

Abstract
MicroRNAs are known to control Toll like receptor activation in phagocytes. We have shown that leukotriene (LT) B4 (LTB4) positively regulates macrophage MyD88 expression by decreasing suppressor of cytokine signaling-1 (SOCS-1) mRNA stability. Here, we investigated the possibility that LTB4 control of MyD88 expression involves the generation of microRNAs. Our data show that LTB4, via its receptor B leukotriene receptor 1 (BLT1) and Gαi signaling, increased macrophage expression of inflammatory microRNAs, including miR-155, miR-146b, and miR-125b. LTB4-mediated miR-155 generation was attributable to AP-1 activation. Furthermore, macrophage transfection with antagomirs against miR-155 and miR-146b prevented both the LTB4-mediated decrease in SOCS-1 and increase in MyD88. Transfection with miR-155 and miR-146b mimics decreased SOCS-1 levels, increased MyD88 expression, and restored TLR4 responsiveness in both WT and LT-deficient macrophages. Together, our data unveil a heretofore unrecognized role for the GPCR BLT1 in controlling expression of microRNAs which regulate MyD88-dependent activation of macrophages.
Introduction
Activation of pattern recognition receptors (PRRs) by either pathogen-associated molecular pattern molecules (PAMPs) or by endogenous damage-associated molecular pattern molecules (DAMPs) elicits proinflammatory programs characterized by the production of inflammatory mediators that are important for the control of microbial infection (1). However, excessive production of such mediators also results in tissue injury.(2) Among the PRRs, Toll like receptors (TLRs) are the best studied. TLR family members and the IL-1β receptor (IL-1βR) share a conserved cytoplasmic Toll–IL-1R (TIR) domain that binds adaptor proteins, including myeloid differentiation factor 88 (MyD88) (3). MyD88 mediates signaling through all of the known TLRs except TLR3. MyD88 is necessary for host defense against a variety of experimental infections (4, 5), but also promotes development of atherosclerosis, autoimmune responses, diabetes, colitis, and familial Mediterranean fever (4, 5). MyD88 directs a variety of signaling programs that culminate in the activation of the master transcription factor NFκB, leading to the expression of proinflammatory genes (6). Among the mediators produced by activated macrophages, the 5-lipoxygenase (5-LO)-derived bioactive lipid leukotriene B4 (LTB4) is conventionally known as a potent phagocyte chemoattractant, and its role in eliciting acute inflammatory responses as well as maintaining chronic inflammation is well established (7). We and others have shown that LTB4 signaling via its cognate receptor B leukotriene receptor 1 (BLT1) through a Gαi-mediated decrease in cyclic AMP levels enhances phagocytosis and killing of both opsonized and non-opsonized (8-12) microbes. LTB4 activates NFκB (13) and stimulates production of cytokines including TNF-α (14), and enhances the production of microbicidal molecules including ROIs (9), RNIs (15, 16), and antimicrobial peptides. We have also shown that LTB4 has a permissive role in TLR activation by mediating both basal and inducible MyD88 expression and NFκB activation in macrophages in vitro and in vivo (13). Suppressor of cytokine signaling (SOCS-1) is an important inhibitor of JAK2 and STAT1(17) – a transcription factor for MyD88 (13) – and also impairs TLR activation by increasing degradation of MyD88-like adaptor (Mal) and IRAKs (18, 19). A key mechanism of LTB4/BLT1-mediated effects is enhanced degradation of mRNA encoding SOCS-1 (13). Reducing the SOCS-1 level represents an important step in macrophage activation (19). SOCS-1 mRNA levels can be controlled both at the levels of transcription and via microRNA-mediated inhibition. MicroRNAs are small oligonucleotides (~ 22 nt) that bind complementary sequences in mRNAs, usually resulting in gene silencing via translational repression or target degradation (20). They are potent regulators of homeostasis and of pathological conditions. Recent studies have shown that microRNAs are key determinants of macrophage activation, controlling the expression of a variety of molecules involved in PRR signaling and NFκB activation (20). There are several reports showing that SOCS-1 is targeted by different microRNAs, particularly miR-155 but also miR-30b and miR-150 (21-23). The mechanisms underlying expression of microRNAs in macrophages are poorly understood. It remains to be determined if microRNA expression is modulated by G protein-coupled receptors. We hypothesized that LTB4/BLT1/Gαi signaling enhances the expression of microRNAs that will in turn control macrophage TLR activation. Here, we report on the role of the GPCR BLT1 in the generation of microRNAs involved in SOCS-1 mRNA degradation, resulting in increased MyD88 expression and TLR-mediated NFκB activation. We also identified miR-125b as a novel regulator of MyD88 degradation.
Materials and Methods
Animals
8-week-old female 5-LO−/− (B6.129-Alox5tm1Fun; (24)), BLT1−/− (B6.129S4-Ltb4r1tm1Adl/J; (25)) and strain-matched WT C57BL/6 mice (The Jackson Laboratory) were maintained according to NIH guidelines for the use of experimental animals with the approval of the University of Michigan and Indiana University Committees for the Use and Care of Animals.
Cell harvest
Elicited macrophages were harvested from the peritoneal cavities of mice by lavage with PBS 4 days after the injection of 2 ml 3% thioglycollate as described previously (11).
Immunoblotting
Western blots were performed as previously described (11, 13). Protein samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with commercially available primary antibodies against MyD88, SOCS-1 (both 1:500; Abcam), or β-actin (1:10,000; Sigma-Aldrich). Densitometric analysis was performed as described previously (11, 13).
Measurement of nitrite and TNF-α
Levels of TNF-α were determined by ELISA (R&D Duoset; R&D Systems) as suggested by the manufacturer. Nitrite, the stable oxidized derivative of NO, was determined using the Griess reaction (13).
RNA isolation and semiquantitative real-time RT-PCR
Total RNA from cultured cells was isolated using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Real-time RT-PCR was performed as previously described (11, 13).
AP-1 activity assay
WT and 5-LO −/− macrophages were stimulated for 24 h with 100 nM LTB4 in the presence or absence of the AP-1 inhibitor SR11302 (10 μM), and DNA binding activity in nuclear extracts (10 μg of protein) was assayed using a transcription factor ELISA for AP-1 (Panomics), according to the manufacturer's instructions.
