary structure of the protein (Figure 3A). The same result was obtained using either phosphate or TrisHCl buffers, independently of the presence of TCEP.
Figure 3.

(A) Far-UV CD spectrum of HpUreF in 20 mM phosphate buffer in the absence (hollow circles) and in the presence of two equivalents of Ni2+ (hollow squares) per protein dimer. The best fit calculated for apo-HpUreF is represented as a solid line. Mean residue ellipticity units are degrees cm2 dm−1 residue−1; (B) Representative 1H-15N TROSY-HSQC spectrum of apo-HpUreF acquired at 900 MHz and 298 K; (C) Plot of the molar mass distribution of HpUreF in the absence (thick dots and line) and in the presence (thin dots and line) of two equivalents of Ni2+ per protein dimer. The solid lines indicate the Superdex-75 elution profiles monitored by the refractive index detector, while the dots are the weight-averaged molecular masses for each slice, measured every second.
The 900 MHz 1H-15N TROSY-HSQC NMR spectrum of apo-HpUreF provided information on the structural features of the protein backbone. The spectrum shows a number of signals consistent with the number of amino acids in the protein sequence (Figure 3B), suggesting the presence of a single conformational form of the protein in solution, or of a protein that undergoes conformational equilibria with sub-millisecond inter-conversions among various possible conformers. The presence of essentially all the expected signals further indicates that the protein is well folded in solution, a result confirmed by the overall dispersion of 1H chemical shifts, spanning the 5.5–9.5 ppm range. The line width of the observed signals varies, indicating that different parts of the protein undergo backbone conformational changes at different rates. Interestingly, in the 7.6–8.0 ppm range of the 1H dimension and in the 125.5–132.5 ppm range in the 15N dimension it is possible to observe ca. 20 peaks with small line widths and narrow dispersion. The number and properties of these peaks suggest that they belong to the 21-residue C-terminal portion, which is ordinarily quickly lost when the protein is prepared from bacteria grown in rich LB medium. In the case of the NMR samples prepared using minimal M9 medium, the presence of these peaks was often observed, with their intensity varying from one preparation to another. This is consistent with SDS-PAGE gels of the NMR samples, which showed the presence of two bands corresponding to the protein cleaved and uncleaved at the C-terminus, and suggests that the C-terminal portion of HpUreF is quickly removed by the higher proteolytic activity of cells grown in rich media, probably because of the larger amount of protease-activating metal ions. On the other hand, the C-terminal portion of recombinant HpUreF more frequently survives the preparation in minimal medium and is slowly hydrolyzed to varying degrees during NMR sample preparation and data collection. The NMR spectrum does not show any significant variation in the presence of Ni2+, suggesting a diamagnetic, low-spin Ni2+ site (four-coordinate planar or five-coordinate pyramidal geometry) and the absence of large and observable changes of the backbone conformation.
The crystal structures of HpUreF, obtained in two independent studies [27, 31], consistently show the protein in a dimeric form in the solid state. However, the physiological relevance of this oligomeric form in the urease activation process has been questioned on the basis of small-angle X-ray scattering data obtained on K. aerogenes proteins, which were interpreted as suggesting the presence of a (UreABCDF)3 complex that contrasts with a functional UreF dimerization [25]. The oligomeric state of isolated apo-HpUreF in solution has been previously investigated using multi-angle light scattering (MALS) in line with size-exclusion chromatography (SEC) [27]. In those experiments, HpUreF was eluted as a single peak with a molecular mass (43 kDa), a value close to, but lower than, the mass estimated for the truncated dimer (53 kDa), suggesting that the protein exists primarily as a dimer in solution in rapid equilibrium with a small amount of monomer. In the same study, and in agreement with the crystal structure, the complex of HpUreD and HpUreF was eluted as a single peak with a molecular mass consistent with that of the HpUreF2-HpUreD2 hetero-tetramer [27].
In order to investigate whether nickel binding affects the quaternary structure of isolated HpUreF, we performed a SEC-MALS-QELS experiment in the absence or presence of two equivalents of Ni2+ per protein dimer. For the apo-protein, a molecular mass of 52.8 ± 0.1 kDa and a hydrodynamic radius of 3.2 ± 0.1 nm were measured for the protein eluted from the column in a single peak (Figure 3C). This value corresponds to that predicted for the dimeric protein. In the presence of two equivalents of Ni2+ ions per protein dimer, a single peak eluted with the same retention volume and similar molecular mass as in the case of the apo-protein (Figure 3C). These results, together with the NMR results that show that no change in fold or oligomeric state occurs upon addition of Ni2+, indicate that the presence of bound Ni2+ does not affect the oligomeric state of the HpUreF dimer.
