

Abstract
Staphylococcus aureus is a Gram-positive pathogen with an unusual mode of cell division in that it divides in orthogonal rather than parallel planes. Through selection using moenomycin, an antibiotic proposed to target peptidoglycan glycosyltransferases (PGTs), we have generated resistant mutants containing a single point mutation in the active site of the PGT domain of an essential peptidoglycan (PG) biosynthetic enzyme, PBP2. Using cell free polymerization assays, we show that this mutation alters PGT activity so that much shorter PG chains are made. The same mutation in another S. aureus PGT, SgtB, has a similar effect on glycan chain length. Moenomycin-resistant S. aureus strains containing mutated PGTs that make only short glycan polymers display major cell division defects, implicating peptidoglycan chain length in determining bacterial cell morphology and division site placement.
Introduction
Peptidoglycan (PG) is a cross-linked carbohydrate polymer that forms a protective shell around bacterial cells, enabling them to withstand fluctuations in osmotic pressure. The PG polymer is assembled on the bacterial cell surface in a process requiring two families of enzymes, the peptidoglycan glycosyltransferases (PGTs) and transpeptidases (TPs) (1–3). The PGTs make the carbohydrate chains of PG from a disaccharide-peptide precursor known as Lipid II (Figure 1A), and the TPs form peptide crosslinks between newly synthesized carbohydrate chains (4, 5). S. aureus contains three PGTs: PBP2, which also contains a TP domain, and two monofunctional PGTs, SgtA and SgtB (also called MGT) (4, 6, 7). Even though the full length protein PBP2 is essential, its PGT activity is not since inactivation of this domain can be complemented by SgtB (8, 9). The other PGT, SgtA, cannot complement loss of PBP2 PGT activity. Both SgtA and SgtB individually are non-essential and can be deleted without affecting growth (7).
Figure 1.

S. aureus peptidoglycan glycosyltransferases (PGTs) catalyze elongation of the glycan chain. a) Schematic of the PGT reaction to produce nascent (uncrosslinked) peptidoglycan (nPG) from the precursor Lipid II. There are three PGTs in S. aureus: SgtA, SgtB, and PBP2. b) Structure of the antibiotic moenomycin A (MoeA), a PGT inhibitor, with ring nomenclature indicated.
S. aureus has an unusual mode of division: each successive division plane forms perpendicularly to the previous plane. It was observed that rings of thickened PG surround S. aureus sacculi and apparently mark the old division plane (10, 11). It was therefore hypothesized that these rings serve as epigenetic determinants that enable the cell division machinery to orient formation of new division planes orthogonal to previous planes (11, 12). This model implies that defects in enzymes that synthesize the PG rings should lead to cell division defects. We hypothesized that it would be possible to obtain S. aureus mutants with altered PG synthesis activity by selecting for resistance to an antibiotic proposed to target the PGTs. Such mutants might provide insight into the importance of PGT activity in establishing orthogonal cell division in S. aureus.
Moenomycin A (MoeA, Figure 1B) is a natural product antibiotic shown to inhibit PGT activity in vitro (13–17). PGTs contain an extended active site cleft and carry out a reaction in which Lipid II adds to the reducing end of the growing polymer chain in a processive manner such that coupling is followed by translocation of product in the active site to position the new reducing end for further elongation (Figure 1A) (18). Crystal structures of several different PGTs containing MoeA have been reported and show that the compound binds in the same region of the active site where the elongating polymer binds (19–22). The antibiotic activity of MoeA has been attributed to PGT inhibition (23–24), although no resistant mutants containing PGT substitutions have been previously identified to confirm this mechanism (17). In fact, no specific mechanisms for MoeA resistance have been described, although it was suggested that upregulation of PBP genes may help overcome MoeA inhibition (25). Here we report MoeA-resistant S. aureus mutants that contain point mutations in the PGT domain of PBP2, providing the first genetic evidence that PGTs are the lethal targets of MoeA. The altered amino acids are located in the proposed polymer binding site of PBP2, and we show that the substitutions interfere with processive chain elongation, resulting in the formation of short glycan chains. Installing the same mutation in the monofunctional transglycosylase SgtB similarly reduces glycan chain length. We further show that cells expressing PGTs capable of making only short glycan chains display septal abnormalities, consistent with an important role for PG strand synthesis in determining the location of the division planes.
Results and Discussion
Isolation of S. aureus PBP2 mutants that confer resistance to Moenomycin A
We plated several S. aureus strains on agar plates containing 1 to 100 μg mL−1 antibiotic and obtained resistant mutants at frequencies ranging from 10−9 to 10−10 (Table 1, Supplementary Table S3). Resistance could be transferred by ϕ85-mediated bacteriophage transduction from a methicillin sensitive S. aureus (MSSA) clinical isolate, Newman, to several other strains, including the laboratory strain RN4220 and two methicillin resistant S. aureus (MRSA) clinical strains, MW2 and COL. The resistance frequencies and phage transduction results are consistent with a specific point mutation in a single gene (26, 27).
Table 1.
