

Abstract
Clinical stroke induces inflammatory processes leading to cerebral and splenic injury and profound peripheral immunosuppression. IL-10 expression is elevated during major CNS diseases and limits inflammation in the brain. Recent evidence demonstrated that absence of B-cells led to larger infarct volumes and CNS damage after middle cerebral artery occlusion (MCAO) that could be prevented by transfer of IL-10+ B-cells. The purpose of this study was to determine if the beneficial immunoregulatory effects on MCAO of the IL-10+ B-cell subpopulation also extends to B-cell-sufficient mice that would better represent stroke subjects.
CNS inflammation and infarct volumes were evaluated in male C57BL/6J (WT) mice that received either RPMI or IL-10+ B-cells and underwent 60 min of middle cerebral artery occlusion (MCAO) followed by 96 hours of reperfusion.
Transfer of IL-10+ B-cells markedly reduced infarct volume in WT recipient mice when given 24 hours prior to or 4 hours after MCAO. B-cell protected MCAO mice had increased regulatory subpopulations in the periphery, reduced numbers of activated, inflammatory T-cells, decreased infiltration of T-cells and a less inflammatory milieu in the ischemic hemispheres of the IL-10+ B-cell-treated group. Moreover, transfer of IL-10+ B-cells 24 hours before MCAO led to a significant preservation of regulatory immune subsets in the IL-10+ B-cell protected group presumably indicating their role in immunomodulatory mechanisms, post-stroke.
Our studies are the first to demonstrate a major immunoregulatory role for IL-10+ regulatory B-cells in preventing and treating MCAO in WT mice and also implicating their potential role in attenuating complications due to post-stroke immunosuppression.
Keywords: MCAO, inflammatory cells, regulatory B-cells, IL-10
Introduction
Stroke remains the third leading cause of death in adults worldwide and the most frequent cause of permanent disability in the world (Donnan et al. 2008). Ischemic stroke, that occurs as a result of an obstruction within a blood vessel supplying blood to the brain, accounts for 87 percent of all stroke cases, in the United States alone (Go et al. 2013). While reperfusion of the ischemic brain is clearly desirable, tissue damage often results from both the transient ischemic insult and the reperfusion process. Reperfusion frequently induces an inflammatory response that either causes additional injury to the cerebral microcirculation and adjacent brain tissue (Arumugam et al. 2005) causing substantial secondary brain damage or may lead to potential repair mechanisms (Iadecola and Anrather 2011).
Key features of the neuroimmunological response to brain ischemia are early microglial activation (Mabuchi et al. 2000) and subsequent recruitment of circulating leukocytes to the ischemic brain (Iadecola and Anrather 2011; Macrez et al. 2011). Inhibition of this early inflammatory response improves outcome in experimental stroke, but clinical trials aimed at preventing leukocyte trafficking into the ischemic brain were unsuccessful (Investigators 2001). Recent experimental data demonstrate the complexities of modulating the immune response after stroke and suggest that immune cells can have both beneficial and detrimental effects. With the experimental model influencing the time course and number of infiltrating immune cells as well as the degree of microglial activation (Zhou et al. 2013), it becomes obvious that the timing and duration of interventions aimed at modulating inflammation are critical. Hence, one of the rapidly evolving areas of focus in stroke research involves defining the molecular and cellular basis for the augmented tissue injury and inflammation associated with transient cerebral ischemia.
Our lab has been actively investigating these post-ischemic inflammatory mechanisms and the interaction of activated immune cells with the ischemic brain tissue. Recently, we demonstrated that B-cells are critical in governing infarct size and that MCAO-induced changes were prevented in B-cell-deficient (μMT−/−) mice after transfer of highly purified wild-type (WT) B-cells, but not IL-10-deficient B-cells, thus implicating IL-10-secreting B-cells as a major regulatory cell type in stroke (Offner and Hurn 2012; Ren et al. 2011). In fact in our preceding study (Bodhankar et al. 2013a), we demonstrated that when B-cell-deficient mice were replenished with IL-10-rich B-cells 24 hours prior to MCAO, there was a significant decrease in infarct volume. We also demonstrated that the proinflammatory responses in the B-cell-deficient recipient mice were inhibited not only in brain but also in the periphery. With the important question about sufficiency of the IL-10+ B-cells in regulating infarct size in the B-cell-deficient mice being addressed, we decided to take our studies further, since results obtained in B-cell-deficient mice do not directly translate to a clinical perspective.
The primary purpose of the present study was to assess the immunoregulatory role of IL-10-rich B-cells in impacting infarct size in B-cell-sufficient mice. Hence, these studies were carried out in C57BL/6J (wild-type, WT) mice. The goal of these studies was to test the efficacy of IL-10-rich B-cells in controlling infarct size when given prophylactically (i.e. 24 hours before the induction of stroke) or therapeutically (i.e. 4 hours after MCAO-induction). Our results clearly demonstrate that the transferred IL-10-rich B-cells influence the T-cells in the periphery, which acquire decreased proinflammatory characteristics. The transfer of IL-10-producing B-cells also led to an increase in the regulatory sub-populations in the periphery. The regulation of peripheral immune responses eventually led to decreased infiltration of immune cells into the ischemic hemispheres of the IL-10+ B-cell-treated group. The transfer of IL-10+ B-cells was also able to impact the inflammatory milieu of the ischemic hemisphere. Our studies are the first to demonstrate a major immunoregulatory role for IL-10+ B-cells in inhibiting reperfusion based cerebral injury and also implicating their potential role in attenuating complications due to post-stroke immunosuppression.
Materials and Methods
Animals
C57BL/6J (wild-type, WT) mice 8 to 12 weeks of age and weighing 20 to 25 g (Jackson Laboratory, Sacramento, CA, USA) were used as recipients for all adoptive transfers and induction of middle cerebral artery occlusion (MCAO) and were housed at the Oregon Health and Science University in accordance with institutional guidelines. Male IL10-GFP reporter mice (on a C57BL/6J background) were used at 8–10 weeks of age as donors for adoptive transfers and were bred and housed in the Animal Resource Facility at the Portland Veterans Affairs Medical Center in accordance with institutional guidelines. The IL10-GFP reporter mice have a floxed neomycin-IRES eGFP cassette (Madan et al. 2009) inserted between the endogenous stop site and the poly(A) site of the Il10 gene to help track IL-10 producing cells in vivo. The mice designated as Vert-X are homozygous, develop normally and are viable and fertile without any obvious phenotype. All experimental protocols were approved by Portland Veteran Affairs Medical Center and Oregon Health and Science University Animal Care and Use Committees.
Cell sorting and adoptive transfer of B-cells
Male IL-10 GFP reporter mice served as donors of B-cells. Splenic CD19+ B-cells were purified using paramagnetic bead-conjugated antibodies (Abs) from the CD19 cell isolation kit and subsequently separated by AutoMACS (Miltenyi Biotec, Auburn, CA). The negative fraction of the cells thus separated were CD19+ B-cells with a purity of ≥ 92%. CD19+ B-cells were suspended in RPMI 1640 medium with 2% Fetal Bovine Serum (FBS) and cultured in the presence of 1 μg/mL lipopolysaccharide (LPS, E. coli strain K12) for 48 hours. After 48 hours of culture, B-cells were harvested from culture plates, washed free of LPS and viable cells were counted using a hemocytometer with trypan blue exclusion method. Five million purified IL-10-GFP+ B-cells from the donor mice were suspended in 100 μL RPMI 1640 medium and were transferred intravenously (i.v.) into WT mice (experimental group) 24 hours before MCAO for one set of experiments and 4 hours after MCAO for a second set of experiments. Each WT mouse either received 5×106/100 μL purified IL-10-GFP+ B-cells or 100 μL RPMI 1640 medium (control group).