Identification of nickel binding residues using X-ray absorption spectroscopy
In order to identify the metal coordination geometry in Ni2+-bound HpUreF and the type of residues involved in metal binding, we employed X-ray absorption spectroscopy (XAS). The XANES spectrum of HpUreF incubated with Ni2+ exhibits two pre-edge features (Figure 4A) at 8331.8 eV and at 8337.5 eV that are associated with 1s→3d and 1s→4pz electronic transitions, respectively. The intensity of the 1s→3d transition (peak 1, 0.11 ± 0.02 eV) falls outside the range typically observed for centrosymmetric coordination geometries, while the distinct shoulder corresponding to a 1s→4pz transition (peak 2, 0.90 ± 0.10 eV) is consistent with tetragonal geometries missing one or both axial ligands (see Table SI-1 for full fitting parameters). Overall, the XANES spectra of Ni-HpUreF are consistent with the presence of a five-coordinate pyramidal geometry for the nickel center [43]. This is consistent with NMR-based data suggesting the presence of diamagnetic Ni2+.
Figure 4.

HpUreF Ni K-edge XAS. (A) XANES spectrum; the inset reports the data (circles) vs. the fitted sum (line) from the individual XANES spectrum pre-edge features (peaks 1 to 5; see SI-1). (B) Fourier-transformed EXAFS spectrum (circles) and best fit (line), shown without phase correction (FT window = 2–12.5 Å−1). Inset: k3-weighted unfiltered EXAFS spectrum and best fit. The fit shown is for the (N/O)1(Namide)1(NHis)1(OCO2)2 model from Table 1.
The Fourier-transformed EXAFS spectrum of Ni-HpUreF has an intense feature at r = 1.5 Å, suggesting that a single shell of N/O-donor ligands is present (Figure 4B). However, single-scattering fits show the presence of at least two shells of N/O-donor ligands, which may account for the broadness of the main feature in r-space. Fits that include sulfur or bromide atoms in the first coordination shell yield unreasonable bond distances or large σ2 values. Large σ2 values result when a dampening of the EXAFS amplitude in the fitting equation is required by the fit, indicating that even incorporating 1 S/Br atom in the model is too much. The intensity found in the 2 – 4 Å range of the Fourier-transformed spectrum is due to the presence of multiple-scattering from ligands such as histidine imidazoles. Scattering paths for histidine imidazole ligands were generated and fit using a rigid model, as previously described [34, 46]. These fits suggest the presence of two shells of scattering atoms consisting of a combination of histidine imidazole ligands and other N/O-donors (Tables SI-2 and SI-3). The best fits are for a five-coordinate nickel center, and are consistent with the XANES analysis (Table 1). The models show contributions from one or two histidine ligands. Even though the model consisting of two different shells of N/O ligands and a single histidine imidazole has a slightly better goodness of fit than the analogous model with two histidine ligands, the single histidine model has a rather small value of σ2 (Table 1). Considering our fitting model, this indicates that the intensity of the Fourier-transformed spectrum in the 2 – 4 Å range, is not fully represented by multiple-scattering from a single histidine. However, the geometry determined from the XANES region of the spectrum might offer a clue. We have previously observed five-coordinate Ni binding sites in proteins, such as Ni superoxide dismutase (NiSOD), where the metal center is coordinated in a chelating manner by a histidine and an amide nitrogen [63], which might account for the amplitude achieved from a two-histidine model (Table 1). Such a fit does indeed give comparable statistical values to those containing one or two histidine ligands with the added benefit of more appropriate σ2 values, and is consistent with the mutation studies (vide infra) that suggest that only one histidine is present in the UreF metal binding site. As the Ni binding site in UreF is, according to our mutational studies, in a region rich in aspartic and glutamic acid residues, we looked at improving the fits by adding contributions from carboxylate groups (Figure 4B, Table 1). Despite the additional fitting parameters for the fits with a carboxylate group, a drop in the χ2ν value by a factor of ~1.7 make them statistically relevant. These follow-up fits strongly suggest the presence of one or two coordinating carboxylate groups. Again the difficulty in modeling the amide scattering in the (N/O)1(Namide)1(NHis)1(OCO2)2 model alludes to a well structured metal binding site with significant multiple-scattering interactions. Due to the error associated with EXAFS fits and the difficulty in distinguishing between N- and O- donors, the best description for the structure of the HpUreF nickel site that is consistent with the mutagenesis studies is a penta-coordinated Ni2+ ion with four N/O-donors at 1.8–1.9 Å, and presumably a longer apical ligand at 2.16 Å. The coordination shell consists of a histidine, most probably binding the nickel center in a chelating manner involving one amide nitrogen, similar to NiSOD, and one or two carboxylate-like groups, possibly from nearby aspartic and glutamic acid residues. Site-directed mutagenesis studies outlined below support a Ni binding site that contains one histidine, which lends support to the EXAFS multiple-scattering models involving only one histidine ligand. To the best of our knowledge, this is the first example of a biological five-coordinate low-spin Ni2+ site without a S-based ligand [64].