Frequency of MoeA resistant mutants for four different strains of S. aureus and distribution of the mutant alleles of pbpB, which encodes PBP2.
| S. aureus strain | Resistant mutant frequency | pbpB mutant alleles | |
|---|---|---|---|
| Y196D | P234Q | ||
| RN4220 | 2.3 × 10−9 | 4/4 | 0/4 |
| Newman (MSSA) | 2.2 × 10−10 | 37/40 | 3/40 |
| MW2 (MRSA) | 1.1 × 10−10 | 3/3 | 0/3 |
| COL (MRSA) | 5.2 × 10−10 | 3/3 | 0/3 |
Because we hypothesized that a likely MoeA resistance mechanism was mutation of the putative target proteins, we sequenced the three known genes for S. aureus PGTs, SgtA, SgtB, and PBP2, from ten MoeA-resistant colonies of strain Newman obtained from plating independent cultures. No mutations were found in the open reading frames or flanking regions of sgtA or sgtB; however, in all 10 mutants, pbpB, the gene encoding PBP2, contained a point mutation altering a single amino acid in the active site cleft of the PGT domain (19–21). In nine strains, Tyr196 was replaced by Asp (PBP2Y196D); in the remaining strain, Pro234 was substituted by Gln (PBP2P234Q). Thirty additional resistant Newman mutants and several resistant MRSA strains were subsequently sequenced and all had the same point mutations (Table 1). Residues 196 and 234 are located in the region of the active site cleft where MoeA binds, but they do not contact the proposed MoeA pharmacophore, which comprises the E-F disaccharide and phosphoglycerate moiety. Instead, they are proximal to the A-B rings of MoeA, which are thought to contribute to its inhibitory activity through non-specific binding interactions (Figure 2A).
Figure 2.

MoeA resistance mutations are located in the catalytic cleft of the PGT domain of S. aureus PBP2. a) Surface representation of the PGT domain of PBP2 with bound MoeA (19). The catalytic glutamate, Glu114, defines the center of the active site and is shown in red. Tyr196 is shown in cyan and Pro234 in green. PDB ID: 2OLV. Image generated by PyMOL. b) Gel electrophoresis showing the length distributions of PG polymers produced from Lipid II in vitro by wild type PBP2 and the PBP2Y196D mutant enzymes as a function of time. c) Model illustrating how different product distributions can arise in wild type and mutant enzymes. PBP2 polymerizes Lipid II in a processive manner, while PBP2Y196D polymerizes by a distributive mechanism. In the key, GlcNAc is N-acetylglucosamine and MurNAc is N-acetylmuramic acid.
To confirm that the mutations in PBP2 are responsible for the resistance phenotype, we constructed merodiploid strains in which wild type PBP2 or the two mutant PGTs, PBP2Y196D and PBP2P234Q, are expressed on a multicopy plasmid under control of their native promoter in S. aureus RN4220. No changes in MoeA susceptibility were observed for the strains expressing wild type PBP2 or the empty vector control; however, expression of the mutated proteins caused a 25-fold increase in the minimum inhibitory concentrations (MICs), from 0.5 μg mL−1 to 12.5 μg mL−1, which is similar to the increase observed in the spontaneous mutants (Supplementary Table S4 and Supplementary Figure S1). These results establish that the mutations observed in the PGT domain of PBP2 are responsible for MoeA resistance. Dominant resistance in the merodiploid strains suggests that the mutant enzymes remain catalytically competent in the presence of MoeA.
PGT domain mutants make shorter glycan polymers than their wild type counterparts
Since the Y196D substitution is much more common, we examined the enzymatic activity of PBP2Y196D. The full-length mutant protein was expressed and purified, and its ability to polymerize synthetic Lipid II was compared to that of wild type full-length PBP2 (28, 29). Reaction mixtures were analyzed by SDS-PAGE, which separates products to single disaccharide resolution (Figure 2B) (30, 31). As demonstrated previously, the wild type enzyme reacted to produce long polymers. The mutated enzyme was still able to couple Lipid II but made much shorter products. Hence, the mutation conferring MoeA resistance substantially alters the product distribution of oligosaccharide polymers in vitro. To examine if the same amino acid substitution in other PGTs (Supplementary Figure S2) would result in the same outcome, we replaced the corresponding tyrosine (Y181) in SgtB with an aspartate (Supplementary Figure S3A). Short glycan chains accumulated in polymerization reactions carried out with purified SgtBY181D, showing that it had a similar elongation defect as PBP2Y196D (Supplementary Figure S3B). Since the tyrosine residue is in the active site cleft where the elongating polymer binds, we suggest that substituting it with aspartate reduces affinity for the growing polymer chain, which increases the probability of product release and results in an accumulation of short glycan chains (Figure 2C) (30).
We also compared the ability of wild type and mutant PBP2Y196D enzymes to polymerize Lipid II in the presence of MoeA. SDS-PAGE analysis showed polymer formation when Lipid II was incubated with PBP2Y196D in the presence of MoeA (1:1 enzyme: MoeA) but not when it was incubated with wild type enzyme and MoeA (Supplementary Figure S4A). Analogous experiments using membranes from sensitive and resistant strains showed similar results in that MoeA fully inactivated polymer synthesis in wild type membranes at a concentration that permitted glycan chain synthesis in membranes from resistant strains (Supplementary Figure S4B). The PBP2 mutation thus reduces inhibition by MoeA, most likely by reducing binding.
The mutant strain is defective in the presence, but not absence, of moenomycin
Since the mutated PBP2Y196D enzyme has defective PGT activity and produces only short glycan chains, we examined several phenotypes of the corresponding mutant Newman strain. In the absence of MoeA, the growth rate of the mutant was identical to wild type (Figure 3A), and mutant cells imaged via light microscopy showed no changes with respect to overall morphology, septal placement, or daughter cell separation (Figure 3B, Supplementary Figure S5). Susceptibility to a wide range of antibiotics, including other cell wall active antibiotics such as beta-lactams and vancomycin, was also identical to wild type (Supplementary Table S4). In vitro and in vivo fitness was also unaffected since similar numbers of mutant and wild type cells were recovered from subcutaneous abscesses in a mouse model (Supplementary Tables S5, S6) (32, 33).
Figure 3.