Targeted miRNA inhibition and overexpression
For inhibition of miR-155, -125b, and -146b, macrophages were transfected using lipofectamine siRNA max transfection agent with appropriate target antagomirs or anti-miR-negative control 1 (anti-miR-control). For overexpression, macrophages were transfected with pre-miR-155, -125b, -146b, or pre-miR-negative control 1 (pre-miR-control). Transfection reagents, antagomirs, and control miRNAs were purchased from Invitrogen. Knockdown efficiency and overexpression efficiency was assessed by determining mature miR-155 and -125b levels in transfected cells. Transfected cells were treated with or without 30 nM microRNA mimic or antagomir for 48 h and, where indicated, treated for indicated times with LTB4 (100 nM) or LPS (100 ng/ml) before collecting supernatants or preparing cell lysates for isolation of RNA (RNeasy kit, Qiagen) or proteins.
Luciferase assays
The plasmids containing 3′UTR of murine MyD88 were purchased from Genecopeia, and those containing the 3’UTR of murine SOCS1 were kindly provided by Christos Tsatsanis, University of Crete, Heraklion, Greece. Plasmids were packaged in the pEZX-MT01 Vector (for MyD88) or pMIR-REPORT-luciferase vector (for SOCS-1, Ambion) as described (21). Constructs were transfected in Raw 264.7 cells in 6-well plates using lipofectamine siRNA max (Invitrogen). Firefly luciferase reporter gene constructs (0.1 μg per well) were co-transfected together with 30 nM of microRNA (miR-155, miR-125b and miR-146b or scrambled-miR control), cells were lysed 24 h after transfection, and luciferase activity was measured. Each sample was assayed in triplicate.
RNA analysis
Quantitative RT-PCR analyses for miR-125b, miR-146a, miR-155, and RNU6 (used as normalization control) were performed using TaqMan miRNA assays with reagents, primers, and probes obtained from Qiagen. In brief, a stem loop primer was used for reverse transcription (30 min, 16°C; 30 min, 42°C; 5 min 85°C) followed by qPCR employing TaqMan probes and primers in a Bio-rad CFX96 Mastercycler. For assessing expression of SOCS-1, MyD88, b-actin, and the primary microRNAs (pri-miRs) mRNAs, cDNA was synthesized using a reverse transcription system (miScript II - Qiagem). qPCR was performed on the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories) as described (13). Primers and pri-miRNAs were purchased from Qiagen. Relative expression was calculated using the comparative threshold cycle (Ct) and expressed relative to control or WT (ΔΔCt method).
Focused miRNA arrays
To determine the expression of inflammatory microRNAs, WT and 5-LO−/− macrophages were treated with or without LTB4 for 24 h. RNA was then extracted from the cells using a Qiagen RNeasy mini kit according to the manufacturer’s instructions. cDNA was synthesized using the RT2 microRNA First Strand Kit (Qiagen) and applied to PCR focused immunopathology microRNA array plates (Qiagen). Plates were processed in a BioRad CFX96 Connect Real-Time PCR System using automated baseline and threshold cycle detection.
Array normalization and statistical analysis
Normalization and statistical analysis of miRNA expression were conducted using SABiosciences’ Online PCR Array Data Analysis Web Portal. miRNA expression was compared between WT and 5-LO−/− cells or between WT cells treated or not with LTB4. The ΔΔCt method was used to calculate fold change (FC). The endogenous control was derived from the average FC of four control genes in the PCR arrays, snoRNA251, snoRNA202, snoRNA142, and U6. The vehicle control group was used as the external control to normalize each sample. The formula: FC = 2^ [-(mean of ΔCt values of treated samples - mean of ΔCt values of control samples)] was used to calculate FC for up-regulated genes, and FC = − 2^ (mean of ΔCt values of treated samples - mean of ΔCt values of control samples) was used to calculate FC for down-regulated genes. T-tests were used to calculate significance of differences in miRNA expression between the control and the treatment groups for each miRNA at each time point; p < 0.01 was considered significant. miRNAs that displayed threshold cycles (Ct) >35 were excluded from the analysis. Data were interpreted using SABiosciences’ web-based PCR array data analysis tool (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
Statistics
Data are presented as mean ± SEM. Comparisons among groups were assessed with ANOVA followed by Bonferroni analysis; P < 0.05 was considered significant.
Results
LTB4 controls the expression of pro-inflammatory microRNAs in macrophages
We have previously shown that both 5-LO- and BLT1-deficient macrophages manifested a selective decrease in expression of MyD88 mRNA and protein (13), but not of other adaptors, and this deficiency was reversed by LTB4 in 5-LO−/− but not in BLT1−/− cells. Decreased MyD88 was a consequence of higher SOCS-1 expression in LTB4- and BLT1-deficient animals (13). The increased SOCS-1 mRNA expression in 5-LO−/− macrophages was associated with an increase in its mRNA stability, while exogenous LTB4 enhanced degradation of its mRNA (13). Here we sought to determine the molecular mechanism responsible for LTB4-mediated degradation of SOCS-1 mRNA. We specifically addressed the capacity of 5-LO metabolites to enhance the expression of microRNAs known to be involved in control of expression of both SOCS-1 and of TLRs and/or their adaptors. We performed a focused microRNA qPCR array (Immunopathogenesis - Qiagen) in WT and 5-LO−/− macrophages, and found that of 88 microRNAs studied, 22 were downregulated at least 2.5 fold in LT-deficient cells, including the following microRNAs that amplify TLRs: Let-7g, miR-155, miR-125b, miR-19a, miR-146a, and miR-146b. 11 microRNAs were upregulated at least 2.5 fold in 5-LO−/− macrophages and 54 microRNAs were not changed (Fig. 1A). To confirm these changes identified in the array, we analyzed the expression of individual microRNAs by qPCR (Fig. 1B). Compared to WT cells, 5-LO−/− macrophages exhibited lower basal expression of miR-155, miR-125b, and miR-146a b but unchanged expression of Let-7g.