Table 1.
EXAFS determined Ni-ligand bond distances for HpUreF
| Ligand | R (Å) | σ2 (×103 Å2) | %R | χ2ν |
|---|---|---|---|---|
|
(N/O)1(N/O)3(NHis)1
| ||||
| 1 N/O | 2.18(4) | 9(2) | 5.81 | 10.8 |
| 3 N/O | 1.88(1) | 9(2) | ||
| 1 NHisa | 1.88(1) | 0(1) | ||
|
| ||||
|
(N/O)1(N/O)2(NHis)2
| ||||
| 1 N/O | 2.17(3) | 6(3) | 5.88 | 10.9 |
| 2 N/O | 1.89(1) | 6(3) | ||
| 2 NHisa | 1.89(1) | 4(2) | ||
|
| ||||
|
(N/O)1(N/O)1(Namide)1(NHis)1(NHis)1
| ||||
| 1 N/O | 2.18(2) | 4.3(5) | ||
| 1 N/O | 1.89(1) | 1.7(6) | 5.80 | 11.2 |
| 1 NHisa | 1.89(1) | 1.7(6) | ||
| 1 NHisb | 1.97(1) | 1.7(6) | ||
| 1 Namide | 1.82(1) | 1.7(6) | ||
|
| ||||
|
(N/O)2(NHis)2(OCO2)1
| ||||
| 2 N/O | 1.89(1) | 8(3) | 3.67 | 7.2 |
| 2 NHisa | 1.89(1) | 2.2(8) | ||
| 1 OCO2 | 2.16(2) | 6(2) | ||
|
| ||||
|
(N/O)1(Namide)1(NHis)1(OCO2)2
| ||||
| 1 N/O | 2.15(3) | 4(3) | ||
| 1 Namide | 1.95(2) | 0(2) | 2.58 | 6.3 |
| 1 NHisb | 1.84(3) | 5(3) | ||
| 2 OCO2 | 1.87(1) | 2(1) | ||
The histidine is bound through the ε nitrogen with α of 10°
The histidine is bound through the δ nitrogen with α of 0°
Identification of nickel binding residues by site-directed mutagenesis
The analysis of the XAS data reveals details of the metal binding site for Ni-HpUreF. The Ni2+ complex shows no contributions from either cysteine ligands or bromide derived from the buffer, and is consistent with a Ni(N/O)5 center with one or two histidine ligands and no solvent exposed sites. Based on the crystal structure of dimeric apo-HpUreF protein [27, 31] (Figure 5), we initially focused our attention on the pair of His180 residues, located in the highly conserved dimer interface, as possible Ni2+ ligands, and addressed the role of these residues in metal binding using the H180A-HpUreF mutant protein. H180A-HpUreF binds Ni2+ ions with a substantially identical calorimetric curve to the wild-type protein (Figure SI-1A), indicating that the pair of His180 residues are not ligands in the nickel site. In particular, a fit of the thermodynamic data (Figure SI-1B) showed that the mutant protein binds two Ni2+ ions per protein dimer with dissociation constant Kd(Ni)H180A= 5.9 ± 0.6 μM, ΔH(Ni)H180A= −5.3 ± 0.2 kcal mol−1 and ΔS(Ni)H180A = + 6.2 cal mol−1K−1.
Figure 5.
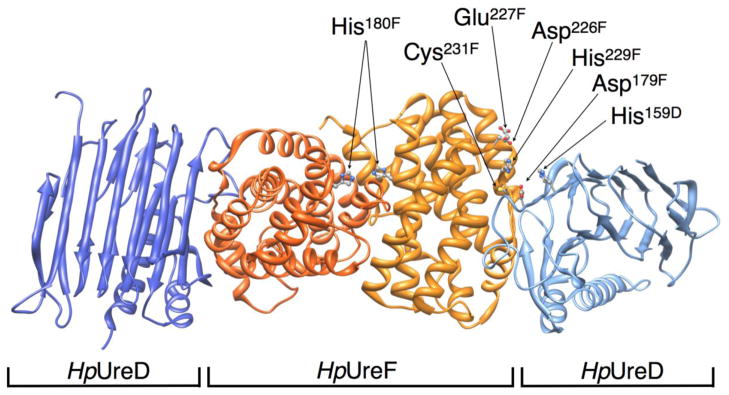
Ribbon scheme of the H. pylori UreD-UreF complex (PDB code 3SF5), highlighting His180, His229, Cys231 on UreF and the nearby residues potentially involved in metal binding and trafficking. UreF monomers are depicted in dark and light orange, UreD monomers in dark and light blue.