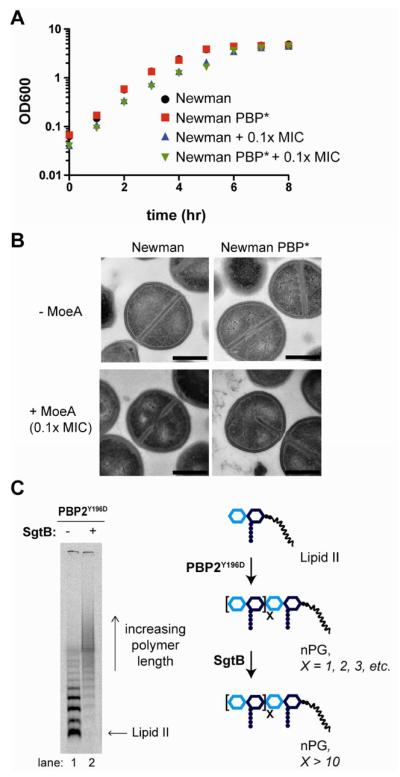
Moenomycin affects SgtB activity in the PBP2Y196D mutants. a) Growth of wild type S. aureus Newman and PBP2Y196D mutant (PBP*) cells in the absence and presence of 0.1X MIC MmA. Concentration of MmA is based on the MIC of each strain shown in Table S4. b) Representative transmission electron microscopy (TEM) images of S. aureus Newman and PBP* cells treated with 0.1X MIC of MoeA. The half septum defect is highlighted in the PBP* strain with added MoeA. Quantification of division defects for each strain is shown in Figure S5A. Scale bars indicate 500 nm. c) Gel electrophoresis shows that short PG polymers produced by PBP2Y196D are elongated by SgtB in vitro, as indicated by the scheme on the right. PBP2Y196D reactions (1 hr) were incubated in the absence or presence of StgB (30 min) prior to heat quenching.
Although appearing normal in the absence of MoeA, the mutant strain showed significant morphological aberrations in its presence, even at 0.1X MIC (1.5 μg mL−1; see Supplementary Table S4; Figure 3B, Supplementary Figure S5). While they grew well (Figure 3A), a substantial fraction of mutant cells treated with MoeA displayed numerous defects, including a thickened cell wall and an inability to separate efficiently. Multicellular clusters containing 3–8 cells were common and many ghost cells were observed (Supplementary Figure S5B). Perhaps more striking, in 1% of treated cells the division plane extended only halfway through the cell (Figure 3B), a phenotype never seen in the wildtype strain. In contrast, untreated mutant cells, like wild type, showed either complete septa or partial septa in which the division plane extends a similar distance from either edge of the cell when viewed in cross section (Figure 3B, 4B–C, Supplementary Figures S5 and S6). The S. aureus division plane is thought to grow centripetally inward from enzyme complexes of unknown composition that form a ring around the cells (10, 34). A hemi-septum suggests that these complexes assemble only along one half of the cell surface, ultimately forming a division plane that extends only half-way. Cell division defects were not observed in the wild type strain treated with MoeA at 0.1X its MIC (0.1 μg mL−1) (Figure 3B), but we note that the absolute MoeA concentration used is ten-fold lower than for the mutant strain. A comparison of the two strains at the same absolute concentration of MoeA is not possible since the wild type cells do not survive, presumably because the activity of all PGTs is inhibited. We suggest that the defects observed in the mutant strain at 1.5 μg mL−1 of MoeA result from inhibition of the monofunctional PGTs, leaving the compromised PBP2Y196D enzyme as the sole functional PGT.
Figure 4.

Inability to produce long PG strands leads to aberrant cell division. a) Light microscopy quantification of cell division states during early exponential growth of the wild-type S. aureus Newman and RN4220 strains, of the mutant PBP* strains, and of the double mutant RN4220 PBP2*ΔsgtB strain. The percentage of cells with division aberrations is underestimated as only cells with clearly visible septa could be counted; cells buried in clumps, which may be the most affected, could not be counted. See Figure S6 for more details. b) Light microscopy of wild type S. aureus RN4220 and the double mutant RN4220 PBP2*ΔsgtB. The cell outline and septa are visualized with a membrane dye (TMA-DPH). The arrows indicate the groups described in (A): full septum (blue); forming septum (red), hemi-septum (purple); parallel septa (green); and otherwise aberrant septum (orange). Scale bar represents 2 μm. c) TEM of wild-type S. aureus RN4220 (left) and the double mutant RN4220 PBP2*ΔsgtB. The double mutants exhibit aberrant division phenotypes including chaining phenotype (middle) and hemi-septa formation in (right, indicated with arrow). Scale bar represents 1 μm. d) Model illustrating cell division of wild type versus mutant S. aureus cells. Dysregulated PG synthesis in the mutant cells leads to formation of chaining and hemi-septum phenotypes.
SgtB can compensate for defective activity of PBP2Y196D
The preceding studies showing normal division of the PBP2Y196D mutant strain in the absence, but not presence, of MoeA imply that a functional, chromosomally-encoded monofunctional PGT compensates for defective PBP2 activity in the mutant strains, resulting in a wild type phenotype. A monofunctional PGT may extend short products made by the PBP2Y196D mutant strain and/or may supply long glycan polymers for cell wall synthesis itself. To address the first possibility, we used a gel electrophoresis assay to examine the ability of SgtB to elongate the reaction products of radiolabeled Lipid II and PBP2Y196D (30, 31). Addition of SgtB and unlabeled Lipid II to a heat-inactivated PBP2Y196D reaction mixture led to the complete disappearance of radiolabeled short products and the formation of long polymers (Figure 3C). Hence, the short products made by PBP2Y196D are good substrates for elongation by SgtB. Consistent with these experiments, we found that only short glycan polymers are produced when Lipid II is added to particulate membranes prepared from the PBP2 mutant strain in a low pH buffer in which PBP2, but not SgtB, is optimally active (Supplementary Figure S7, lanes 1 and 2) (31, 35). Upon incubation at neutral pH under conditions in which SgtB shows good activity, the mutant membranes produce long glycan polymers, although they are still shorter than the polymers made by wild type membranes in the same buffer (Supplementary Figure S7, lanes 3 and 4). We have previously observed that purified SgtB produces longer PG polymers than purified PBP2 in vitro (31). SgtB is upregulated during cell wall stress and its ability to synthesize long polymers might play a protective role (36, 37). Experiments using purified SgtA were precluded since we were unable to reconstitute its activity. However, the genetic experiments described below suggest that SgtB, but not SgtA, compensates for the defective activity of the PBP2Y196D mutant in cells.