Figure 1. Role of 5-LO metabolites in the expression of pro-inflammatory microRNAs in macrophages.
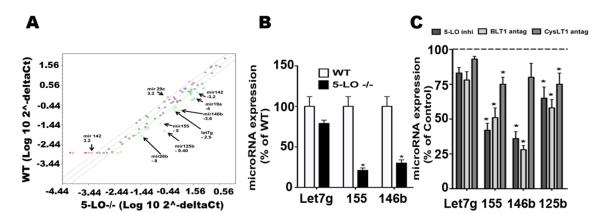
(A) Thioglycollate-elicited macrophages from WT and 5-LO−/− mice were harvested, and total RNA was isolated. MicroRNAs were isolated and subjected to miScript cDNA synthesis and to real time PCR array as described in Materials and Methods. Data are representative of 5 individual experiments. (B) Detection of individual microRNAs in WT and 5-LO−/− macrophages. Detection of miR-155, Let-7g and miR-146b was performed as described in Materials and Methods. (C) WT MΦs treated with the 5-LO inhibitor AA-861, the BLT1 antagonist U7532 and the cysLT1 antagonist MK571 (all at 1 μM) for 24 h, followed by determination of the indicated microRNAs. Data are mean ± SEM from 3 individual experiments, each performed in triplicate. *P < 0.05 versus untreated WT; #P < 0.05 versus WT control or LTB4 alone.
Since the 5-LO enzyme generates a variety of metabolic products, including both LTB4 and cysteinyl LTs, we investigated which class of LTs was involved in the regulation of expression of specific microRNAs. We employed pharmacological means to inhibit 5-LO activation and to selectively antagonize BLT1 and the cysteinyl LT receptor 1 (cysLT1), and treated cells with these agents for 24 h prior to microRNA determination. 5-LO inhibition markedly and significantly decreased the expression of miR-146b and miR-155, and modestly but significantly decreased that of miR-19a and miR125b (Fig. 1 C). As was also true for 5-LO−/− cells, no differences were seen in Let-7g expression. BLT1 antagonism followed the same pattern as observed in 5-LO-inhibited cells (Fig. 1 C). The cysLT1 antagonist MK571 was less effective than BLT1 in decreasing expression of all of the microRNAs studied with the exception of miR125b. Since endogenous LTB4 appeared to exhibit a greater effect on microRNA expression than did endogenous cysLTs, we directly investigated the ability of LTB4 to modulate expression of inflammatory microRNAs known to target SOCS-1 as well as the resulting consequences with respect to TLR activation.
PCR array analysis of WT macrophages treated with LTB4 for 24 h revealed enhanced expression of those microRNAs that were downregulated in 5-LO−/− cells, as well as of additional microRNAs involved in TLR responses (Fig. 2A). We validated the enhancement by LTB4 of the levels of miR-155 and miR-146b, but not of Let-7g, in both WT and 5-LO−/− macrophages (Fig. 2B). We also assessed the expression of inflammatory microRNAs in macrophages from BLT1−/− mice, and found markedly lower expression of miR-155, miR-146b, and miR-125b than in WT mice (Fig. 2C). Since BLT1 can be coupled to both Gαi and Gαq in macrophages (8), we sought to determine which G protein accounted for BLT1 effects on microRNA expression. Basal expression of all the microRNAs studied except Let-7g was diminished by treatment with the Gαi inhibitor pertussis toxin (PTX) (Fig. 2D). These data show that Gαi signaling controls basal expression of microRNAs. To determine whether Gαi actions were also involved in the enhancing effects of LTB4/BLT1 signaling, cells were pretreated with PTX for 18 h, followed by treatment with LTB4 for another 24 h. PTX treatment substantially decreased LTB4-induced expression of miR-155 and miR-146b, but only partially attenuated LTB4-induced expression of miR-125b, suggesting a contribution of Gαq signaling in BLT1-mediated expression of this microRNA (Fig. 2D). Together, these data show a stimulatory effect of the LTB4/BLT1/Gαi axis on the expression of microRNAs involved in both SOCS-1 expression and TLR activation.
Figure 2. LTB4/BLT1 enhances expression of microRNAs in macrophages.
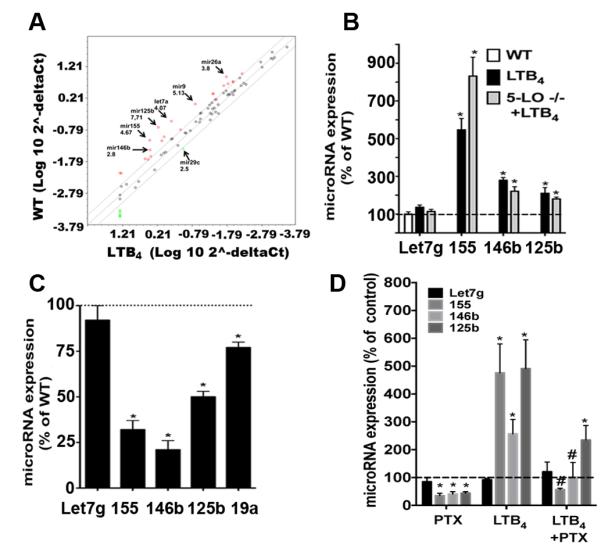
(A) WT macrophages were stimulated or not with 100 nM LTB4 for 24 h and total RNA was isolated. microRNAs were isolated and subjected to miScript cDNA synthesis. A real time PCR array was performed as described in Materials and Methods. Data are representative of 5 individual experiments. (B) Detection of individual microRNAs in WT and 5-LO−/− macrophages stimulated with 100 nM LTB4 for 24 h and in unstimulated WT controls. Detection of miR-155, Let-7g and miR-146b was performed as described in Materials and Methods. Data represent 3-5 independent experiments performed in triplicate. (C) microRNAs were isolated from WT and BLT1−/− macrophages, and the expression of Let-7g, miR-155, miR-146b, miR-19a, and miR-125b was determined by real time RT-PCR. Dashed line shows the level of microRNAs detected in WT macrophages normalized to 100%. Data represent 3-5 independent experiments performed in triplicate. (D) WT macrophages were pretreated for 24 hours with or without the Gαi inhibitor PTX (600 ng/ml), followed by 30 minutes of treatment with or without LTB4. The expression of the indicated microRNAs was detected as in B. In B-D data are mean ± SEM from 3 individual experiments, each performed in triplicate. *P < 0.05 versus untreated WT; #P < 0.05 versus WT control or LTB4 alone.