The crystal structure of apo-HpUreF [27, 31] further reveals a pair of closely spaced and surface-exposed His229 and Cys231 residues, potentially constituting a nickel-binding site, similar to that established for HpUreG [18]. In order to verify the involvement of these putative ligands in nickel binding, they were mutated to alanine, and the metal binding properties of the single and double mutants were determined by ITC. In the H229A/C231A-HpUreF double mutant, Ni2+ binding was substantially abolished (Figure 2B). The fact that mutation of H229 and C231 is sufficient to fully abrogate Ni binding to HpUreF indicates that the histidine present in the GSH sequence retained at the N-terminus after removal of the His-tag is not involved in Ni binding to the protein. This observation prompted us to dissect the exact role of His229 and Cys231 for Ni2+ binding by producing the two single mutants H229A-HpUreF and C231A-HpUreF and titrating Ni2+ into the mutant protein solutions (Figure 2B). When His229 is mutated to alanine, Ni2+ binds the protein (n = 1.5 ± 0.1) with lower affinity (Kd,H229A= 8.1 ± 0.5 μM) with respect to the wild-type protein and with similar enthalpic and entropic factors (ΔHH229A= −4.6 ± 0.1 kcal mol−1; ΔSH229A = +7.9 cal mol−1K−1). This result, coupled with the total abolishment of the Ni2+ binding capability of the double mutant H229A/C231A-HpUreF, suggests that another nearby residue compensates the mutation of His229. This residue is located in a protein region that is rich in aspartic acid residues, consistent with the multiple scattering features in the EXAFS spectrum of Ni-HpUreF interpreted as arising also from carboxylate ligands. When Cys231 is mutated to alanine, Ni2+ binding occurs with similar affinity (Kd,C231A= 12.2 ± 0.1 μM), but with different stoichiometry (n= 0.4 ± 0.1) and thermodynamic factors as compared to the wild type, and in particular with a unfavorable entropy (ΔSC231A = −27.7 cal mol−1 K−1) compensated by a largely negative enthalpy of binding (ΔHC231A= −14.9 ± 0.3 kcal mol−1). XAS data are consistent with the presence of at least one histidine residue, most likely His229, in the Ni2+ coordination environment, while they are inconsistent with sulfur binding to the metal ion. Taken together, these observations indicate that Cys231, although not being a direct Ni2+ binding residue, is important to maintain the integrity of the metal binding site. Considering the structural vicinity of the two residues, this could in principle be achieved through H-bonding interactions between the thiol group of the cysteine and the non-coordinating nitrogen atom of the metal binding histidine imidazole ring.
Function of metal binding residues for HpUreF activity in the cellular milieu
In the recently published structure of HpUreF-HpUreD complex, [27] the His229 residue on HpUreF, established in this study as a Ni2+ binding ligand, faces His159 in HpUreD (Figure 5), suggesting that this protein region is functionally important for metal trafficking, possibly by being involved in protein-protein interactions or in Ni2+ delivery to the urease active site. For this reason, the functional role of His229 and Cys231 was explored by testing the effect of the mutation of these residues on the urease activation process in the cellular milieu (Figure 6). This experiment was performed by transforming and culturing E. coli BL21(DE3) cells containing a novel plasmid (pGEM::ureOP), produced by cloning the 6.1 kbp urease operon by PCR on the H. pylori genome, and inserting the PCR fragment into a commercial vector (Figure 6A). The operon was inserted in the same orientation of the T7 promoter found in the vector, allowing urease overexpression to be induced by IPTG in bacteria grown in the presence of nickel in the medium. The urease activity produced in the soluble fraction of the cellular extract was quantitated using the pH-stat method [52, 53]. Site-specific HpUreF mutation was performed on the pGEM::ureOP plasmid and urease activities of the strains carrying the plasmid with the single mutations H229A-HpUreF and C231A-HpUreF, as well as the double mutation H229A/C231A-HpUreF, was similarly analyzed and compared to that of the wild-type pGEM::ureOP strain. A similar protocol was applied to the non-recombinant BL21(DE3) strain as a negative control, which showed undetectable urease activity. In the case of cells transformed with wild type pGEM::ureOP, a significantly higher urease activity was detected as compared to the case in which the bacteria were transformed with the mutant plasmids containing the single or double mutants H229A, C231A and H229A/C231A, for which the activity represents only ca. 20–25% of that observed for the wild type (Figure 6B). These data demonstrate that mutation of either or both His229 and Cys231 significantly decreases urease maturation in the cellular milieu.