Middle cerebral artery occlusion model
Transient focal cerebral ischemia was induced in male WT mice for 60 minutes by reversible right middle cerebral artery occlusion (MCAO) under isoflurane anesthesia followed by 96 hours of reperfusion as previously described (Chen et al. 2012). The surgeon was blinded to treatment group. Head and body temperature were controlled at 36.5 ± 1.0°C throughout MCAO surgery with warm water pads and a heating lamp. Occlusion and reperfusion were verified in each animal by laser Doppler flowmetry (LDF) (Model DRT4, Moor Instruments, Inc., Wilmington, DE, USA). Occlusion was accomplished by introducing a 6-0 nylon monofilament (ETHICON, Inc., Somerville, NJ, USA) with a silicone-coated (Xantopren comfort light, Heraeus, Germany) tip through an external carotid artery stump distal to the internal carotid artery to the origin of the middle cerebral artery. Adequacy of artery occlusion was confirmed by monitoring cortical blood flow at the onset of the occlusion with a LDF probe affixed to the skull. Animals were excluded if intra-ischemic LDF was greater than 25% pre-ischemic baseline. After the occlusion, the incision was closed with 6-0 surgical sutures (ETHICON, Inc., Somerville, NJ, USA). Then each animal was awakened during occlusion and was placed in a separate cage with a warm water pad and heating lamp. At the end of the 60 minute ischemic period, mice were briefly re-anesthetized, the laser Doppler probe was repositioned over the same site on the skull, the occluding filament was withdrawn for reperfusion, and the incision was closed with 6-0 surgical sutures (ETHICON, Inc., Somerville, NJ, USA). Each animal was then awakened and recovered in a separate cage with a warm water pad.
Neurological deficit scores
Neurological deficit scores were determined at 1, 24, 48, 72, and 96 hours of reperfusion to confirm ischemia and the presence of ischemic injury using a 0 to 4 point scale as follows: 0, no neurological dysfunction; 1, failure to extend left forelimb fully when lifted by tail; 2, circling to the contralateral side; 3, falling to the left; and 4, no spontaneous movement or in a comatose state (Chen et al. 2012). Any animal without a deficit at 1 h of reperfusion was excluded from the study.
Infarct volume analysis
Individual performing infarct volume analysis was blinded to treatment group. Mice were euthanized and brains collected at 96 hours of reperfusion for 2,3,5-triphenyltetrazolium chloride histology and then digital image analysis of infarct volume as previously described (Chen et al. 2012). Images were analyzed using Sigma Scan Pro 5.0 Software (Systat, Inc., Point Richmond, CA). To control for edema, regional infarct volume (cortex, striatum, and hemisphere) was determined by subtraction of the ipsilateral non-infarcted regional volume from the contralateral regional volume. This value was then divided by the contralateral regional volume and multiplied by 100 to yield regional infarct volume as a percent of the contralateral region.
Isolation of leukocytes from spleen and brain
Spleens from individual control and B-cell recipient WT mice were removed and a single-cell suspension was prepared by passing the tissue through a 100 μm nylon mesh (BD Falcon, Bedford, MA). The cells were washed using RPMI 1640. Red cells were lysed using 1x red cell lysis buffer (eBioscience, Inc., San Diego, CA) and incubated for 3 min. Cells were then washed twice with RPMI 1640, counted, and resuspended in stimulation medium (RPMI, containing 10% FBS, 1% sodium pyruvate, 1% L-glutamine, 0.4% βME). The brain was divided into the ischemic (right) and nonischemic (left) hemisphere, dissociated mechanically through a 100 μm nylon mesh screen, resuspended in 80% Percoll (GE Healthcare, Pittsburgh, PA) overlaid with 40% Percoll and subjected to density gradient centrifugation for 30 min at 1600 rpm, according to a method described previously (Campanella et al. 2002). Inflammatory cells were removed from the interphase for further analysis. Cells were then washed twice with RPMI 1640, counted and resuspended in stimulation medium. Cells from individual brain hemispheres were evaluated by flow cytometry.
Analysis of cell populations by fluorescence-activated cell sorting (FACS)
All antibodies were purchased from BD Biosciences (San Jose, CA) or eBioscience, Inc. (San Diego, CA) as published (Offner et al. 2006b; Offner et al. 2006c). Four-color (FITC, PE, APC and PI/PerCP/PECy7) fluorescence flow cytometry analyses were performed to determine the phenotypes of splenocytes and brain leukocytes as previously published (Offner et al. 2006a). Single-cell suspensions were washed with staining medium (PBS containing 0.1% NaN3 and 0.5% bovine serum albumin (Sigma, Illinois) and incubated with the combinations of the following monoclonal antibodies: CD3 (17A2 BD Pharmingen), CD4 (GK1.5, BD Pharmingen), CD8a (53–6.7, BD Pharmingen), CD11b (M1/70, eBioscience), CD45 (Ly-5, BD Pharmingen), CD19 (1D3, BD Pharmingen), CD1d (1B1, BD Pharmingen), CD122 (TM-β1 BD Pharmingen), ICAM-1 (3E2, BD Pharmingen), LFA-1(M17/4, eBioscience), MHCII (2G9, BD Pharmingen), CD69 (H1.2F3, BD Pharmingen), for 10 min at 4°C. One mL of staining buffer was added to wash the cells. Propidium iodide (PI) was added to identify dead cells whenever only 3 channels on the FACSCalibur were used for detection of fluorescent antibody staining. The FoxP3 staining kit was used according to the manufacturer’s protocol (eBioscience) and as previously described (Zhang et al. 2010). FACS data acquisition was performed on FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and data were analyzed using FCS express software (De Novo Software, Los Angeles, CA).
Intracellular Staining
Intracellular staining was visualized using a published immunofluorescence protocol (Subramanian et al. 2011). Briefly, 2×106 cells/mL were resuspended in complete medium (RPMI 1640 containing 10% fetal calf serum, 1 mM/L pyruvate, 200 μg/mL penicillin, 200 U/mL streptomycin, 4 mM/L L-glutamine, and 5×10−5 mol/L 2-β-ME) with PMA (50 ng/mL), ionomycin (500 ng/mL), and Brefeldin A (10 μg/mL; Sigma-Aldrich) for 4 hours. For intracellular IL-10 detection, a modification was followed for the immunofluorescence staining protocol (Yanaba et al. 2008). Briefly, isolated leukocytes or purified cells were resuspended (2×106 cells/mL) in complete medium and cultured with LPS (10 μg/mL) in addition to PMA (50 ng/mL), ionomycin (500 ng/mL), and Brefeldin A (10 μg/mL) (all reagents are from Sigma-Aldrich) for 4 hours. Fc receptors were blocked with anti-FcR mAb (2.3G2; BD PharMingen) before cell-surface staining, and fixed and permeabilized with the Fixation/Permeabilization buffer (eBioscience) according to the manufacturer’s instructions. Permeabilized cells were washed with 1X Permeabilization Buffer (eBioscience) and were stained with tumor necrosis factor-α (MP6-XT22; BD Pharmingen), IL-17A (TC11-18H10, BD Pharmingen), (Interferon-γ (XMG1.2; eBioscience), and/or APC-conjugated anti-IL-10 mAb (JES5-16E3; eBioscience). Isotype matched mAb served as negative controls to demonstrate specificity and to establish background TNF-α, IL-17, IFN-γ and IL-10-staining levels.
Cytokine detection by Luminex bead array
Single-cell suspensions of spleen and brain mononuclear cells obtained from control (RPMI-treated) and IL-10+ B-cells-treated WT mice 96 hours after MCAO were cultured in 24 well plate coated with plate-bound anti-CD3/CD28 antibodies for 48 hours. Culture supernatants were evaluated for secreted levels of cytokines by using a Luminex Bio-Plex cytokine assay kit according to the instructions by the manufacturer (Bio-Rad, Richmond, CA).
RNA Isolation and Reverse transcription-Polymerase Chain Reaction
Total RNA was isolated from brains of control and IL-10+ B-cell recipient WT mice using the RNeasy mini kit protocol (Qiagen, Valencia, CA, USA) and converted into cDNA using oligo-dT, random hexamers, and Superscript RT II (Invitrogen, Grand Island, NY, USA). Reverse transcription-PCR was performed using TaqMan PCR master mix and pre-designed Taqman primers for MCP-1 (CCL2), MIP-1α (CCL3), RANTES (CCL5), MIF, CXCL2 (MIP-2), CXCL13, CXCR5, IL-1β, TNF-α, TGF-β1 and IL-10 primers (Applied Biosystems, Foster City, CA, USA). mRNA was quantified in reactions conducted on the ABI Prism 7000 Sequence Detection System (Applied Biosystems) and data represented as relative units compared with the GAPDH reference gene.