AP-1 mediates LTB4-induced miR-155 expression
As expression of approximately 25% of the microRNAs was lower in LT-deficient cells than WT cells, we speculated that LTs could be involved in the expression/activation of enzymes responsible for the generation of microRNAs, such as Dicer and Drosha (26). We observed an insignificant decrease in Dicer mRNA expression; LTB4 enhanced the expression of Dicer in 5-LO−/− cells, and no changes in Drosha levels were detected in cells of either genotype, treated or not with LTB4 (Fig. 3A).
Figure 3. LTB4/BLT1-induced AP-1 activation is required for miR-155 expression.

(A) Thioglycollate-elicited macrophages from WT and 5-LO−/− mice were treated or not with 100 nM LTB4 for 24 h, and the expression of Dicer or Drosha were determined by qPCR. (B) Macrophages from WT and 5-LO−/− mice were stimulated as in A and the expression of indicated pri-microRNAs were determined by qPCR. (C) Macrophages from WT and 5-LO−/− mice were stimulated as in A or pretreated with the AP-1 inhibitor SR11302 for 30 min before stimulation with LTB4 and nuclear AP-1 activity was determined as described in Materials and Methods. (D) WT macrophages were incubated with the AP-1 inhibitor SR11302 for 30 min before stimulation with LTB4 for 24 h. miR-155 expression was determined as described in Materials and Methods. Data represent mean ± SEM from 3-5 individual experiments, each performed in triplicate. *P < 0.05 versus unstimulated WT; #P < 0.05 versus stimulated WT.
microRNAs are initially expressed as long primary transcripts termed primary miRNAs (pri-microRNAs) that undergo sequential processing by Drosha and then Dicer to yield mature microRNAs (20). In the absence of any change in the expression of enzymes involved in microRNA biosynthesis, we speculated that LTB4 could be involved in the generation of pri-microRNAs. Therefore, we measured the amounts of pri-miR-155, pri-miR-146b, pri-miR-125b, and pri-Let-7g by qPCR. Indeed, expression of all pri-microRNAs other than pri-Let-7g was lower in both 5-LO/- and BLT1−/− than WT macrophages (Fig. 3B). Lower expression of these primary microRNAs suggested a role for a LT in the control of the transcriptional machinery involved in microRNA expression. Activating protein-1 (AP-1) is a key transcription factor involved in the expression of different primary microRNAs including miR-155, miR-125b, miR-146a, and miR-146b (27, 28). We tested the ability of LTB4 to enhance the activation of AP-1 and the subsequent expression of miR-155. Initially, we performed AP-1 DNA binding assays in WT and 5-LO−/− macrophages incubated with or without LTB4 for 4 h. LT-deficient cells exhibited lower basal AP-1 activation than WT macrophages, and LTB4 greatly enhanced AP-1 DNA binding activity in 5-LO−/− cells (Fig. 3 C). To determine if AP-1 is involved in LTB4-induced miR-155 expression, we pretreated WT macrophages with the AP-1 inhibitor SR11302 , followed by LTB4 challenge for 24 h. AP-1 inhibition decreased basal miR-155 levels and attenuated LTB4-enhanced miR-155 expression (Fig. 3 D). That the AP-1 inhibitor prevents AP-1 DNA binding activity was confirmed in LTB4-treated cells (Fig. 3C). Together, these findings show that LTB4-mediated AP-1 activation accounts for the expression of inflammatory microRNAs which control expression of SOCS-1 and activation of TLR (Fig. 3D).
LTB4-mediated miR-155 and miR-146b expression accounts for increased SOCS-1 mRNA turnover, MyD88 expression, and TLR4 responsiveness
We previously reported that SOCS-1 acts as a brake on expression of MyD88 by inhibiting the MyD88 transcription factor STAT-1 (13). SOCS-1 gene silencing allows STAT-1 to be active and enhances MyD88 expression and improves TLR responsiveness in macrophages (13). We next sought to determine whether LTB4-enhanced microRNA expression leads to changes in SOCS-1 and MyD88 levels, and therefore, macrophage function. We transfected antagomirs known to directly inhibit expression of SOCS-1 (anti-miR-155 and anti-miR-146b) and MyD88 (anti-miR-125b) into macrophages stimulated or not with LTB4 for 24 h. Our data show that miR-155 inhibition decreased basal MyD88 expression and prevented LTB4-enhanced MyD88 expression. By contrast, inhibition of miR-125b and miR-146b enhanced MyD88 above baseline levels, but did not alter levels in the presence of LTB4. Furthermore, miR-125b increased MyD88 to a greater extent than did miR-146b (Fig. 4 A). As expected (13), LTB4 decreased basal SOCS-1 expression, and miR-155 antagonism increased both basal and LTB4-induced SOCS-1 expression (Fig. 4 B). However, miR-125b did not influence SOCS-1 levels, indicating that it instead exerted a direct effect on MyD88 mRNA expression (Fig. 4 B). The miR-146b antagomir enhanced basal SOCS-1 levels and prevented LTB4 inhibitory effects on SOCS-1 expression (Fig. 4B). These findings were confirmed by immunoblot (Fig. 4D). As 5-LO−/− cells exhibited lower microRNA expression, we perform complementary “add-back” experiments, in which miR-155, miR-146b, and miR-125b precursors (miRNA mimic) were transfected into both WT and 5-LO-deficient macrophages, and the expression of both MyD88 and SOCS-1 were determined by qPCR. MyD88 expression was slightly enhanced in WT cells transfected with miR-155 mimic, and the levels of MyD88 expression were elevated in 5-LO−/− cells transfected with miR-155 mimic. MyD88 expression was increased by miR-146b mimic and decreased by miR-125b mimic in both strains studied, again showing that these microRNAs control MyD88 expression (Fig. 4E). We observed higher SOCS-1 expression in 5-LO−/− cells than in WT cells as we have previously shown (13), and both miR-155 and miR-146b transfection decreased SOCS-1 in both WT and 5-LO−/− deficient cells. Transfection of miR-125b decreased SOCS-1 levels in 5-LO−/− macrophages but not in WT, indicating a unique requirement for LTB4 in miR-125b-mediated effects (Fig. 4 E). Together, by inhibiting SOCS-1 levels with miR-155 and miR-146b mimics, we observed an increase in MyD88 expression in both WT and 5-LO−/− cells. Next, we determined whether miR-125b, miR-146b and miR-155 directly target MyD88 and SOCS-1 using Raw 264.7 cells transfected with either empty vector, 3’ UTR MyD88 or 3’UTR SOCS-1 followed by addition of the microRNA mimics mentioned above. Our data confirmed that while miR-125b targets MyD88, miR-146b and miR-155 directly target SOCS-1. The temporal relationship between microRNAs, SOCS-1 and MyD88 expression was also investigated in LTB4-stimulated cells. We observed that LTB4 rapidly enhanced miR-155 expression within 1 h, while the decrease in SOCS-1 expression required 4 h. We only observed significant differences in LTB4-induced MyD88 expression after 24 h, further suggesting that decreased SOCS-1 accounts for the enhanced MyD88. Together, our data show that LTB4 enhances MyD88 expression by increasing the expression of microRNAs that directly target SOCS-1.