Figure 6.

(A) Scheme of the pGEM::ureOP plasmid containing the 6.1 kbp H. pylori urease operon utilized for in cell studies; (B) Urease activity measured for the soluble cellular extract of E. coli BL21(DE3) cells transformed with the pGEM::ureOP plasmid and with its H229A-HpUreF, C231A-HpUreF and H229A/C231A-HpUreF mutants. The enzymatic activities, measured in duplicate on two different cultures, are normalized to the total protein content of bacterial lysate.
Significance of nickel binding to UreF for urease activation
The observation of decreased urease activation in the cellular milieu for the single and double mutants of His229 and Cys231 could be due either to the disruption of the nickel trafficking mediated by this binding site in the UreF-UreD complex, or to an alteration of the structural properties of the protein-protein interaction surface independent of metal binding. The latter can be largely excluded on the basis of differential scanning fluorimetry assay (Thermofluor)[36], which indicated a Tm of 55.3 °C and 54.8 °C for the wild type and the double mutant, respectively (Figure SI-2). After the submission of this article, the crystal structure of the apo UreG2-UreF2-UreD2 complex from H. pylori 26695 strain was released (PDB: 4HI0) [55]. The UreF2-UreD2 portion of the new complex is nearly identical to that observed in the crystal structure without HpUreG [27]. The latter protein, known to exist in a largely disordered ensemble of structures in solution in the absence of its partner proteins [16–19, 65, 66] was observed in a folded state with a secondary and tertiary structure, position of guanine phosphate cofactor and oligomerization state that are essentially identical to the models of folded UreG from different sources previously calculated on the basis of homology modeling [16–19, 46, 66] (overall root mean square deviation of Cα positions = 1.9 Å). The HpUreF region observed to interact with the HpUreG dimer in the crystal structure is in good agreement with that proposed on the basis of mutagenesis studies performed on KaUreG [32]. On the other hand, the HpUreG region that interacts with HpUreF in the solid state was in fact proposed to interact with HpUreE using computational studies based on calorimetric and NMR experimental data obtained in solution [33]. In the crystal structure, the conserved metal binding Cys-Pro-His (CPH) motif, observed to interact with the metal binding site of HpUreE in solution, is instead found in close contact with the dimerization interface of HpUreF. This difference may be reconciled by suggesting a change of partner by UreG, which could interact with UreE as calculated in the theoretical complex in a stage of the process, while making a complex with UreF as observed in the crystal structure in another phase of the metabolic pathway of urease assembly. The crystallized complex features a cavity at the interface between UreF and UreG, containing several inner water molecules that interconnect through a network aligned along the horizontal axis of the UreF2-UreD2 portion of the structure (Figure 7A).
Figure 7.

(A) Longitudinal section of the solvent excluded surface of the apo UreG2-UreF2-UreD2 crystal structure from H. pylori 26695 strain (PDB: 4HI0) [55]. HpUreD, HpUreF and HpUreG chains are colored as in Figure 1. Water molecules are depicted as light blue spheres. (B) Channels departing from the buried CPH motif calculated using the program CAVER 3.0 [53] and the HpUreG2-UreF2-UreD2 crystal structure (PDB: 4HI0) [55]. Protein backbones are reported in cartoons colored as in Figure 1, while the tunnels are reported as light blue spheres. The radius of the sphere is indicative of the thickness of the tunnel. The HpUreG Cys66 residues, as well as HpUreF His229 and Cys231, are reported as balls-and-sticks colored accordingly to atom type. In the bottom panel the structure is rotated by 90° along the horizontal axis. (C) Detail of the position of HpUreF His229 and Cys231 with respect of the calculated tunnel. Protein backbones and the tunnel are colored as in panel B.