Statistical Analysis
Infarct volume data are presented as mean ± SEM. Differences in regional infarct volumes were determined with Student’s t-test. Functional outcomes for neurological deficit scores were analyzed by Mann Whitney Rank Sum test. Statistical significance was p<0.05. Statistical analyses were performed using SigmaStat Statistical Software, Version 9.01 (Systat Software Inc., Chicago, IL, USA). Statistical significance for the differences between percentages of cellular subtypes by FACS analysis and cytokine production by Luminex for splenocytes were analyzed with Student’s t-test. Statistical significance for data obtained by FACS analysis and Luminex for the brain leukocytes were analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Tukey’s test. The criterion for statistical significance was p ≤ 0.05. All values are reported as mean ± SEM. Significant differences are denoted as *p ≤0.05; **p≤ 0.01; ***p≤0.001.
Results
Mortality and Exclusions
Overall mortality from MCAO for infarct volume and immunology studies was 7 mice out of a total of 102 mice, with mortality ranging from 0 to 3 mice within the experimental groups. Overall number of mice excluded due to intra-ischemic LDF greater than 25% pre-ischemic baseline was 5 mice out of a total of 102 mice, with exclusions ranging from 0 to 2 mice within the experimental groups.
Adoptive transfer of IL10+ B-cells, 24 hours before or 4 hours after MCAO, reduces infarct volume in male WT mice
As shown in Fig. 1A, WT mice that received IL10+ B-cells (n=10) 24 hours before MCAO exhibited significantly reduced cortical (p=0.026), striatal (p=0.012) and total hemisphere (p=0.016) infarct volumes after 60 minutes MCAO followed by 96 hours of reperfusion compared to no-cell transferred vehicle (RPMI) controls (n=12). Representative cerebral sections from WT mice treated with RPMI or IL10+ B-cells are shown in Fig. 1B. Distribution of neurological deficit scores within each group at each time point would suggest that adoptive transfer of IL10+ B-cells 24 hours before MCAO had a greater impact on decreasing, and thus improving, the neurological deficit score over time when compared to WT mice treated with RPMI (Table 1). Differences in the median neurological deficit scores between the experimental and control groups were greater than would be expected by chance at 24 hours reperfusion (p=0.004) but not at 1 (p=0.245), 48 (p=0.218), 72 (p=0.103) or 96 (p=0.103) hours reperfusion (Table 1).
Figure 1. Adoptive transfer of IL10+ B-cells, 24 hours before or 4 hours after MCAO, reduces infarct volume in male WT mice.

A) Intravenous transfer of 5 million IL10+ B-cells given 24 hours before surgery to induce middle cerebral artery occlusion (MCAO) reduced infarct volume in C57BL/6J (wild-type, WT) mice (n=10), 96 hours following 60 minutes of MCAO compared to intravenous transfer of RPMI vehicle (no cells) (n=12). *P<0.05 B) Representative 2,3,5 triphenyltetetrazolium chloride stained cerebral sections 96 hours following 60 minutes of MCAO. Localization of the ischemic lesion differed between WT mice receiving intravenous IL10+ B-cells (right column) vs. RPMI vehicle (no cells)(left column) 24 hours before MCAO. C) Intravenous transfer of 5 million IL10+ B-cells 4 hours after surgery to induce MCAO reduced cortical and hemispheric (total) infarct volume in WT mice (n=13), 96 hours following 60 minutes of MCAO compared to intravenous transfer of RPMI vehicle (no cells) (n=14),. *p<0.05; **p<0.01 D) Representative 2,3,5 triphenyltetetrazolium chloride stained cerebral sections 96 hours following 60 minutes of MCAO. Localization of the ischemic lesion differed between WT mice receiving intravenous IL10+ B-cells (right column) vs. RPMI vehicle (no cells)(left column) 4 hours after MCAO.
Table 1.
Neurological deficit score distribution and median scores at various reperfusion time points following 60 minutes of middle cerebral artery occlusion (MCAO) in C57BL/6J (wild-type, WT) treated intravenously with RPMI (vehicle) or 5 million IL10+ B-cells 24 hours before MCAO.
| Experimental Groups | Distribution Of Neurological Deficit Scores During Reperfusion
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 Hour | 24 Hours | 48 Hours | 72 Hours | 96 Hours | |||||||||||||||||||||
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | |
| WT+RPMI (n=12) | 0 | 0 | 6 | 6 | 0 | 0 | 2 | 10 | 0 | 0 | 0 | 7 | 5 | 0 | 0 | 0 | 7 | 5 | 0 | 0 | 0 | 7 | 5 | 0 | 0 |
| WT+IL10+ B-cells (n=10) | 0 | 0 | 8 | 2 | 0 | 0 | 9 | 1 | 0 | 0 | 0 | 9 | 1 | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 10 | 0 | 0 | 0 |
|
| |||||||||||||||||||||||||
| Experimental Groups |
Median Neurological Deficit Scores During Reperfusion
|
||||||||||||||||||||||||
| 1 Hour (P=0.245) | 24 Hours (P=0.004) | 48 Hours (P=0.218) | 72 Hours (P=0.103) | 96 Hours (P=0.103) | |||||||||||||||||||||
|
| |||||||||||||||||||||||||
| WT+RPMI (n=12) | 2.5 | 2 | 1 | 1 | 1 | ||||||||||||||||||||
| WT+IL10+ B-cells (n=10) | 2 | 1# | 1 | 1 | 1 | ||||||||||||||||||||
p<0.01 compared to RPMI
As shown in Fig. 1C, WT mice that received IL10+ B-cells (n=13) 4 hours after MCAO exhibited significantly reduced cortical (p=0.001), striatal (p=0.036) and total hemisphere (p=0.006) infarct volumes after 60 minutes MCAO followed by 96 hours of reperfusion compared to no-cell transferred vehicle (RPMI) controls (n=14). Representative cerebral sections from WT mice treated with RPMI or IL10+ B-cells are shown in Fig. 1D. Distribution of neurological deficit scores within each group at each time point would suggest that adoptive transfer of IL10+ B-cells 4 hours after MCAO had a greater impact on decreasing, and thus improving, neurological deficit score over time when compared to mice treated with RPMI (Table 2). No statistically significant differences were observed in the median neurological deficit scores between the two experimental groups at 1 (p=0.574), 24 (p=0.864), 48 (p=0.231), 72 (p=0.393), and 96 (p=0.574) hours reperfusion (Table 2).
Table 2.
Neurological deficit score distribution and median scores at various reperfusion time points following 60 minutes of middle cerebral artery occlusion (MCAO) in C57BL/6J (wild-type, WT) treated intravenously with RPMI (vehicle) or 5 million IL10+ B-cells 4 hours after MCAO.
| Experimental Groups | Distribution Of Neurological Deficit Scores During Reperfusion
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 Hour | 24 Hours | 48 Hours | 72 Hours | 96 Hours | |||||||||||||||||||||
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | |
| WT+RPMI (n=14) | 0 | 0 | 10 | 4 | 0 | 0 | 8 | 6 | 0 | 0 | 0 | 8 | 6 | 0 | 0 | 0 | 8 | 6 | 0 | 0 | 0 | 10 | 4 | 0 | 0 |
| WT+IL10+ B-cells (n=13) | 0 | 0 | 11 | 2 | 0 | 0 | 8 | 5 | 0 | 0 | 0 | 11 | 2 | 0 | 0 | 0 | 10 | 3 | 0 | 0 | 0 | 11 | 2 | 0 | 0 |
|
| |||||||||||||||||||||||||
| Experimental Groups |
Median Neurological Deficit Scores During Reperfusion
|
||||||||||||||||||||||||
| 1 Hour (P=0.574) | 24 Hours (P=0.864) | 48 Hours (P=0.231) | 72 Hours (P=0.393) | 96 Hours (P=0.574) | |||||||||||||||||||||
|
| |||||||||||||||||||||||||
| WT+RPMI (n=14) | 2 | 1 | 1 | 1 | 1 | ||||||||||||||||||||
| WT+IL10+ B-cells (n=13) | 2 | 1 | 1 | 1 | 1 | ||||||||||||||||||||
B-cell-sufficient (WT) mice transferred with IL-10-GFP+ B-cells before MCAO-induction have significantly reduced splenic atrophy
As demonstrated in Fig. 1, transfer of IL-10+ B-cells, both 24 hours before and 4 hours after MCAO-induction, led to reduced infarct volumes. To further discern the regulatory role of IL-10+ B-cells on the developing infarct, we focused on their role in a prophylactic scenario. Hence, immunological studies were carried out in WT recipients of RPMI- or IL-10+ B-cells 24 hours before MCAO-induction. This was in continuation and allowed direct comparison with our preceding study (Bodhankar et al. 2013a) in which the IL-10+ B-cells were transferred to B-cell-deficient mice 24 hours before MCAO-induction.