An S. aureus pbpB mutant lacking SgtB displays many division defects
Wild type S. aureus divides in three orthogonal planes, typically resulting in clusters of spherical cells (11, 12, 38). Ribs of PG have been implicated in defining the division planes, although no direct evidence linking PGTs to orthogonal cell division has been provided (11). It is known that PBP2 localizes to the septum during division (39) and we show above that the PBP2Y196D mutant exhibits abnormal septum formation in the presence of sublethal MoeA, which we attributed to antibiotic inhibition of the monofunctional PGTs. To test this possibility, we constructed deletions of sgtA and sgtB in S. aureus RN4220 and transduced the MoeA resistance allele (pbpBY196D) into the deletion strains. The ΔsgtA and ΔsgtB RN4220 strains showed no growth defects (7), nor did the pbpBY196D ΔsgtA double mutant; however, the pbpBY196D ΔsgtB double mutant grew very slowly (data not shown), and the size distribution of the cells was broad and included cells that were more than two times larger than average (Supplementary Figure S6A). In addition, a third of the dividing pbpBY196D ΔsgtB cells had pronounced defects (Figures 4A, Supplementary Figure S6), many of them similar to those observed in MoeA-treated PBP2Y196D mutant cells (Figure 3B, Supplementary Figure S5). For example, hemi-septa were common and a notable fraction of cells showed a chaining phenotype, indicating that division occurred in parallel planes (Figure 4, Supplementary Figure S6). Sublethal MoeA treatment (1.5 μg mL−1) did not exaggerate these phenotypes (data not shown). The division defects in the pbpBY196D ΔsgtB cells were due to the loss of SgtB activity rather than a scaffolding function of SgtB since complementation with a plasmid expressing fully functional SgtB restored normal growth and division (data not shown), while complementation with a plasmid expressing the crippled SgtB variant, SgtBY181D, did not correct the defects (Supplementary Figure S8). We conclude that glycan chain length plays a role in determining S. aureus morphology.
Conclusions
We have identified MoeA-resistant S. aureus mutants that contain a point mutation in the PGT domain of PBP2, an essential PG biosynthetic enzyme (4, 5, 19, 20, 28). These resistant mutants provide long sought genetic evidence that PGTs are the lethal targets of MoeA in bacteria. The point mutations alter amino acids in the catalytic cleft of the PGT domain (Figure 2A) (5, 20). Since these amino acids are known to contact the MoeA A-B rings, which contribute to MoeA's affinity for PBP2, the increased MICs of the resistant mutants likely reflect decreased MoeA binding. Consistent with this, we have shown that PBP2Y196D, unlike the wild type enzyme, is not inactivated by stoichiometric MoeA (Supplementary Figure S4A). That all identified resistance mutations were found to map to only two residues in PBP2 implies that a very narrow subset of target mutations can abrogate MoeA binding without abolishing catalytic activity of the enzyme (Figure 2B). Since the residues that contact the MoeA pharmacophore are invariant and critical for enzymatic activity, it is not surprising that the resistance mutations are in residues that contact a non-essential region of MoeA.
While the amino acid substitutions do not abolish PGT activity, we have characterized the effects of the more common mutation and report that it impairs function. PBP2Y196D makes glycan chains that are significantly shorter than normal and we suggest that the amino acid substitution decreases binding of the polymer substrate in the active site, leading to premature release of the elongating chain (Figures 2B and 2C). While the MoeA-resistant mutant does not have a phenotype in the absence of compound, most likely because SgtB compensates for defective activity of PBP2Y196D, it displays severe cell division defects in the presence of sublethal concentrations of MoeA or if SgtB activity is compromised by mutation (Figures 3B and Supplementary Figure S5 versus Figure 4B–C and Supplementary Figure S6, respectively). Two striking phenotypes include a chaining defect, which results from parallel division with incomplete cell separation, and a hemi-septum defect, which to our knowledge has never been observed before. We hypothesized that the hemi-septum phenotype may be due to poor coordination between the timing of new division plane initiation and completion of the previous cross wall (Figure 4D).
It was previously proposed that ribs of PG, observed to mark old division planes, provide an epigenetic scaffold that specifies the location of the biosynthetic machinery involved in forming new planes (Figure 4D) (11, 12). This model for how orthogonal division planes are specified leads to the prediction that defects in PG polymer synthesis will lead to cell division defects. Indeed, we show that cell division is dysregulated in strains containing defective PGTs that make short glycan chains. This work provides experimental support that existing PG is involved in the temporal and spatial regulation of the biosynthetic machinery involved in the formation of new division planes.
Methods
Bacterial strains, growth conditions, antimicrobial agents and reagents
Strains and plasmids used in this work are listed in Table S1. Escherichia coli strains were grown at 37 °C in Luria Bertani (LB) media. The S. aureus strains were grown at 37 °C in LB or Tryptic Soy Broth (TSB) media under aeration with shaking. All chemicals were ordered from Sigma-Aldrich, unless otherwise noted. Antibiotics were used when required. 95–97% pure MoeA was used as a 10 mg mL−1 stock solution in DMSO (40). Unlabeled and [14C]-radiolabeled Lipid II was prepared as previously described (28–31, 41)
Genetic manipulations
To transfer MoeA resistance mutations into different S. aureus strains ϕ85 phage mediated transduction was use (26, 42). Colonies of interest were selected on TSB agar plates containing sodium citrate (10 μg mL−1) and MoeA (2 μg mL−1). Newman and RN4220 strains were used as background. Newman is a clinical isolate (43). RN4220 is a methicillin-susceptible S. aureus (MSSA) strain, which is a restriction-deficient mutant of 8325-4 (26, 39). RN4220 was used for verification of resistance phenotypes by expression of PBP2 alleles from vectors and for mutant SgtA and SgtB construction. Mutant morphological and resistance phenotypes when treated with MoeA were the same for all tested background strains.