This observation prompted us to investigated the presence of channels departing from the buried CPH motif to the solvent in the UreG2-UreF2-UreD2 protein complex, using the program CAVER 3.0 [54], in an effort to discover possible ways for metal ions such as Ni2+ or Zn2+, known to bind to the CPH motif but absent in the crystal structure, to reach a way out into the urease active site. Figure 7B shows the results of this search: two nearly identical and symmetric tunnels were found, going from the central cavity in the complex and exiting in the proximity of HpUreD C-terminal α-helix, passing through HpUreF and HpUreD. The distance through the calculated pathway between the CPH motif and the tunnel end on the HpUreD side is ca. 65 Å. The distance between the ends of the two tunnels is ca. 100 Å. Interestingly, both tunnels pass in close proximity to HpUreF His229 and Cys231 (Figure 7C). This observation based on computational analysis, together with the experimental data obtained in this study, supports a role of UreF in the metal ion transport through these tunnels during urease activation. In particular, the channel surface within HpUreF features several potential metal binding residues (namely Glu55, Glu85, His159) possibly involved in a metal trafficking pathway. This hypothesis is currently under investigation in our laboratories.
Supplementary Material
Acknowledgments
Funding Sources
Portions of this research were performed at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. This work was supported by NIH grant [R01-GM-69696 (MJM)], by Italian PRIN 2009, by the Center for Magnetic Resonance (Italy) and the Consorzio Interuniversitario di Risonanze Magnetiche di Metallo-Proteine (CIRMMP). The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program. FM was supported by Programma Operativo del Fondo Sociale Europeo 2007/2013 of Regione Autonoma Friuli Venezia Giulia (Italy) and by CIRMMP.
Fabio Calogiuri is thanked for acquiring NMR data at the Center for Magnetic Resonance of the University of Florence.
ABBREVIATIONS
- Hp
Helicobacter pylori
- Bp
Bacillus pasteurii
- Ka
Klebsiella aerogenes
- His, H
histidine
- Glu
glutamate
- Asp
aspartate
- Cys, C
cysteine
- Ala, A
alanine
- GTP
Guanosine-5′-triphosphate
- Tris
2-amino-2-hydroxymethyl-propane-1,3-diol
- TCEP
tris(2-carboxyethyl)phosphine
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- ITC
isothermal titration calorimetry
- Kd
equilibrium dissociation constant
- GeV
Giga electron Volt
- XANES
X-ray absorption near-edge spectroscopy
- EXAFS
extended X-ray absorption fine structure
- rmsd
root mean square deviation
- MALS
multiple angle light scattering
- QELS
quasi elastic light scattering
- SAXS
small angle light scattering
- CD
circular dichroism
- NMR
nuclear magnetic resonance
Footnotes
In the case of H. pylori, UreD is traditionally named UreH; in this paper only the UreD denomination is used
XANES data and fits, single- and multiple-scattering EXAFS data and fits for Ni-HpUreF, ITC data and fits for the titration of wild type and mutated HpUreF with Ni2+, Thermofluor assay results.
References
- 1.Polk DB, Peek RM., Jr Nat Rev Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slonczewski JL, Fujisawa M, Dopson M, Krulwich TA. Adv Microb Physiol. 2009;55:1–79. doi: 10.1016/S0065-2911(09)05501-5. [DOI] [PubMed] [Google Scholar]
- 3.Mobley HL, Island MD, Hausinger RP. Microbiol Rev. 1995;59:451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krajewska B. J Mol Catal B-Enzym. 2009;59:9–21. [Google Scholar]
- 5.Jabri E, Karplus PA. Biochemistry. 1996;35:10616–10626. doi: 10.1021/bi960424z. [DOI] [PubMed] [Google Scholar]