Figure 4. miR-155 and miR-146b levels control LTB4-mediated SOCS-1 inhibition and enhanced MyD88 levels.

(A) Thioglycollate-elicited macrophages from WT were transfected with 30 nM of miR-155, miR-146b, or miR-125b antagomir for 24 h, then stimulated with 100 nM LTB4 for another 24, followed by determination of (A) MyD88 or (B) SOCS-1 mRNA levels or protein (C). (D and E) WT and 5-LO−/− macrophages were transfected with 30 nM of the mimics miR-155, miR-146b, or miR-125b for 48 h followed by determination of MyD88 (D) and SOCS-1 (E) mRNA levels as described in Materials and Methods. Inset: The predicted miR-146b and miR-125b binding sites located in the 3′-UTR of SOCS-1 and MyD88, respectively. The sequence alignment of miR-146b/SOCS-1 and miR-125b/MyD88 is shown, and perfect matches are indicated by a line. (F) Raw264.7 macrophages were transfected with a luciferase construct containing the 3′ UTR of SOCS1, 3’ UTR of MyD88 and empty vector expressing luciferase reporter plasmid followed by the microRNA mimics miR-155, miR-146b and miR-125b for 24 h and the luciferase activity was determined as described in the Material and Methods. (G) WT macrophages were stimulated with 100 nM LTB4 for different time points and the expression of miR-155, SOCS-1 and MyD88 were determined by qPCR. Data represent mean ± SEM from at least 3 individual experiments, each performed in triplicate. *P < 0.05 versus unstimulated WT; #P < 0.05 versus stimulated WT.
We next sought to determine if changes in the expression of MyD88 and SOCS-1 influenced TLR signaling and if increased microRNA expression restored 5-LO−/− macrophage responsiveness to LPS, which we have previously reported to be impaired (13). WT and 5-LO−/− macrophages were transfected with mimics of miR-155, miR-125b, or miR-146b for 24 h, followed by LPS stimulation for another 24 h. As expected, 5-LO−/− macrophages generated less TNF-α and nitrite than WT cells in response to LPS. Transfection of both miR-155 and miR-146b mimics enhanced LPS-induced TNF-α (Fig. 5A) and NO (Fig. 5B) production in WT and especially 5-LO−/− cells. In contrast, miR-125b mimic inhibited LPS-enhanced TNF-α levels exclusively in WT macrophages, which is consistent with the findings in Fig. 4B.
Fig. 5. miR-155 and miR-146b mimic overcome LPS responsiveness in LT-deficient macrophages.
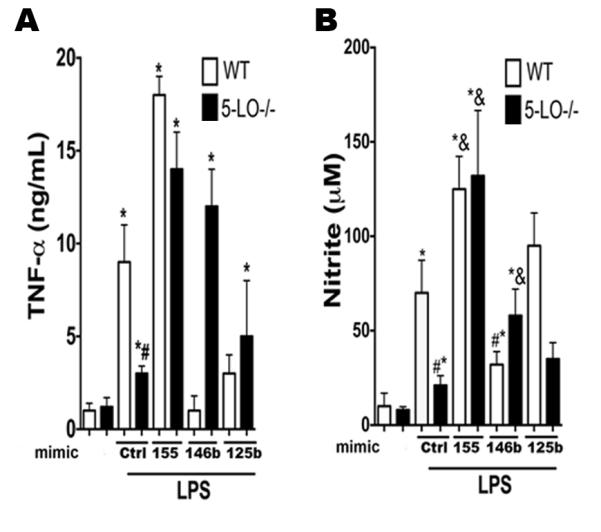
WT and 5-LO−/− macrophages were transfected with 30 nM of the mimics miR-155, miR-146b, or miR-125b for 48 h, stimulated with LPS for 24 h, and TNF-α (A) and NO (B) levels were detected by ELISA and Griess reaction, respectively. Data represent mean ± SEM from 3 individual experiments, each performed in triplicate. *P < 0.05 versus unstimulated WT; #P < 0.05 versus stimulated WT.
Discussion
This study highlights a novel regulatory mechanism of microRNA expression induced after GPCR activation. We have previously shown that BLT1 activation inhibits SOCS-1 expression, leading to enhanced MyD88 expression and consequently increased TLR activation. Here, we extended these findings by investigating the involvement of BLT1 in microRNA generation that influences the molecular programs involved in SOCS-1/MyD88 expression. To our knowledge, this is the first demonstration of an essential role for a GPCR in microRNA generation. More specifically, we found that: 1) LTB4/BLT1/Gαi activation enhances the expression of inflammatory microRNAs; 2) LTB4/BLT1 activation of AP-1 accounts for the enhanced microRNA generation in macrophages; 3) preventing miR-155 and miR-146b actions abolished LTB4-induced SOCS-1 mRNA degradation; 4) adding back miR-155 and miR-146b restored MyD88 levels and responsiveness in LT-deficient mice; 5) miR-125b degrades MyD88 in a manner dependent on LTB4 synthesis.