Stroke-induced splenic atrophy is an established phenomenon (Offner et al. 2006c). We were also able to demonstrate in our preceding study that the transfer of the IL-10+ B-cells to μMT−/− mice rescued stroke-induced splenic atrophy. Hence to ascertain whether reduced ischemic brain injury in the WT mice also entails less splenic atrophy in the B-cell-transferred group, cell numbers in the spleen were evaluated in post-ischemic RPMI- and B-cell-transferred WT mice. As expected and previously demonstrated (Offner et al. 2006c; Bodhankar et al. 2013a), MCAO resulted in large reductions in spleen cell numbers in RPMI-treated WT mice. The reduction in spleen counts was from ~100 million spleen cells/naïve WT mouse (data not shown) to ~7–8 million spleens cells/WT mouse after 60 minutes MCAO and 96 hours of reperfusion, accounting for ~93% reduction in the total splenocytes (Fig. 2A). Interestingly, the viable cell counts were significantly higher (~16–18 million cells/mouse) in spleens of WT mice receiving IL-10 GFP+ B-cells as compared to the RPMI-treated WT mice (p=0.048) 96 hours following MCAO (Fig. 2A). Given the partial restoration of splenic cell numbers in the IL-10+ B-cell-treated group, we further evaluated the frequencies of specific surviving splenic cell types. There was a significant increase in the percentages of both the CD4+ T-cells (CD4+; p=0.002) and CD8+ T-cells (CD8+; p= 0.047) in the spleens of WT mice treated with IL-10-GFP+ B-cells (Fig. 2B). However, there were no differences in the percentages (or absolute numbers; data not shown) of B-cells (CD19+) or monocytes (CD11b+). After 96 hours of reperfusion, the GFP+ B-cells comprised only 2% of the total splenocytes from WT recipient mice (data not shown), indicating that the increase in the cell numbers is not contributed by the transferred cells, but rather may be due to the influence of the transferred cells on the splenocyte distribution.
Figure 2. B-cell-sufficient (WT) mice transferred with IL-10-GFP+ B-cells before MCAO-induction have significantly reduced splenic atrophy.
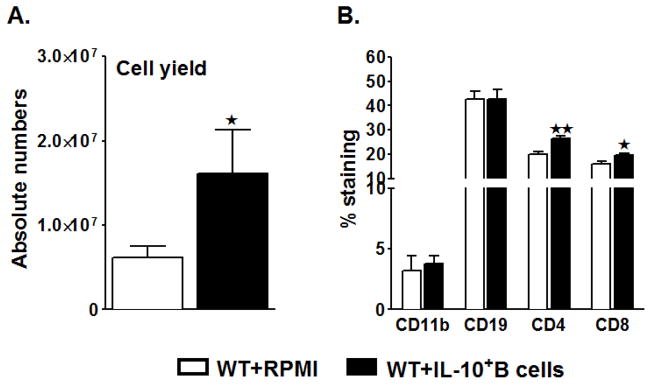
Ninety six hours after MCAO, mononuclear cells were isolated from spleens of RPMI or IL-10-GFP+ B-cell recipient WT mice and analyzed for: A. Total cell count via hemocytometer. Values represent mean numbers (±SEM) of indicated cell subsets from 16–17 mice in each group, from at least 5 separate experiments; B. Comparison of CD11b+ monocytes, CD19+ B-cells, CD4+ and CD8+ T-cell populations. Values represent mean numbers (±SEM) of indicated cell subsets, gated on live leukocytes (by PI exclusion), from 6–7 mice of each group, from at least 2 separate experiments. Statistical analysis was performed with Student’s t-test to compare between RPMI and IL-10-GFP+ B-cell recipient mice. Significant differences between sample means are indicated (*p≤0.05).
IL-10-GFP+ B-cells, transferred 24 hours before MCAO, suppress the pro-inflammatory state and inhibit activation of the T-cells and monocytes in the periphery of recipient mice
It is now well established that the initial insult from stroke is followed by an early induction of inflammatory cytokines and chemokines that attract mononuclear cells and granulocytes, which cause further damage to the ischemic and surrounding areas of brain tissue (Offner et al. 2006b; Offner et al. 2009). To further evaluate the possible regulatory effects of the IL-10-producing B-cells on the resident spleen cells when transferred 24 hours before MCAO-induction, we evaluated the proinflammatory milieu in the spleens by Luminex assay after 60 minutes of MCAO and 96 hours of reperfusion. Amongst the cytokines analyzed, the levels of TNF-α (p=0.013) and IL-17 (p=0.043) were significantly reduced in the spleens of the B-cell-transferred group (Fig. 3A), with a downward trend in MCP-1 as well. To discern the cytokine production by specific cells (i.e. T-cells and monocytes) in the spleens post-MCAO, Flow Cytometry was carried out on ex vivo-activated cells (Fig 3B). Monocytes (CD11b+ cells) demonstrated a trend in the reduction of TNF-α, but there was no difference in TNF-α production by the T-cells (CD3+ cells) between the two recipient groups of WT mice. However, there was a striking reduction in the production of IFN-γ by CD3+ T-cells (p=0.0001) and of IL-17 by CD4+ T-cells (p=0.027) (Fig 3B). Further, the expression of Major Histocompatibility Complex class II and the co-stimulatory molecule CD80 on monocytes were analyzed. On one hand, where the expression of CD80 was significantly reduced (p=0.035) in the B-cell-transferred WT mice, there was no difference in the percent expression of MHCII (Fig. 3C).
Figure 3. IL-10-GFP+ B-cells transferred 24 hours before MCAO suppress the pro-inflammatory state and inhibit activation of the T-cells and monocytes in the periphery of recipient mice.

Splenocytes were isolated from RPMI or IL-10-GFP+ B-cell recipient WT mice, 96 hours after MCAO and A. were stimulated with plate-bound anti-CD3/CD28 antibodies for 48 h. Culture supernatants were evaluated for secreted levels of cytokines by Luminex Bead array. Data are representative of 2 independent experiments (mean ± SEM); B. CD11b+ monocytes were analyzed for TNF-α+ production, CD3+ T-cells for TNF-α+ and IFN-γ+ production and CD4+ T-cells for IL-17+ production by Flow cytometry; C. expression of CD80 and MHC class II on gated CD11b+ monocytes and expression of activation marker, CD69, on gated CD4+ T-cells. Values for B. and C. represent mean percentages (±SEM) of indicated cell subsets from 6 RPMI-pretreated and 7 IL-10+ B cell-pretreated mice, from at least 2 separate experiments. Statistical analysis was performed with Student’s t-test to compare between RPMI and IL-10-GFP+ B-cell recipient WT mice. Significant differences between sample means are indicated (*p≤0.05 and **p≤0.01).
To further correlate the reduction in infarct volumes and splenic atrophy to the activation states of T-cells in spleens that might be influencing the splenic milieu, we evaluated the percent frequencies of CD44 and CD69 expression by CD4+ T-cells in the spleens of control vs. B-cell-replenished WT mice. CD44 is a memory T-cell marker that is involved in T-cell activation, binding to selectins on the endothelium during transmigration across the blood-brain barrier and release of cytokines in brain to amplify the inflammatory response; and CD69 is an ‘activation marker’ that is rapidly induced on mature T-cells after stimulation through the TCR. As shown in Fig. 3C, splenocytes from RPMI-treated WT mice, after MCAO, had increased percentages of CD69+ CD4+ T-cells. Transfer of IL-10 GFP+ B-cells prevented this increase, resulting in a significant decrease in CD69 expression by CD4+ T-cells (p=0.0.017) compared to RPMI-treated mice. The percentage expression of CD44 expression between the two groups was not different (data not shown). These results indicate a dampening role played by the IL-10+ B-cells in limiting both the proinflammatory responses and the activation states of specifically T-cells in the periphery after MCAO.