Selection of Moenomycin A resistant mutants and determination of mutation frequencies
MoeA resistant mutants were recovered by plating aliquots of overnight cultures of respective S. aureus strains onto TSB agar plates with different concentrations of MoeA ranging from 1 to 100 μg mL−1. For RN4220, MW2 and COL strains the mutation frequencies were estimated using total cell counts in each of two independent cultures. For S. aureus Newman the frequency of mutations was determined by a Luria-Delbrück fluctuation test using ten independent cultures (27).
Identification of moenomycin resistance mutation
Genes encoding monofunctional transglycosylases SgtA and SgtB were amplified with the primer pairs Mgt1F/Mgt1R and Mgt2F/Mgt2R respectively (Supplementary Table S2). The pbp2 gene was amplified using two primer sets Pbp2Sa1F/Pbp2Sa1R and Pbp2Sa2F/Pbp2Sa2R (Supplementary Table S2) to generate the entire gene sequence and flanking regions. PCR fragments were sequenced and sequences were analyzed with DNAstar software package, v 5.05 (Lasergene).
Wild type pbp2 gene including promoter region and two mutant alleles of pbp2 were amplified with the Sa4Fbam/Sa4Rkpn primers (Supplementary Table S2) and digested by BamHI and KpnI prior to insertion into a similarly digested pLI50 vector (44). Resulting plasmids pLIwt, pLINE1 and pLINE5–1 were verified by sequencing and introduced into S. aureus RN4220 by electrotransformation (45).
Antibiotics and lysostaphin susceptibility and autolysis activity determination
The in vitro susceptibility (MIC) tests were conducted as described before (46). Briefly, serial MoeA dilutions (1:1) were prepared using DMSO as a solvent prior to aliquoting 3 μL) was used as a positive control (full inhibition of growth), and DMSO was used for the negative control (full growth). Plates were shaken at 30 °C for 16–20 h. The optical densities of the wells were measured on a plate reader and normalized to the positive and negative controls to give percent growth at each concentration. The MIC was defined as the lowest concentration that gave less than 10% of growth. In addition, 5 μl of thiazolyl blue tetrazolium bromide (10 mg mL−1) solution was added to each well after measuring OD600 and plates were incubated at 30 °C for 1 hour. MIC was determined as a concentration of antibiotic in a well where compound did not change the color from yellow to dark-blue. The autolysis activity test and lysostaphin susceptibility assay were performed in the absence of MoeA and in the presence of 0.1X MIC (based on MICs for each strain shown in Supplementary Table S4) of the drug as described (47, 48).
In vitro fitness for MSSA strain
Fitness costs associated with MoeA resistance were determined by pair-wise comparison of the mutant with the wild type strain. Both mutants and S. aureus Newman were transformed with pLIgfp and pLImCherry plasmids purified from S. aureus RN4220 to overcome the host restriction system. A 3 mL flask of TSB media was inoculated with a mixture of overnight cultures of the wild type strain and the respective mutant in a 1:1 ratio. Dilutions were plated on TSB agar plates immediately and after 12 and 24 hr of growth at 37°C. Plates were scanned with a Typhoon 9400 Scanner (GE Healthcare). As a control, a mixture of the wild type strain expressing EGFP and mCherry was used. Relative competitive fitness was determined as the wild type to mutant output ratio normalized by the input ratio. The determinations were conducted in triplicates.
In vivo and in vitro fitness assay for MW2 strain
Swiss Webster male mice that were 4 to 6 weeks old were used for the subcutaneous abscess model (32, 33). Exponential-phase cells were prepared by 100x dilution of overnight cultures into TSB media and incubation at 37 °C until the OD600 was 0.8. S. aureus MW2 and MW2 PBP2Y196D cells were mixed in equal volumes, washed and 20x diluted in phosphate buffered saline (PBS). The cell suspension was then mixed with an equal volume of autoclaved Cytodex-1 beads (131 to 220 μm, Sigma) in PBS. 0.2 mL of the mixture was injected subcutaneously into the shaved flank of a mouse anesthetized with ketamine and xylazine. The inoculum for each abscess typically contained 1×106 to 3×106 CFU. After 48 hr, the mice were euthanized, and the subcutaneous abscesses were removed aseptically and homogenized in 5 mL PBS. The input ratio of wild type and mutant was estimated with FISHMAN-R microfluidic flow cytometer (On-Chip Biotechnologies) and colony counts. The output ratio was estimated by counting the number of colonies resistant to 1 μg mL−1 MoeA. The final ratio is defined as the wild type/mutant output ratio normalized by the input ratio. For in vitro competition assays, 100 μL of a suspension of cells was inoculated into 4 mL of TSB broth and grown for 24 hr at 37 °C with agitation. The input and output ratios of wild type and mutant were determined as above.
Cloning, expression, purification and characterization of wild type and mutant PBP2
The mutant allele (PBP2D196Y) of the full-length pbpB gene was amplified with the primers PBP2exF/PBP2exR (Supplementary Table S2) using NE1 chromosomal DNA as a template. Each fragment was digested using NheI and XhoI, and inserted into a similarly digested pET24b vector to generate pET24bNE1. pET21bPBP2Sa (28) encoding wild type PBP2 and pET24bNE1 were transformed into E. coli BL21(DE3) for overexpression. Growth and purification was performed as described previously for PBP2 (28) using 1.5 L of LB each for growth. For Figure 2B, PBPs (800 nM) were incubated with radiolabeled Lipid II (8 μM) in 50 mM CHES (N-cyclohexyl-2-aminoethanesulfonic acid), 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 50 mM acetic acid, 50 mM MES (2-(N-morpholino)ethanesulfonic acid) (pH = 5), 10 mM CaCl2 and 20% (v/v) DMSO at 37 °C for indicated time points prior to heat quenching and gel electrophoresis using our previously reported method (30, 31).