- 6.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. Structure Fold Des. 1999;7:205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 7.Ha N-C, Oh S-T, Sung JY, Cha KA, Lee MH, Oh B-H. Nat Struct Biol. 2001;8:505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 8.Balasubramanian A, Ponnuraj K. J Mol Biol. 2010;400:274–283. doi: 10.1016/j.jmb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Balasubramanian A, Durairajpandian V, Elumalai S, Mathivanan N, Munirajan AK, Ponnuraj K. Int J Biol Macromol. 2013;58:301–309. doi: 10.1016/j.ijbiomac.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 10.Ciurli S, Benini S, Rypniewski WR, Wilson KS, Miletti S, Mangani S. Coord Chem Rev. 1999;190–192:331–355. [Google Scholar]
- 11.Callahan BP, Yuan Y, Wolfenden R. J Am Chem Soc. 2005;127:10828–10829. doi: 10.1021/ja0525399. [DOI] [PubMed] [Google Scholar]
- 12.Zambelli B, Musiani F, Benini S, Ciurli S. Acc Chem Res. 2011;44:520–530. doi: 10.1021/ar200041k. [DOI] [PubMed] [Google Scholar]
- 13.Farrugia MA, Macomber L, Hausinger RP. J Biol Chem. 2013;288:13178–13185. doi: 10.1074/jbc.R112.446526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moncrief MB, Hausinger RP. J Bacteriol. 1997;179:4081–4086. doi: 10.1128/jb.179.13.4081-4086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soriano A, Hausinger RP. Proc Natl Acad Sci, USA. 1999;96:11140–11144. doi: 10.1073/pnas.96.20.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zambelli B, Musiani F, Savini M, Tucker P, Ciurli S. Biochemistry. 2007;46:3171–3182. doi: 10.1021/bi6024676. [DOI] [PubMed] [Google Scholar]
- 17.Zambelli B, Stola M, Musiani F, De Vriendt K, Samyn B, Devreese B, Van Beeumen J, Turano P, Dikiy A, Bryant DA, Ciurli S. J Biol Chem. 2005;280:4684–4695. doi: 10.1074/jbc.M408483200. [DOI] [PubMed] [Google Scholar]
- 18.Zambelli B, Turano P, Musiani F, Neyroz P, Ciurli S. Proteins. 2009;74:222–239. doi: 10.1002/prot.22205. [DOI] [PubMed] [Google Scholar]
- 19.Musiani F, Ippoliti E, Micheletti C, Carloni P, Ciurli S. Biochemistry. 2013;52:2949–2954. doi: 10.1021/bi4001744. [DOI] [PubMed] [Google Scholar]
- 20.Colpas GJ, Hausinger RP. J Biol Chem. 2000;275:10731–10737. doi: 10.1074/jbc.275.15.10731. [DOI] [PubMed] [Google Scholar]
- 21.Chang Z, Kuchar J, Hausinger RP. J Biol Chem. 2004;279:15305–15313. doi: 10.1074/jbc.M312979200. [DOI] [PubMed] [Google Scholar]
- 22.Heimer SR, Mobley HL. J Bacteriol. 2001;183:1423–1433. doi: 10.1128/JB.183.4.1423-1433.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park IS, Carr MB, Hausinger RP. Proc Natl Acad Sci U S A. 1994;91:3233–3237. doi: 10.1073/pnas.91.8.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musiani F, Bellucci M, Ciurli S. J Chem Inf Model. 2011;51:1513–1520. doi: 10.1021/ci200183n. [DOI] [PubMed] [Google Scholar]
- 25.Quiroz-Valenzuela S, Sukuru SC, Hausinger RP, Kuhn LA, Heller WT. Arch Biochem Biophys. 2008;480:51–57. doi: 10.1016/j.abb.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voland P, Weeks DL, Marcus EA, Prinz C, Sachs G, Scott D. Am J Physiol Gastrointest Liver Physiol. 2003;284:G96–G106. doi: 10.1152/ajpgi.00160.2002. [DOI] [PubMed] [Google Scholar]
- 27.Fong YH, Wong HC, Chuck CP, Chen YW, Sun H, Wong KB. J Biol Chem. 2011;286:43241–43249. doi: 10.1074/jbc.M111.296830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moncrief MC, Hausinger RP. J Bacteriol. 1996;178:5417–5421. doi: 10.1128/jb.178.18.5417-5421.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salomone-Stagni M, Zambelli B, Musiani F, Ciurli S. Proteins. 2007;68:749–761. doi: 10.1002/prot.21472. [DOI] [PubMed] [Google Scholar]
- 30.Donovan S, Shannon KM, Bollag G. Biochim Biophys Acta. 2002;1602:23–45. doi: 10.1016/s0304-419x(01)00041-5. [DOI] [PubMed] [Google Scholar]
- 31.Lam R, Romanov V, Johns K, Battaile KP, Wu-Brown J, Guthrie JL, Hausinger RP, Pai EF, Chirgadze NY. Proteins. 2010;78:2839–2848. doi: 10.1002/prot.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boer JL, Hausinger RP. Biochemistry. 2012;51:2298–2308. doi: 10.1021/bi3000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellucci M, Zambelli B, Musiani F, Turano P, Ciurli S. Biochem J. 2009;422:91–100. doi: 10.1042/BJ20090434. [DOI] [PubMed] [Google Scholar]