Transferred IL-10-GFP+ B-cells lead to an increase in the regulatory cell populations and increased anti-inflammatory status of the spleens
With the demonstration of attenuation of the proinflammatory milieu and activation states of the T-cells and monocytes, we next sought to determine whether the IL-10-rich B-cells, when transferred 24 hours before MCAO-induction, influence the frequencies of the various regulatory subpopulations in the periphery. Our earlier studies demonstrate that MCAO induced a 3 fold increase in the percentage of Foxp3+CD4+ Tregs in the spleens as compared to sham or naïve mice (Offner et al. 2006c). In our preceding study (Bodhankar et al. 2013a) we also demonstrated a significant increase in the frequencies of a recently identified regulatory T-cell population, CD8+CD122+. Hence, we assessed these known regulatory cells in the MCAO-protected IL-10-GFP+ B-cell recipients by Flow Cytometry. We observed significant increases in the frequency of both Foxp3+CD4+ (p=0.014) and CD8+CD122+ (p=0.003) Tregs, as well as the CD1dhiCD19+ Bregs (p=0.044, Fig. 4) in the B-cell recipient vs. control mice (Fig. 4). These results further confirm the regulatory role of the donor IL-10 GFP+ B-cells.
Figure 4. Transferred IL-10-GFP+ B-cells lead to an increase in the regulatory cell populations.

Splenocytes from RPMI and IL-10-GFP+ B-cell transferred WT recipient mice were harvested 96 hours after MCAO and assessed for expression of: FoxP3+CD4+ T-cells, CD8+CD122+ T-cells and CD1dhighCD19+ regulatory B-cells. Data are representative of 2 independent experiments with spleens processed from 6 RPMI-pretreated and 7 IL-10+ B cell-pretreated mice (mean ± SEM). Significant differences between the groups were determined using Student’s t-test. (*p≤0.05, **p≤0.01 and ***p≤0.001).
Unlike our earlier study (Bodhankar et al. 2013a), where the IL-10-GFP+ B-cells were transferred to B-cell-deficient (μMT−/−) mice, our present study involved B-cell sufficient WT recipients. Having previously demonstrated that B-cells are the major producers of IL-10 post-MCAO (Ren et al. 2011), we here similarly evaluated IL-10 production by the various cells in the spleens by the Luminex assay. As demonstrated in Fig. 5A, splenocyte supernatants from the B-cell-recipient WT mice had significantly increased levels of IL-10 production. Further comparison of IL-10-producing capacities of recipient (GFP−) vs. donor (GFP+) cells by Flow Cytometry revealed a significant increase in the IL-10 production by the GFP− splenic cells in the B-cell-transferred WT recipient mice. A low percentage of GFP+ transferred cells was also found in the spleens of the recipient mice. As demonstrated in Fig. 5B, the total IL-10 GFP− percentage expressed by all cells in spleens was significantly higher (p=0.012) in the B-cell-recipient WT mice as compared to the RPMI-treated mice, and we further demonstrated a significant increase in the IL-10-producing capacity of the CD8+CD122+ and the CD1dhiCD19+ sub-populations in the recipients of regulatory B-cells (p=0.043 and p=0.017 respectively). These results confirm that the IL-10-GFP+ B-cells play a protective role in the induction of regulatory T-cells and B-cells, which in turn could contribute to immunomodulation in the recipient mice after stroke.
Figure 5. Transferred IL-10-GFP+ B-cells lead to increased anti-inflammatory status of the spleens.
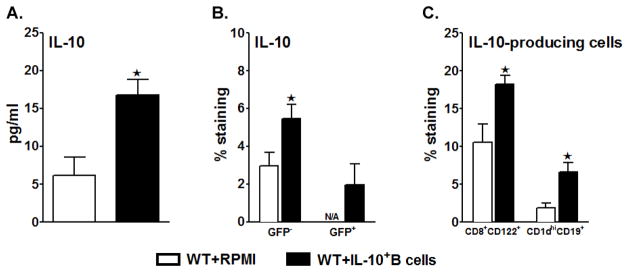
Splenocytes were isolated from RPMI or IL-10-GFP+ B-cell recipient WT mice 96 hours after MCAO and were A. stimulated with plate-bound anti-CD3/CD28 antibodies for 48 h. Culture supernatants were evaluated for secreted levels of IL-10 by Luminex Bead array. Data are representative of 2 independent experiments (mean ± SEM); B. analyzed for IL-10-production on GFP− cells using IL-10 APC mAb for detection and/or detecting the GFP+CD19+ cells (i.e. IL-10-GFP+ transferred B-cells) by Flow cytometry; C. assessed for expression of IL-10 (GFP−) on gated CD8+CD122+ T-cells and CD1dhighCD19+ regulatory B-cells. Values for B. and C. represent mean percentages (±SEM) of indicated cell subsets from 6 RPMI-pretreated and 7 IL-10+ B cell-pretreated mice, from at least 2 separate experiments. Significant differences between the groups were determined using Student’s t-test. (*p≤0.05).
Transfer of IL-10 GFP+ B-cells, 24 hours before MCAO reduces LFA-1 expression, possibly preventing migration of activated T-cells outside of spleens
The intracellular adhesion molecule-1 (ICAM-1) binds to the lymphocyte function–associated antigen-1 (LFA-1). The interaction of ICAM-1 with LFA-1 is required for adhesion of leukocytes to endothelium and their transmigration into tissues of inflammation (Yang et al. 2005). Because the influx of leukocytes into ischemic brain tissue contributes to brain damage, it seemed logical to investigate expression of these molecules in the periphery as agents that could be involved in regulating this inflammatory response. As demonstrated in Fig. 2B, the frequencies of CD4+ and CD8+ T-cells were significantly higher in the spleens of IL-10+ B-cell recipients. To discern whether this difference is due to less apoptotic cells or due to the retention of the cells in the periphery, the adhesion molecules (ICAM-1 and LFA-1, respectively) on monocytes and T-cells were determined by Flow Cytometry. While there was no difference in the expression levels of ICAM-1 by the monocytes between the two group of recipients (Fig. 6A), the recipients of regulatory B-cells demonstrated a significant reduction in LFA-1 expression by both CD4+ and CD8+ T-cells (p=0.046 and p=0.043; respectively) (Fig. 6B). These data suggest that down-regulation of adhesion molecules in the periphery of the B cell-recipients may plausibly prevent a plausible extravasation of activated/proinflammatory cells from the periphery to the target tissue of injury.
Figure 6. Transfer of IL-10 GFP+ B-cells 24 hours before MCAO prevents migration of activated T-cells outside of spleens.

Splenocytes from RPMI and IL-10-GFP+ B-cell transferred WT recipient mice were harvested 96 hours after MCAO and assessed for expression of ICAM-1 by CD11b+ monocytes and LFA-1 by CD4+ and CD8+ T-cells. Data are representative of 2 independent experiments with spleens processed from 6 RPMI-pretreated and 6 IL-10+ B cell-pretreated mice (mean ± SEM). Significant differences between the groups were determined using Student’s t-test. (*p≤0.05).