Cloning, expression, purification and characterization of wild type and mutant SgtB
The SgtBY181D expression plasmid was generated by introduction of the respective mutation into the pMgt1 plasmid, encoding wild type SgtB (35), using a QuikChange II Site-Directed Mutagenesis Kit (Agilent) and primers Mgt2Y181DF/Mgt2Y181DR (Table S2). The obtained plasmid, pET24bSgtBY181D, was verified by sequencing and transformed into E. coli BL21(DE3). Flasks containing 1.5 L of LB were inoculated with overnight BL21 cultures containing wild type or mutant SgtB, and were grown at 37 °C until OD600 0.6, followed by a shift to 20°C. Expression was induced with addition of 0.5 mM IPTG. Cultures were incubated at 20 °C overnight, and then cells were harvested by centrifugation (5000 × g, 4°C, 20 min) and lysed in BugBuster protein extraction solution (Novagen) as described previously (29). PGT activity was assessed by PG gel electrophoresis using our previously reported method (30, 31). For Figure 3C, PBP2D196Y (800 nM) was incubated with radiolabeled Lipid II (8 μM) in 50 mM CHES, 50 mM HEPES, 50 mM acetic acid, 50 mM MES (pH = 5), 10 mM CaCl2 and 20% (v/v) DMSO prior to heat quenching and addition of SgtB (80 nM) and unlabeled Lipid II (4 μM) followed by heat quenching and gel analysis (30, 31). All reactions were performed at 37 °C.
Generation and analysis of sgtB deletion mutant of S. aureus
sgtB gene flanking regions were amplified from the chromosome of S. aureus RN4220 using Mgt2F1B/Mgt2R1H (Mgt2L fragment) and Mgt2F2H/Mgt2R2Xb (Mgt2R fragment) primer pairs (Supplementary Table S2) and cloned into pSTBlue-1 AccepTor vector (Novagen). Flanking regions were assembled by cloning of the Mgt2R fragment into pSTMgt2L following HindIII and XbaI digestion. An aadA spectinomycin resistance cassette was inserted into the HindIII site of the resulting plasmid. Flanking regions of the sgtA gene were amplified with Mgt1F1B/Mgt1R1H (Mgt1L fragment) and Mgt1F2H/Mgt1R2Xb (Mgt1R fragment) primer pairs (Supplementary Table S2), cloned into a pSTBlue-1 AccepTor vector and assembled as described for the sgtB deletion plasmid, except the aadA cassette was not used. Resulting constructs were cloned into a pKOR1 temperature sensitive vector and final plasmids were transformed into S. aureus RN4220. Selection of the mutant strains was performed as described (49). Deletion of sgtB and sgtA in the chromosome of RN4220 were verified by Southern hybridization using Amersham ECL Direct Nucleic Acid Labeling And Detection Systems (GE Healthcare). The PBP2Y196D mutation was transduced into obtained mutant strains using ϕ85 phage as described above.
The complementation plasmid was constructed as follows: sgtB gene was amplified from the chromosome of S. aureus RN4220 using Mgt2F/Mgt2R primers (Supplementary Table S2) and blunt-cloned into pLI50 digested with SmaI. SgtBY181D allele was generated using Mgt2Y181DF/Mgt2Y181DR primer pair (Supplementary Table S2) and a QuikChange II Site-Directed Mutagenesis Kit (Agilent) using pLI50 containing wild type SgtB as a template.
Electron microscopy
The overnight cultures were diluted 1/100 with fresh TSB media without or 0.1X MIC of MoeA. The strains were grown for 4 hr at 37°C and fixed with formaldehyde. The negative stained samples were prepared, cut and analysis was performed using JEOL 1200EX microscope at Conventional Electron Microscopy facility, Harvard Medical School as described (46).
Isolation of particulate membranes of S. aureus
The crude membrane fractions were prepared according to Attia et al. (50). Briefly, 500 mL cultures of Newman wild type or PBP2Y196D mutant strains were grown to mid-log phase and then centrifuged (5000 × g, 10 min). The pellet was resuspended in 10 mL of 100 mM Tris-HCl (pH 7.4), 500 mM sucrose, 10 mM MgCl2 supplemented with lysostaphin (10 mg mL−1) and incubated for 1 hr at 37 °C. The mixture was then centrifuged (16,000 × g, 15 min) to remove the cell wall fraction. The pellet was resuspended in 10 mL of membrane buffer (50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 60 mM KCl) and subjected to sonication for a total of 1 min (with alternating 1 sec on and 1 sec off). The membrane fraction was pelleted by centrifugation (100,000 × g, 45 min) and resuspended in 500 μL of membrane buffer. The protein concentration was quantified by the DC protein assay (BioRad) using BSA as a standard. The protein concentrations in Newman wild type and mutant membrane fractions were normalized for SDS-PAGE experiments.