TLR activation is dictated by fine-tuning events that culminate in important changes in inflammatory response and host defense (20). Among the “fine-tuners”, microRNAs are known to inhibit the expression of many key components of the TLR signaling program, including MyD88 (29, 30), IRAKs (31-33) and TRAF-6 (33-35). microRNAs can be produced constitutively or be upregulated by a variety of inflammatory stimuli, including LPS (21, 36-38), Poly(I:C) (37, 38) and TNF-α (36). However, whether GPCRs also participate in the generation of microRNAs is unknown. We have shown that the other major class of 5-LO metabolite, the CysLTs, did not exert effects on most microRNAs, excluding miR-125b, emphasizing the specific and nonredundant role of LTB4 in regulating microRNAs that control SOCS-1 expression in macrophages. Also striking was the fact that the upregulatory influence on microRNA expression was observed not only following the addition of exogenous LTB4, but also under basal conditions, in which levels of constitutive endogenous LTB4 generation would be expected to be quite low. Basal release of LTB4 by elicited peritoneal macrophages from WT animals over 90 min was approximately 100 pg/ml, which is equivalent to 0.5 nM (13). This concentration is indeed sufficient to activate BLT1, since we have shown that LTB4 amplifies AM antimicrobial functions at concentrations as low as 0.01 nM (8). These findings support the conclusion that LTB4 is a homeostatic regulator of microRNA expression that controls numerous aspects of macrophage biology.
We also identified Gαi-independent microRNA expression, such as that of miR-125b and miR19a (not shown). The relative role of other G proteins, such as Gαq and Gαs, in modulating expression of microRNAs that regulate macrophage biology is an active area of research in our laboratory.
The fact that LT deficiency decreased the expression of a myriad of microRNAs suggested the possibility that LTs enhance the expression/activity of the enzymes involved in microRNA biogenesis, such as Dicer and Drosha (20, 39). Although we did not observe any effect of LTs on the expression of these enzymes, we cannot exclude possible actions of LTB4 on Dicer or Drosha activation. It remains to be determined whether GPCRs also influence the expression or activation of enzymes involved in the microRNA machinery. A variety of transcription factors have been implicated in microRNA generation, such as NFκB (40), Egr (41, 42) and AP-1 (27, 43-46). We further investigated whether LTB4/BLT1 could enhance the generation of microRNAs by controlling activation of AP-1 since this transcription factor is responsible for the expression of miR-155 and also miR-146b (27, 28). AP-1 regulates gene expression in cells activated by a myriad of stimuli, including proinflammatory cytokines, TLR ligands, growth factors, and stress (45). Upon cell activation, regulation of gene expression by AP-1 is dependent mainly on activation of MAP kinases, including ERK1/2 and p38 (45). We and others have shown that LTB4 activates ERK1/2 and p38 in macrophages (47, 48). However, whether LTB4 enhances AP-1 activation is controversial. Stankova, et. al., have shown that LTB4 enhances expression and DNA binding activity of the AP-1 member c-fos (49), while Brach, et al showed that AP-1 is not affected by LTB4 in stimulated human monocytes (50). Here, we showed that LTB4 activates AP-1 to enhance miR-155 expression. AP-1 binding to the BIC promoter of these microRNAs has been previously reported in both human and mouse cells (27, 44, 51, 52). Furthermore, McCoy et. al., (53) have shown that Ets3 activation inhibits miR-155 expression. Whether LTB4 enhances miR-155 expression by activating transcription factors other than AP-1, such as NFκB, or by inhibiting Ets3 actions, remains to be determined.
We further investigated whether changes in microRNA levels account for changes in the expression of SOCS-1 and MyD88 in macrophages from LT-deficient mice, and found that miR-155 and miR-146b decrease SOCS-1 levels and miR-125b inhibits MyD88 only. We recognize that these microRNAs could be modulating the expression of SOCS-1 and MyD88 indirectly. Therefore, we employed different complementary approaches such as antagomir treatment of LTB4-treated cells and 3’-UTR luciferase assay to further confirm our findings. Our findings confirm that miR-155 inhibits SOCS-1 expression and newly identify that miR-125b targets MyD88 and miR-146b targets SOCS-1. A caveat of our studies is that we cannot exclude a potential indirect effect of the microRNA mimics in the degradation of SOCS-1 and MyD88 3’UTR constructs in RAW 264.7 macrophages, since we did not transfect macrophages with the mutants lacking the 3’UTR microRNA binding sites. The temporal relationship in the expression of these molecules is consistent with a causal role for miR-155 in the degradation of SOCS-1 and MyD88, as the increase in miR-155 expression (noted 1 h after LTB4 treatment) preceded the decrease in SOCS-1 levels (noted after 4 h), which in turn preceded the increase in MyD88 levels (noted after 24 h). Although we did not measure the temporal expression of the other microRNAs, the microRNA mimic and antagomir data supporting the roles of miR-146b in control of SOCS-1 and of miR-125b in the control of MyD88 strongly imply that these microRNAs too would be expected to precede changes in levels of their target genes. Furthermore, we cannot definitively exclude the possibility that the microRNAs studied here might target other genes that also influence TLR activation. The fact that miR-125b does not further inhibit MyD88 expression and TNF-α levels in 5-LO−/− cells is intriguing, and the molecular programs involved in these events will be studied in the future. Another intriguing fact is that miR-125b suppresses LTB4-induced SOCS1 mRNA without affecting its UTR. We speculate that the effect of miR-125b on SOCS-1 is indirect, by targeting either transcription factors involved in SOCS-1 mRNA or by changing the levels of other microRNAs that further degrade SOCS-1 in LTB4-treated cells.