Transfer of IL-10-GFP+ B-cells to WT mice, 24 hours before MCAO, leads to reduced cerebral inflammatory cell infiltration post-MCAO
Our earlier study demonstrated that with a reduction in the infarct volumes there was also a reduction in infiltrating inflammatory cells into the ischemic brain hemispheres of the μMT−/− mice transferred with IL-10-GFP+ B-cells (Bodhankar et al. 2013a). To determine whether the transfer of IL-10-GFP+ B-cells to WT mice also potentially influences cellular infiltration leading to a similar reduction in the infiltration of the MCAO-affected hemisphere, we enumerated the total number of live leukocytes obtained from each of the hemispheres. RPMI-treated WT mice demonstrated a massive infiltration of leukocytes in the ischemic hemispheres after 96 hours of reperfusion. This cellular infiltration was significantly reduced (p=0.008) in the MCAO-affected hemispheres of the IL-10-GFP+ B-cells-recipient WT mice (Fig. 7A). The total cell yield from the contralateral hemispheres of each of RPMI-transferred or B-cell-transferred WT recipients remained similar, confirming the cellular infiltration on the ipsilateral side to be reperfusion-related. We further evaluated the leukocyte composition in the brains of the RPMI-treated (control) vs. IL-10-GFP+ B-cell-treated (experimental) mice. Percentage of activated microglia/infiltrating macrophages (CD11b+CD45high), total T-cells (CD3+) and total B-cells (CD19+) were evaluated by Flow Cytometry. After 96 hours of reperfusion, the absolute numbers of CD11b+CD45high (p=0.006), infiltrating CD3+ T-cells (p=0.016) and CD19+ (p=0.026) subpopulations were significantly reduced in the ipsilateral hemispheres of the MCAO-induced B-cell-transferred group as compared to the control WT mice (Fig. 7B). Transfer of IL-10+ B-cells also led to significant reductions in percentages of total TNF-α producing leukocytes (Fig. 7C, TNF-α+CD45+; p=0.025). Upon further characterizing the TNF-α-producing capacity, CD3+ T-cells from the B-cell transferred group demonstrated a significantly reduced capacity to produce TNF-α after 96 hours of reperfusion (Fig. 7C, p= 0.006). However, this was not true for the CD11b+ subpopulation (p= 0.908) in the ischemic hemispheres of B-cell vs. control recipient mice. These results confirmed that regulatory effects of the IL-10-GFP+ B-cells in B-cell-sufficient (WT) mice were similar to those demonstrated in μMT−/− mice. In both cases, it appears that the major regulatory effects occur in the periphery rather than in the brain, given that transferred IL-10-GFP+ B-cells could not be found in the left (non-ischemic) or right (ischemic) hemispheres of the brains of the experimental group (not shown).
Figure 7. Transfer of IL-10-GFP+ B-cells to WT mice 24 hours before MCAO leads to reduced cerebral inflammatory cell infiltration post-MCAO.

Ninety six hours after MCAO, mononuclear cells were isolated from brains of RPMI and IL-10 GFP+ B-cell recipient WT mice and were analyzed for: A. Total cell count via hemocytometer. Values for A. represent mean numbers (±SEM) of indicated cell subsets from 11 mice per group, from at least 3 separate experiments. B. Absolute numbers of CD11b+CD45high activated microglia (MG)/monocytes, CD3+ T-cells and CD19+ B-cells obtained from the non-ischemic (left) and ischemic (right) hemispheres of WT recipient mice by Flow cytometry. Values represent mean numbers (±SEM) of indicated cell subsets, gated on live leukocytes (by PI exclusion), from 6 mice each from the RPMI-pretreated and IL-10+ B cell-pretreated groups, from at least 2 separate experiments. C. total TNF-α production by CD45+ cells; TNF-α+CD11b+ CD45high activated microglia (MG)/monocytes and TNF-α+CD3+ T-cells in the nonischemic (left) and ischemic (right) hemispheres from WT mice transferred with medium or IL-10-GFP+ B-cells after 96 hours of MCAO. Values represent mean numbers (±SEM) of indicated cell subsets from 6 mice, each from the RPMI-pretreated and IL-10+ B cell-pretreated groups, from at least 2 separate experiments. Statistical analysis was performed with ANOVA followed by Tukey’s multiple comparison post-hoc test. Significant differences between sample means are indicated (#p ≤0.05; ##p ≤0.01; ###p ≤0.001 as compared to their respective left hemisphere and *p ≤0.05; **p ≤0.01 and ***p≤0.001 as compared to the ischemic right hemisphere of RPMI-treated WT recipient mice.
Transfer of IL-10-GFP+ B-cells 24 hours before MCAO induction reduces the overall pro-inflammatory milieu in the ischemic brain hemisphere
It is now known that ischemic stroke leads to secretion of pro-inflammatory cytokines and chemokines in spleen that can result in local injury and also trigger a massive influx of leukocytes from the periphery into the affected brain hemisphere, causing further damage. Hence, we determined the cytokine-secreting ability of leukocytes in brain by culturing mononuclear cells obtained from MCAO-affected ipsilateral vs. unaffected contralateral hemispheres from experimental and control mice. Culture supernatants were collected after 48 hours and cytokine concentrations determined by the Luminex assay. The results demonstrated a significant increase in the levels of secreted proinflammatory factors (IL-1β, TNF-α and MCP-1) from cells obtained from the ipsilateral vs. the contralateral hemispheres of the RPMI-treated control group. However, there was a significant reduction in levels of the proinflammatory factors but an increase in anti-inflammatory IL-10 in the ipsilateral hemisphere of IL-10-GFP+ B-cell treated mice vs. control mice (Fig. 8A).
Figure 8. Transfer of IL-10 GFP+ B-cells, 24 hours before MCAO-induction, reduces the overall pro-inflammatory milieu in the ischemic brain hemisphere.

Ninety-six hours after MCAO, mononuclear cells were isolated from the brains of RPMI and IL-10 GFP+ B-cell recipient WT mice and A. were stimulated with plate-bound anti-CD3/CD28 antibodies for 48 h. Culture supernatants were evaluated for secreted levels of IL-1β, TNF-α, MCP-1 and IL-10 by Luminex Bead array. Data are representative of 2 independent experiments (mean ± SEM); Brains were collected from RPMI and IL-10 GFP+ B-cell recipient WT mice 96 hours after occlusion, and mRNA prepared from ipsilateral (right) hemispheres of brain tissues for RT-PCR analysis. Relative expression (R.E.) of mRNA levels is presented for B. inflammatory cytokines and chemokines/receptors and C. anti-inflammatory cytokines. Values for B. and C. represent mean percentages (±SEM) from 4 RPMI-pretreated and 5 IL-10+ B cell-pretreated mice, from at least 2 separate experiments. Significant differences between the right ischemic hemispheres of the RPMI-treated and IL-10+ B-cell-treated groups were determined using Student’s t-test. (*p≤0.05 and **p≤0.01).
Additionally, we determined expression of proinflammatory cytokine and chemokine genes in the MCAO-affected ipsilateral hemisphere by real time PCR 96 hours after reperfusion. As expected, mice transferred with IL-10-GFP+ B-cells demonstrated significantly decreased levels of proinflammatory cytokine and chemokine mRNA expression as compared to mice receiving RPMI (Fig. 8B), with striking differences for the chemokines MIP-1α (p=0.032), MCP-1 (p=0.049) and MIF (p=0.022), but not for RANTES and MIP-2. Interestingly, the expression level of CXCL13, known to play a role in the recruitment of B-cells to the central nervous system (CNS) compartment during neuroinflammation, was significantly lower in the B-cell transferred group. There was no difference, however, in expression of CXCR5, the receptor for CXCL13, between the two groups. However, there was a significant reduction (p=0.010) in expression of matrix metalloproteinase (MMP)-2 (T cell derived zinc-containing endoproteinase that degrades the extracellular matrix and facilitates leukocyte migration through the basement membranes (Korpos et al. 2010)) in the ischemic hemisphere of IL-10+ B-cell-transferred vs. RPMI-treated mice.
Amongst the proinflammatory cytokines analyzed, there was a significant reduction in the expression levels of IL-1β (p=0.001) and TNF-α (p=0.047) in the MCAO-affected right hemispheres of the mice that received B-cells. We also analyzed the expression levels of TGF-β1 and IL-10 that are known as anti-inflammatory cytokines. As expected, there was a significant increase (p=0.032) in the mRNA levels of IL-10 in the right hemispheres of the mice that received B-cells (p<0.05). However, the relative expression levels of mRNA for TGF-β1 were significantly reduced (p=0.008) in the experimental group. These findings further confirm that the transferred IL-10-GFP+ B-cells create an anti-inflammatory environment, thus impacting not only the immune cells in periphery, but also leading to an anti-inflammatory milieu in the MCAO-affected brain hemisphere, despite the 96 hour reperfusion period.