- 34.Banaszak K, Martin-Diaconescu V, Bellucci M, Zambelli B, Rypniewski W, Maroney MJ, Ciurli S. Biochem J. 2012;441:1017–1026. doi: 10.1042/BJ20111659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biagi F, Musiani F, Ciurli S. J Biol Inorg Chem. 2013;18:571–577. doi: 10.1007/s00775-013-1002-8. [DOI] [PubMed] [Google Scholar]
- 36.Boivin S, Kozak S, Meijers R. Protein Expres Purif. 2013;91:192–206. doi: 10.1016/j.pep.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Stola M, Musiani F, Mangani S, Turano P, Safarov N, Zambelli B, Ciurli S. Biochemistry. 2006;45:6495–6509. doi: 10.1021/bi0601003. [DOI] [PubMed] [Google Scholar]
- 38.Charlwood PA. J Am Chem Soc. 1957;79:776–781. [Google Scholar]
- 39.Yang D, Kay LE. J Biomol NMR. 1999;13:3–10. doi: 10.1023/A:1008329230975. [DOI] [PubMed] [Google Scholar]
- 40.Nietlispach D. J Biomol NMR. 2005;31:161–166. doi: 10.1007/s10858-004-8195-7. [DOI] [PubMed] [Google Scholar]
- 41.Webb SM. Physica Scripta. 2005;T115:1011–1014. [Google Scholar]
- 42.Ravel B, Newville M. J Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 43.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Baidya N, Mascharak PK. Inorg Chem. 1991;30:920–928. [Google Scholar]
- 44.Newville M. J Synchrotron Radiat. 2001;8:96–100. doi: 10.1107/s0909049500016290. [DOI] [PubMed] [Google Scholar]
- 45.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Phys Rev B Condens Matter. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 46.Martin-Diaconescu V, Bellucci M, Musiani F, Ciurli S, Maroney MJ. J Biol Inorg Chem. 2012;17:353–361. doi: 10.1007/s00775-011-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bunker G. Introduction to XAFS. Cambridge Univ Pr; 2010. [Google Scholar]
- 48.Engh RA, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 49.Blackburn NJ, Hasnain SS, Pettingill TM, Strange RW. J Biol Chem. 1991;266:23120–23127. [PubMed] [Google Scholar]
- 50.Ferreira GC, Franco R, Mangravita A, George GN. Biochemistry. 2002;41:4809–4818. doi: 10.1021/bi015814m. [DOI] [PubMed] [Google Scholar]
- 51.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 52.Blakeley RL, Webb EC, Zerner B. Biochemistry. 1969;8:1984–1990. doi: 10.1021/bi00833a031. [DOI] [PubMed] [Google Scholar]
- 53.Benini S, Gessa C, Ciurli S. Soil Biol Biochem. 1996;28:819–821. [Google Scholar]
- 54.Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J. PLoS Comp Biol. 2012;8:1–12. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fong YH, Wong HC, Yuen MH, Lau PH, Chen YW, Wong K-B. PLoS Biology. 2013;11:1–16. doi: 10.1371/journal.pbio.1001678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 57.Herbst RW, Perovic I, Martin-Diaconescu V, O’Brien K, Chivers PT, Pochapsky SS, Pochapsky TC, Maroney MJ. J Am Chem Soc. 2010;132:10338–10351. doi: 10.1021/ja1005724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sydor AM, Liu J, Zamble DB. J Bacteriol. 2011;193:1359–1368. doi: 10.1128/JB.01333-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng T, Li H, Xia W, Sun H. J Biol Inorg Chem. 2011;58:1672–1681. [Google Scholar]
- 60.Tottey S, Harvie DR, Robinson NJ. Acc Chem Res. 2005;38:775–783. doi: 10.1021/ar0300118. [DOI] [PubMed] [Google Scholar]
- 61.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 62.Frishman D, Argos P. Proteins: Struct, Funct, Gen. 1997;27:329–335. doi: 10.1002/(sici)1097-0134(199703)27:3<329::aid-prot1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 63.Ryan KC, Johnson OE, Cabelli DE, Brunold TC, Maroney MJ. J Biol Inorg Chem. 2010;15:795–807. doi: 10.1007/s00775-010-0645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin-Diaconescu V, Maroney MJ. In: Comprehensive Inorganic Chemistry II. Reedijk J, Poppelmeier KR, editors. Elsevier; Oxford: 2013. [Google Scholar]
- 65.Neyroz P, Zambelli B, Ciurli S. Biochemistry. 2006;45:8918–8930. doi: 10.1021/bi060227s. [DOI] [PubMed] [Google Scholar]
- 66.Real-Guerra R, Staniscuaski F, Zambelli B, Musiani F, Ciurli S, Carlini CR. Plant Mol Biol. 2012;78:461–475. doi: 10.1007/s11103-012-9878-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.