In summary, our findings show that LTB4, via its cognate receptor BLT1 and Gαi protein activation, enhances the expression of microRNAs known to degrade SOCS-1 levels and thereby enhances MyD88 protein expression and TLR responsiveness. We also showed for the first time that miR-125b targets MyD88 directly. These data demonstrate that GPCRs amplify innate immune and inflammatory responses by modulating the expression of important microRNAs that further control macrophage activation. It will be of interest to dissect the interplay between GPCR signaling and microRNAs in in vivo host defense and inflammatory responses. Our findings have direct translational importance, as unveiling the LTB4/BLT1/microRNA axis to control TIR-dependent macrophage activation would be expected to attenuate excessive NFκB activation which contributes to tissue injury (4, 6) or of unchecked IL-1βR activation in autoinflammatory conditions (54). Moreover, states of immunosuppression might be overcome by administration of exogenous LTB4 plus microRNA mimics, such as miR-155 and miR-146b.
Acknowledgements
We thank Alexandra Medeiros for the critical input.
This work was supported by National Institutes of Health Grants HL-058897 (to M. P-G.) and HL-103777–01 (C. H. S.). This work was also supported by the Fundação de Amparo a Pesquisa do Estado de São Paulo.
Footnotes
Competing interests. The authors have no conflict of interest
References
- 1.Medzhitov R, Janeway C., Jr. The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8:452–456. doi: 10.1016/s0966-842x(00)01845-x. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Damage control in host-pathogen interactions. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15525–15526. doi: 10.1073/pnas.0908451106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 4.Netea MG, Wijmenga C, O'Neill LA. Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol. 2012;13:535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- 5.Connolly DJ, O'Neill LA. New developments in Toll-like receptor targeted therapeutics. Curr Opin Pharmacol. 2012;12:510–518. doi: 10.1016/j.coph.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 7.Flamand N, Mancuso P, Serezani CH, Brock TG. Leukotrienes: mediators that have been typecast as villains. Cell Mol Life Sci. 2007;64:2657–2670. doi: 10.1007/s00018-007-7228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peres CM, Aronoff DM, Serezani CH, N. Flamand L. H. Faccioli, Peters-Golden M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J Immunol. 2007;179:5454–5461. doi: 10.4049/jimmunol.179.8.5454. [DOI] [PubMed] [Google Scholar]
- 9.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood. 2005;106:1067–1075. doi: 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serezani CH, Aronoff DM, Sitrin RG, Peters-Golden M. FcgammaRI ligation leads to a complex with BLT1 in lipid rafts that enhances rat lung macrophage antimicrobial functions. Blood. 2009;114:3316–3324. doi: 10.1182/blood-2009-01-199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serezani CH, Kane S, Collins L, Morato-Marques M, Osterholzer JJ, Peters-Golden M. Macrophage dectin-1 expression is controlled by leukotriene B4 via a GM-CSF/PU.1 axis. J Immunol. 2012;189:906–915. doi: 10.4049/jimmunol.1200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters-Golden M, Henderson WR., Jr. Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 13.Serezani CH, Lewis C, Jancar S, Peters-Golden M. Leukotriene B4 amplifies NF-kappaB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. The Journal of clinical investigation. 2011;121:671–682. doi: 10.1172/JCI43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kihara Y, Yokomizo T, Kunita A, Morishita Y, Fukayama M, Ishii S, Shimizu T. The leukotriene B4 receptor, BLT1, is required for the induction of experimental autoimmune encephalomyelitis. Biochemical and biophysical research communications. 2010;394:673–678. doi: 10.1016/j.bbrc.2010.03.049. [DOI] [PubMed] [Google Scholar]
- 15.Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. J Immunol. 2006;177:3201–3208. doi: 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 16.Medeiros AI, Sa-Nunes A, Turato WM, Secatto A, Frantz FG, Sorgi CA, Serezani CH, Deepe GS, Jr., Faccioli LH. Leukotrienes are potent adjuvant during fungal infection: effects on memory T cells. J Immunol. 2008;181:8544–8551. doi: 10.4049/jimmunol.181.12.8544. [DOI] [PubMed] [Google Scholar]
- 17.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 18.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O'Neill LA, Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 20.O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 21.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CC, Zhang QY, Liu Z, Clynes RA, Suciu-Foca N, Vlad G. Downregulation of inflammatory microRNAs by Ig-like transcript 3 is essential for the differentiation of human CD8(+) T suppressor cells. J Immunol. 2012;188:3042–3052. doi: 10.4049/jimmunol.1102899. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, A. Hasni S, Perez P, Tandon M, Jang SI, Zheng C, Kopp JB, Austin H, 3rd, Balow JE, Alevizos I, Illei GG. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. Journal of the American Society of Nephrology : JASN. 2013;24:1073–1087. doi: 10.1681/ASN.2012080849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen XS, R. Sheller J, N. Johnson E, Funk CD. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature. 1994;372:179–182. doi: 10.1038/372179a0. [DOI] [PubMed] [Google Scholar]
- 25.Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, Carafone AD, Gerszten RE, Luster AD. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–990. doi: 10.1038/ni970. [DOI] [PubMed] [Google Scholar]
- 26.Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–298. doi: 10.1111/j.1365-2567.2009.03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin Q, Wang X, McBride J, Fewell C, Flemington E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. The Journal of biological chemistry. 2008;283:2654–2662. doi: 10.1074/jbc.M708218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curtale G, Citarella F, Carissimi C, Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V, Macino G. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010;115:265–273. doi: 10.1182/blood-2009-06-225987. [DOI] [PubMed] [Google Scholar]