Discussion
Ischemic stroke induces neurological deficits in almost one-third of the patients, leading to increased mortality and long-term functional disability (Castillo et al. 1997; Davalos et al. 1999). Among the many detrimental cascades (Lo et al. 2003) that have been characterized to be elicited following ischemic stroke, inflammatory mechanisms have come into focus in the latest research. These inflammatory processes are known to contribute substantially to secondary brain damage (Dirnagl 2004; Hurn et al. 2007; Wang et al. 2007), although the exact underlying mechanisms have not been completely unraveled. Studies in the transient MCAO model (Kleinschnitz et al. 2010) indicate that the reconstitution of RAG1−/− mice with B cells led to the development of significantly smaller brain infarcts as compared to the WT controls. However, since the end-point of these studies was as early as 22 h post-stroke, it was necessary to investigate the role of B cells that can eventually affect the T-cell activation pathway beyond the 22 h post-MCAO window. In our laboratory’s efforts in deciphering the different detrimental and/or beneficial arms of the immune system in post-ischemic stroke, we were able to demonstrate the immunoregulatory role of IL-10-rich B-cells in B-cell-deficient mice (Bodhankar et al. 2013a). Thus, our current study was initiated as an extension of this previous study, in taking into consideration the caveat that in a clinical scenario, stroke subjects would not likely have a complete B-cell-deficiency. The current study addresses the hypothesis that the IL-10+ regulatory B-cells would also play an immunoregulatory role in a B-cell-sufficient environment. As shown above, we here demonstrate the crucial role of IL-10+ B-cells in regulating infarct size in a B-cell-sufficient environment, both at 24 hours before and 4 hours after the induction of MCAO.
Inflammatory cascades in the course of the evolving ischemic brain damage are not restricted to resident cells of the brain but also involve recruited immune cells from systemic immune compartment (Iadecola and Anrather 2011; Liesz et al. 2011). It is now clear that human stroke creates not just a single organ insult, but a complex interaction between the CNS and the peripheral immune system. Several mediators of these inflammatory cascades have been previously described and T lymphocytes are central to the development of sustained inflammatory response. Cytokines involved in the proinflammatory response include IL-1β, IL-12, and IL-23, as well as interferon-γ (IFN- γ), IL-17A, and TNF-α. In contrast, IL-4, TGF-β, and IL-10 predominantly contribute to protective pathways. However, the specific integration of each cell type and cytokine in the post-ischemic inflammatory network still needs to be elucidated. Also, there remain major unanswered questions regarding mechanisms of antigen-independent T cell activation within hours after stroke. In ischemia-related inflammation, IL-17A can be crucial for chemokine induction. Importantly, IL-17A has been characterized to be rapidly released by γδT-cells in response to cytokine activation and/or engagement of innate receptors, in the absence of TCR activation. Similarly, IFN-γ pathways have also been implicated in ischemia/reperfusion injury. In lieu of the current literature, our studies demonstrated a significant decrease in IFN-γ and IL-17 production by splenic T-cells, in addition to reductions in MCP-1 and TNF-α (Figs. 3A and B) in the IL-10+ B-cell-treated mice. That the regulatory impact of the IL-10+ B-cells included inflammatory T-cells was further confirmed by a significant reduction in the activation marker, CD69, on T helper cells (CD4+ T-cells) in the periphery (Fig. 3C).
Infiltration of inflammatory leukocytes is also a well-described feature of human stroke (Mena et al. 2004). T-cells are known to accumulate in the post-ischemic brain within 24 hours of focal cerebral ischemia (Brait et al. 2010), with peak levels observed 72 hours after cerebral ischemia (Gelderblom et al. 2009). Consistent with these findings, our results also demonstrate a significant infiltration of T-cells in the ischemic hemispheres of the control mice, but reduced T cell and proinflammatory cytokine levels in the IL-10+ B-cell-transferred mice (Figs. 7 and 8). The potential importance of B-cell-dependent neutralization of T cell infiltration and cytokine secretion is supported by previous studies demonstrating an association between decreased T-cell cytokine levels (e.g., IL-17, IL-12, IL-23 and IFN-γ) and reduced infarct volume and improved neurologic outcome scores over 7 days post-stroke (Konoeda et al. 2010; Shichita et al. 2009). Our study further demonstrated reduced mRNA expression of various proinflammatory factors known to be important in promoting cerebral damage (Tuttolomondo et al. 2008) (Kowarik et al. 2012) in the ischemic hemispheres of the IL-10+ B-cell-treated mice (e.g. MIP-1α, MCP-1, MIF, MMP-2, CXCL13, IL-1β and TNF-α) vs. the RPMI-treated control mice. Of note, B-cell-dependent reduction of MMP-2 expression could reduce its enzymatic activity that promotes breakdown of the extracellular matrix and blood brain barrier, producing hemorrhage and inflammation.
Our results validate the anti-inflammatory milieu generated in the stroke-protected B-cell-recipients by demonstrating a significant increase in the IL-10 levels in the ischemic hemispheres of these mice. However, TGF-β expression was significantly decreased. Although TGF-β from microglia and macrophages may facilitate tissue repair by promoting the resolution of inflammation and exerting direct cytoprotective effects on surviving cells in the ischemic territory (Woodruff et al. 2011), it is also well known for its proinflammatory effects. One plausible explanation for our result might be that increased IL-10 levels observed in the IL-10+ B-cell-recipients could reduce the need for the resident cells to produce TGF-β to dampen the inflammation.
Recent studies indicate that the Foxp3+CD4+ Treg cell sub-population plays multiple key roles as immunomodulators of post-ischemic CNS injury, including regulating the immune inflammatory response, limiting lesion development and promoting tissue repair (Chen et al. 2013; Liesz et al. 2009; Stubbe et al. 2013; Offner et al. 2006d; Bodhankar et al. 2013b). Additionally, naturally occurring CD8+CD122+ regulatory T-cells (Rifa’i et al. 2004) can effectively suppress the proliferation and IFN-γ production of both CD8+ and CD4+ T-cells by virtue of their IL-10 production (Rifa’i et al. 2008). Moreover, regulatory B-cells (Bregs) are known to mediate protection against other inflammatory CNS conditions (Mann et al. 2007; Matsushita et al. 2010; Yanaba et al. 2008) and the ability of Bregs to limit CNS injury is associated with anti-inflammatory effects of IL-10 (Fillatreau et al. 2002). As shown above, we here demonstrate the potent multi-faceted ability of transferred IL-10+ B-cells to increase all three regulatory cell sub-populations in the periphery of the stroke-protected WT recipient mice (Fig. 4).
In summary, the current study demonstrates conclusively the beneficial effects of IL-10+ regulatory B-cells in reducing ischemic brain injury in B-cell-sufficient WT mice when administered 24 hours prior to or 4 hours after MCAO. B-cell treated mice had significantly smaller lesion volumes after 96 hours of reperfusion. When B-cells were transferred 24 hours before MCAO-induction, reduction in infarct volume was attributed to the significant reduction in the brain infiltrating cells as well as the attenuated proinflammatory state in the stroke-affected brain hemisphere. However, what makes our study of even broader interest is the demonstration that the regulatory B-cells bring about their effect not only by directly impacting the ischemic site but also by beneficially regulating peripheral immune responses. This peripheral regulation involved 1) preservation of splenocytes, 2) suppression of inflammatory T-cells and prevention of their subsequent activation and extravasation from the spleen, eventually leading to reduced cellular infiltration of the ischemic cerebral hemisphere, and 3) augmentation of three different T/Breg subpopulations, presumably indicating their role in immunomodulatory mechanisms, post-stroke. Thus, our novel findings are the first to implicate IL-10-producing B-cells as a major regulatory mediator in WT mice with ischemic stroke.