Light microscopy and cell counting
For septa counting, strains were grown in TSB at 37 °C to an OD600 of 0.2 (Figure 4A, Supplementary Figure S5A and S6) or diluted 1 in 100 from an overnight culture and grown for 4 hr with our without addition of 0.1X MIC MoeA (Supplementary Figure S5A). Cells were pelleted (2 min, 7500 × g) and resuspended in 5 mM TMA-DPH (trimethylammonium diphenylhexatriene) in TBS, placed on a 2% agarose in TBS pad, and imaged in the DAPI channel with an exposure time of 600–1000 ms. To break up clumps formed by strain RN4220 pbpBY196D ΔsgtB, the cell suspension was sonicated briefly, so that the cells would not be damaged. Images were acquired with a Hamamatsu digital camera model ORCA-ER connected to a Nikon Eclipse TE2000-U microscope with an X-cite 120 illumination system. Image manipulation was limited to adjusting brightness and contrast using ImageJ (51). For septa counting, approximately 700–1000 clearly visible cells were counted, for cell dimension measurements, 100 cells were measured.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (GM076710), the Harvard-Wide Antibiotic Resistance Program (AI083214), and NERCE (AI057159). YQ gratefully acknowledges Agency for Science, Technology and Research (A*STAR) of Singapore for a PhD scholarship. KS gratefully acknowledges the Deutsche Forschungsgemeinschaft (DFG) for a research fellowship. We would like to thank M. Lazarus for assistance with generating PyMOL images.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 2.Sun SX, Jiang H. Physics of bacterial morphogenesis. Microbiol. Mol. Biol. Rev. 2011;75:543–565. doi: 10.1128/MMBR.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 5.Lovering AL, Gretes M, Strynadka NCJ. Structural details of the glycosyltransferase step of peptidoglycan assembly. Curr. Opin. Struct. Biol. 2008;18:534–543. doi: 10.1016/j.sbi.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Terrak M, Nguyen-Distèche M. Kinetic characterization of the monofunctional glycosyltransferase from Staphylococcus aureus. J. Bacteriol. 2006;188:2528–2532. doi: 10.1128/JB.188.7.2528-2532.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed P, Veiga H, Jorge AM, Terrak M, Pinho MG. Monofunctional Transglycosylases Are Not Essential for Staphylococcus aureus Cell Wall Synthesis. J. Bacteriol. 2011;193:2549–2556. doi: 10.1128/JB.01474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinho MG, de Lencastre H, Tomasz A. An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10886–10891. doi: 10.1073/pnas.191260798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinho MG, Filipe SR, de Lencastre H, Tomasz A. Complementation of the essential peptidoglycan transpeptidase function of penicillin-binding protein 2 (PBP2) by the drug resistance protein PBP2A in Staphylococcus aureus. J. Bacteriol. 2001;183:6525–6531. doi: 10.1128/JB.183.22.6525-6531.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Touhami A, Jericho MH, Beveridge TJ. Atomic force microscopy of cell growth and division in Staphylococcus aureus. J. Bacteriol. 2004;186:3286–3295. doi: 10.1128/JB.186.11.3286-3295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner RD, Ratcliffe EC, Wheeler R, Golestanian R, Hobbs JK, Foster SJ. Peptidoglycan architecture can specify division planes in Staphylococcus aureus. Nat. Commun. 2010;1:1–9. doi: 10.1038/ncomms1025. [DOI] [PubMed] [Google Scholar]
- 12.Dmitriev BA, Toukach FV, Holst O, Rietschel ET, Ehlers S. Tertiary structure of Staphylococcus aureus cell wall murein. J. Bacteriol. 2004;186:7141–7148. doi: 10.1128/JB.186.21.7141-7148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Heijenoort Y, Derrien M, Van Heijenoort J. Polymerization by transglycosylation in the biosynthesis of the peptidoglycan of Escherichia coli K 12 and its inhibition by antibiotics. FEBS Lett. 1978;89:141–144. doi: 10.1016/0014-5793(78)80540-7. [DOI] [PubMed] [Google Scholar]
- 14.van Heijenoort Y, Leduc M, Singer H, Van Heijenoort J. Effects of moenomycin on Escherichia coli. J. Gen. Microbiol. 1987;133:667–674. doi: 10.1099/00221287-133-3-667. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welzel P. Syntheses around the transglycosylation step in peptidoglycan biosynthesis. Chem. Rev. 2005;105:4610–4660. doi: 10.1021/cr040634e. [DOI] [PubMed] [Google Scholar]
- 17.Ostash B, Walker S. Moenomycin family antibiotics: chemical synthesis, biosynthesis, and biological activity. Nat. Prod. Rep. 2010;27:1594–1617. doi: 10.1039/c001461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perlstein DL, Zhang Y, Wang TS, Kahne DE, Walker S. The Direction of Glycan Chain Elongation by Peptidoglycan Glycosyltransferases. J. Am. Chem. Soc. 2007;129:12674–12675. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lovering AL, de Castro LH, Lim D, Strynadka NCJ. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science. 2007;315:1402–1405. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]
- 20.Lovering AL, De Castro L, Strynadka NCJ. Identification of dynamic structural motifs involved in peptidoglycan glycosyltransfer. J. Mol. Biol. 2008;383:167–177. doi: 10.1016/j.jmb.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Yuan Y, Fuse S, Ostash B, Sliz P, Kahne D, Walker S. Structural analysis of the contacts anchoring moenomycin to peptidoglycan glycosyltransferases and implications for antibiotic design. ACS Chem. Biol. 2008;3:429–436. doi: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuse S, Tsukamoto H, Yuan Y, Wang TSA, Zhang Y, Bolla M, Walker S, Sliz P, Kahne D. Functional and structural analysis of a key region of the cell wall inhibitor moenomycin. ACS Chem. Biol. 2010;5:701–711. doi: 10.1021/cb100048q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gampe CM, Tsukamoto H, Doud EH, Walker S, Kahne D. Tuning the moenomycin pharmacophore to enable discovery of bacterial cell wall synthesis inhibitors. J. Am. Chem. Soc. 2013;135:3776–3779. doi: 10.1021/ja4000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welzel P. A long research story culminates in the first total synthesis of moenomycin A. Angew. Chem. Int. Ed. Engl. 2007;46:4825–4829. doi: 10.1002/anie.200700765. [DOI] [PubMed] [Google Scholar]