- 29.Wei J, Huang X, Zhang Z, Jia W, Zhao Z, Zhang Y, Liu X, Xu G. MyD88 as a target of microRNA-203 in regulation of lipopolysaccharide or Bacille Calmette-Guerin induced inflammatory response of macrophage RAW264.7 cells. Molecular immunology. 2013;55:303–309. doi: 10.1016/j.molimm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Wendlandt EB, W. Graff J, L. Gioannini T, P. McCaffrey A, Wilson ME. The role of microRNAs miR-200b and miR-200c in TLR4 signaling and NF-kappaB activation. Innate immunity. 2012;18:846–855. doi: 10.1177/1753425912443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nahid MA, Yao B, Dominguez-Gutierrez PR, Kesavalu L, Satoh M, Chan EK. Regulation of TLR2-mediated tolerance and cross-tolerance through IRAK4 modulation by miR-132 and miR-212. J Immunol. 2013;190:1250–1263. doi: 10.4049/jimmunol.1103060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chassin C, Hempel C, Stockinger S, Dupont A, Kubler JF, Wedemeyer J, Vandewalle A, Hornef MW. MicroRNA-146a-mediated downregulation of IRAK1 protects mouse and human small intestine against ischemia/reperfusion injury. EMBO Mol Med. 2012;4:1308–1319. doi: 10.1002/emmm.201201298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramkaran P, Khan S, Phulukdaree A, Moodley D, Chuturgoon AA. miR-146a Polymorphism Influences Levels of miR-146a, IRAK-1, and TRAF-6 in Young Patients with Coronary Artery Disease. Cell Biochem Biophys. 2013 doi: 10.1007/s12013-013-9704-7. [DOI] [PubMed] [Google Scholar]
- 34.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 35.Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol. 2011;8:388–403. doi: 10.1038/cmi.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 37.Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swaminathan G, Rossi F, Sierra LJ, Gupta A, Navas-Martin S, Martin-Garcia J. A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS pathogens 8: e1002937. 2012 doi: 10.1371/journal.ppat.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bi Y, Liu G, Yang R. MicroRNAs: novel regulators during the immune response. J Cell Physiol. 2009;218:467–472. doi: 10.1002/jcp.21639. [DOI] [PubMed] [Google Scholar]
- 40.Cremer TJ, Fatehchand K, Shah P, Gillette D, Patel H, Marsh RL, Besecker BY, Rajaram MV, Cormet-Boyaka E, Kanneganti TD, Schlesinger LS, Butchar JP, Tridandapani S. MiR-155 induction by microbes/microbial ligands requires NF-kappaB-dependent de novo protein synthesis. Frontiers in cellular and infection microbiology. 2012;2:73. doi: 10.3389/fcimb.2012.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giallourakis CC, Benita Y, Molinie B, Cao Z, Despo O, Pratt HE, Zukerberg LR, Daly MJ, Rioux JD, Xavier RJ. Genome-wide Analysis of Immune System Genes by Expressed Sequence Tag Profiling. J Immunol. 2013;190:5578–5587. doi: 10.4049/jimmunol.1203471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma F, Liu X, Li D, Wang P, Li N, Lu L, Cao X. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J Immunol. 2010;184:6053–6059. doi: 10.4049/jimmunol.0902308. [DOI] [PubMed] [Google Scholar]
- 43.Kappelmann M, Kuphal S, Meister G, Vardimon L, Bosserhoff AK. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene. 2013;32:2984–2991. doi: 10.1038/onc.2012.307. [DOI] [PubMed] [Google Scholar]
- 44.Marsolier J, Pineau S, Medjkane S, Perichon M, Yin Q, Flemington E, Weitzman MD, Weitzman JB. OncomiR addiction is generated by a miR-155 feedback loop in Theileria-transformed leukocytes. PLoS pathogens 9: e1003222. 2013 doi: 10.1371/journal.ppat.1003222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis 70 Suppl 1: i109-112. 2011 doi: 10.1136/ard.2010.140533. [DOI] [PubMed] [Google Scholar]
- 46.Yin Q, McBride J, Fewell C, Lacey M, Wang X, Lin Z, Cameron J, Flemington EK. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. Journal of virology. 2008;82:5295–5306. doi: 10.1128/JVI.02380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bates ME, Green VL, Bertics PJ. ERK1 and ERK2 activation by chemotactic factors in human eosinophils is interleukin 5-dependent and contributes to leukotriene C(4) biosynthesis. The Journal of biological chemistry. 2000;275:10968–10975. doi: 10.1074/jbc.275.15.10968. [DOI] [PubMed] [Google Scholar]
- 48.Campos MR, Serezani CH, Peters-Golden M, Jancar S. Differential kinase requirement for enhancement of Fc gammaR-mediated phagocytosis in alveolar macrophages by leukotriene B4 vs. D4. Molecular immunology. 2009;46:1204–1211. doi: 10.1016/j.molimm.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 49.Stankova J, Rola-Pleszczynski M. Leukotriene B4 stimulates c-fos and c-jun gene transcription and AP-1 binding activity in human monocytes. Biochem J 282 ( Pt 3): 625-629. 1992 doi: 10.1042/bj2820625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brach MA, de Vos S, Arnold C, Gruss HJ, Mertelsmann R, Herrmann F. Leukotriene B4 transcriptionally activates interleukin-6 expression involving NK-chi B and NF-IL6. Eur J Immunol. 1992;22:2705–2711. doi: 10.1002/eji.1830221034. [DOI] [PubMed] [Google Scholar]
- 51.Onyeagucha BC, Mercado-Pimentel ME, Hutchison J, Flemington EK, Nelson MA. S100P/RAGE signaling regulates microRNA-155 expression via AP-1 activation in colon cancer. Exp Cell Res. 2013 doi: 10.1016/j.yexcr.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao B, Liu Z, Li BS, Tang B, Li W, Guo G, Shi Y, Wang F, Wu Y, Tong WD, Guo H, Mao XH, Zou QM. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J Infect Dis. 2009;200:916–925. doi: 10.1086/605443. [DOI] [PubMed] [Google Scholar]
- 53.McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, O'Neill LA. IL-10 inhibits miR-155 induction by toll-like receptors. The Journal of biological chemistry. 2010;285:20492–20498. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgard U, Cowen EW, Pham TH, Booty M, Estes JD, Sandler NG, Plass N, Stone DL, Turner ML, Hill S, Butman JA, Schneider R, Babyn P, El-Shanti HI, Pope E, Barron K, Bing X, Laurence A, Lee CC, Chapelle D, Clarke GI, Ohson K, Nicholson M, Gadina M, Yang B, Korman BD, Gregersen PK, van Hagen PM, Hak AE, Huizing M, Rahman P, Douek DC, Remmers EF, Kastner DL, Goldbach-Mansky R. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]