Acknowledgments
The authors wish to thank Andrew Lapato for technical assistance and Melissa S. Barber for assistance with manuscript submission. This work was supported by NIH/NINDS 1RO1 NS075887. This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- CNS
Central nervous system
- MCAO
Middle cerebral artery occlusion
- WT
wild-type
- TNF-α
Tumor necrosis factor α
- INF-γ
Interferon γ
- CD
Cluster of Differentiation
- MHC II
Major Histocompatibility Complex II
- RPMI
Roswell Park Memorial Institute
- IL
Interleukin
- PBS
Phosphate-buffered saline
- DNase I
Deoxyribonuclease I
- FACS
Fluorescence Activated Cell Sorter
- PI
propidium iodide
Footnotes
Competing interests: The authors declare no competing financial interests
Authors’ contribution: SB designed, performed the immunology experiments, carried out statistical analyses, prepared graphics and wrote the manuscript; YC performed the MCAO procedures, carried out statistical analyses, prepared the graphics and wrote the methods and results for infarct volume data; AAV critiqued and edited the manuscript; SJM directed study design and data analysis of the MCAO experiments and edited the manuscript; HO directed the overall study, supervised the immunological studies and data analysis and edited the manuscript. All authors read and approved the final version of the manuscript
References
- Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromolecular Med. 2005;7 (3):229–242. doi: 10.1385/NMM:7:3:229. [DOI] [PubMed] [Google Scholar]
- Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis. 2013a;28 (3):375–386. doi: 10.1007/s11011-013-9413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PD-L1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J Neuroinflammation. 2013b;10 (1):111. doi: 10.1186/1742-2094-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brait VH, Jackman KA, Walduck AK, Selemidis S, Diep H, Mast AE, Guida E, Broughton BR, Drummond GR, Sobey CG. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30 (7):1306–1317. doi: 10.1038/jcbfm.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33 (2):586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Noya M. Progression of ischaemic stroke and excitotoxic aminoacids. Lancet. 1997;349 (9045):79–83. doi: 10.1016/S0140-6736(96)04453-4. S0140-6736(96)04453-4 [pii] [DOI] [PubMed] [Google Scholar]
- Chen S, Wu H, Klebe D, Hong Y, Zhang J, Tang J. Regulatory T cell in stroke: a new paradigm for immune regulation. Clin Dev Immunol. 2013;2013:689827. doi: 10.1155/2013/689827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bodhankar S, Murphy SJ, Vandenbark AA, Alkayed NJ, Offner H. Intrastriatal B-cell administration limits infarct size after stroke in B-cell deficient mice. Metab Brain Dis. 2012;27 (4):487–493. doi: 10.1007/s11011-012-9317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A, Toni D, Iweins F, Lesaffre E, Bastianello S, Castillo J. Neurological deterioration in acute ischemic stroke: potential predictors and associated factors in the European cooperative acute stroke study (ECASS) I. Stroke. 1999;30 (12):2631–2636. doi: 10.1161/01.str.30.12.2631. [DOI] [PubMed] [Google Scholar]
- Dirnagl U. Inflammation in stroke: the good, the bad, and the unknown. Ernst Schering Res Found Workshop. 2004;(47):87–99. doi: 10.1007/978-3-662-05426-0_5. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371 (9624):1612–1623. doi: 10.1016/S0140-6736(08)60694-7. S0140-6736(08)60694-7 [pii] [DOI] [PubMed] [Google Scholar]
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–950. doi: 10.1038/ni833. ni833 [pii] [DOI] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40 (5):1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127 (1):e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27 (11):1798–1805. doi: 10.1038/sj.jcbfm.9600482. 9600482 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17 (7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Investigators EAST. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. (2001/10/24) 2001;57 doi: 10.1212/wnl.57.8.1428. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115 (18):3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- Konoeda F, Shichita T, Yoshida H, Sugiyama Y, Muto G, Hasegawa E, Morita R, Suzuki N, Yoshimura A. Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem Biophys Res Commun. 2010;402 (3):500–506. doi: 10.1016/j.bbrc.2010.10.058. [DOI] [PubMed] [Google Scholar]
- Korpos E, Wu C, Song J, Hallmann R, Sorokin L. Role of the extracellular matrix in lymphocyte migration. Cell Tissue Res. 2010;339 (1):47–57. doi: 10.1007/s00441-009-0853-3. [DOI] [PubMed] [Google Scholar]
- Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, Berthele A, Hemmer B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation. 2012;9:93. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15 (2):192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, Sun L, Bruder D, Stegemann S, Cerwenka A, Sommer C, Dalpke AH, Veltkamp R. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134 (Pt 3):704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4 (5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T, Hori M, Matsumoto M. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke. 2000;31 (7):1735–1743. doi: 10.1161/01.str.31.7.1735. [DOI] [PubMed] [Google Scholar]
- Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 2011;10 (5):471–480. doi: 10.1016/S1474-4422(11)70066-7. [DOI] [PubMed] [Google Scholar]
- Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, Boespflug N, Fogolin MB, Grobe L, Greweling M, Finkelman FD, Cardin R, Mohrs M, Muller W, Waisman A, Roers A, Karp CL. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183 (4):2312–2320. doi: 10.4049/jimmunol.0900185. jimmunol.0900185 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007;178(6):3447–3456. doi: 10.4049/jimmunol.178.6.3447. 178/6/3447 [pii] [DOI] [PubMed] [Google Scholar]
- Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185 (4):2240–2252. doi: 10.4049/jimmunol.1001307. jimmunol.1001307 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena H, Cadavid D, Rushing EJ. Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol. 2004;108 (6):524–530. doi: 10.1007/s00401-004-0918-z. [DOI] [PubMed] [Google Scholar]
- Offner H, Hurn PD. A Novel Hypothesis: Regulatory B Lymphocytes Shape Outcome from Experimental Stroke. Transl Stroke Res. 2012;3 (3):324–330. doi: 10.1007/s12975-012-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006a;26 (5):654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006b;26 (5):654–665. doi: 10.1038/sj.jcbfm.9600217. 9600217 [pii] [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006c;176(11):6523–6531. doi: 10.4049/jimmunol.176.11.6523. 176/11/6523 [pii] [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006d;176 (11):6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Offner H, Vandenbark AA, Hurn PD. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience. 2009;158 (3):1098–1111. doi: 10.1016/j.neuroscience.2008.05.033. S0306-4522(08)00765-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, Offner H. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011;31 (23):8556–8563. doi: 10.1523/JNEUROSCI.1623-11.2011. 31/23/8556 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifa’i M, Kawamoto Y, Nakashima I, Suzuki H. Essential roles of CD8+CD122+ regulatory T cells in the maintenance of T cell homeostasis. J Exp Med. 2004;200 (9):1123–1134. doi: 10.1084/jem.20040395. jem.20040395 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifa’i M, Shi Z, Zhang SY, Lee YH, Shiku H, Isobe K, Suzuki H. CD8+CD122+ regulatory T cells recognize activated T cells via conventional MHC class I-alphabetaTCR interaction and become IL-10-producing active regulatory cells. Int Immunol. 2008;20 (7):937–947. doi: 10.1093/intimm/dxn052. dxn052 [pii] [DOI] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15 (8):946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Stubbe T, Ebner F, Richter D, Randolf Engel O, Klehmet J, Royl G, Meisel A, Nitsch R, Meisel C, Brandt C. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33 (1):37–47. doi: 10.1038/jcbfm.2012.128. jcbfm2012128 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Yates M, Vandenbark AA, Offner H. Oestrogen-mediated protection of experimental autoimmune encephalomyelitis in the absence of Foxp3+ regulatory T cells implicates compensatory pathways including regulatory B cells. Immunology. 2011;132 (3):340–347. doi: 10.1111/j.1365-2567.2010.03380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14 (33):3574–3589. doi: 10.2174/138161208786848739. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184 (1–2):53–68. doi: 10.1016/j.jneuroim.2006.11.014. S0165-5728(06)00469-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6 (1):11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28 (5):639–650. doi: 10.1016/j.immuni.2008.03.017. S1074-7613(08)00193-3 [pii] [DOI] [PubMed] [Google Scholar]
- Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106 (2):584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H, Hurn PD. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol. 2010;184 (8):4087–4094. doi: 10.4049/jimmunol.0902339. jimmunol.0902339 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, Grabe N, Veltkamp R. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23 (1):34–44. doi: 10.1111/j.1750-3639.2012.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]