- 25.Salzberg LI, Luo Y, Hachmann A-B, Mascher T, Helmann JD. The Bacillus subtilis GntR family repressor YtrA responds to cell wall antibiotics. J. Bacteriol. 2011;193:5793–5801. doi: 10.1128/JB.05862-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novick RP. Genetic systems in staphylococci. Methods Enzymol. 1991;204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 27.Maloy SR, Cronan JR, Freiflder D. Microbial Genetics. 2nd ed. Jones and Bartlett; Boston: 1994. [Google Scholar]
- 28.Barrett D, Leimkuhler C, Chen L, Walker D, Kahne D, Walker S. Kinetic characterization of the glycosyltransferase module of Staphylococcus aureus PBP2. J. Bacteriol. 2005;187:2215–2217. doi: 10.1128/JB.187.6.2215-2217.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang TSA, Lupoli TJ, Sumida Y, Tsukamoto H, Wu Y, Rebets Y, Kahne DE, Walker S. Primer preactivation of peptidoglycan polymerases. J. Am. Chem. Soc. 2011;133:8528–8530. doi: 10.1021/ja2028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett D, Wang T-SA, Yuan Y, Zhang Y, Kahne D, Walker S. Analysis of glycan polymers produced by peptidoglycan glycosyltransferases. J. Biol. Chem. 2007;282:31964–31971. doi: 10.1074/jbc.M705440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang TSA, Manning SA, Walker S, Kahne D. Isolated Peptidoglycan Glycosyltransferases from Different Organisms Produce Different Glycan Chain Lengths. J. Am. Chem. Soc. 2008;130:14068–14069. doi: 10.1021/ja806016y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bunce C, Wheeler L, Reed G, Musser J, Barg N. Murine model of cutaneous infection with gram-positive cocci. Infect. Immun. 1992;60:2636–2640. doi: 10.1128/iai.60.7.2636-2640.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ford CW, Hamel JC, Stapert D, Yancey RJ. Establishment of an experimental model of a Staphylococcus aureus abscess in mice by use of dextran and gelatin microcarriers. J. Med. Microbiol. 1989;28:259–266. doi: 10.1099/00222615-28-4-259. [DOI] [PubMed] [Google Scholar]
- 34.Steele VR, Bottomley AL, Garcia-Lara J, Kasturiarachchi J, Foster SJ. Multiple essential roles for EzrA in cell division of Staphylococcus aureus. Mol. Microbiol. 2011;80:542–555. doi: 10.1111/j.1365-2958.2011.07591.x. [DOI] [PubMed] [Google Scholar]
- 35.Heaslet H, Shaw B, Mistry A, Miller AA. Characterization of the active site of S. aureus monofunctional glycosyltransferase (Mtg) by site-directed mutation and structural analysis of the protein complexed with moenomycin. J. Struct. Biol. 2009;167:129–135. doi: 10.1016/j.jsb.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Steidl R, Pearson S, Stephenson RE, Ledala N, Sitthisak S, Wilkinson BJ, Jayaswal RK. Staphylococcus aureus cell wall stress stimulon gene-lacZ fusion strains: potential for use in screening for cell wall-active antimicrobials. Antimicrob. Agents Chemother. 2008;52:2923–2925. doi: 10.1128/AAC.00273-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balibar CJ, Shen X, McGuire D, Yu D, McKenney D, Tao J. cwrA, a gene that specifically responds to cell wall damage in Staphylococcus aureus. Microbiology. 2010;156:1372–1383. doi: 10.1099/mic.0.036129-0. [DOI] [PubMed] [Google Scholar]
- 38.Tzagoloff H, Novick R. Geometry of cell division in Staphylococcus aureus. J. Bacteriol. 1977;129:343–350. doi: 10.1128/jb.129.1.343-350.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinho MG, Errington J. Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol. Microbiol. 2004;55:799–807. doi: 10.1111/j.1365-2958.2004.04420.x. [DOI] [PubMed] [Google Scholar]
- 40.Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE. Degradation and Reconstruction of Moenomycin A and Derivatives: Dissecting the Function of the Isoprenoid Chain. J. Am. Chem. Soc. 2006;128:14012–14013. doi: 10.1021/ja065905c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S. Better substrates for bacterial transglycosylases. J. Am. Chem. Soc. 2001;123:3155–3156. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]
- 42.Truong-Bolduc QC, Dunman PM, Strahilevitz J, Projan SJ, Hooper DC. MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J. Bacteriol. 2005;187:2395–2405. doi: 10.1128/JB.187.7.2395-2405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duthie ES, Lorenz LL. Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 1952;6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- 44.Lee CY, Buranen SL, Ye ZH. Construction of single-copy integration vectors for Staphylococcus aureus. Gene. 1991;103:101–105. doi: 10.1016/0378-1119(91)90399-v. [DOI] [PubMed] [Google Scholar]
- 45.Schenk S, Laddaga RA. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 1992;73:133–138. doi: 10.1016/0378-1097(92)90596-g. [DOI] [PubMed] [Google Scholar]
- 46.Campbell J, Singh AK, Santa Maria JPJ, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 2011;6:106–116. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jonge BL, de Lencastre H, Tomasz A. Suppression of autolysis and cell wall turnover in heterogeneous Tn551 mutants of a methicillin-resistant Staphylococcus aureus strain. J. Bacteriol. 1991;173:1105–1110. doi: 10.1128/jb.173.3.1105-1110.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kusuma CM, Kokai-Kun JF. Comparison of four methods for determining lysostaphin susceptibility of various strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 2005;49:3256–3263. doi: 10.1128/AAC.49.8.3256-3263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Attia AS, Benson MA, Stauff DL, Torres VJ, Skaar EP. Membrane damage elicits an immunomodulatory program in Staphylococcus aureus. PLoS Pathog. 2010;6:e1000802. doi: 10.1371/journal.ppat.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rasband WS. ImageJ. U.S. National Institutes of Health, Bethesda; Maryland, USA: imagej.nih.gov/ij/,1997–2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.