

Abstract
In metastatic ovarian cancer, resistance to platinum chemotherapy is common. Although the orphan nuclear receptor TR3 (nur77/NR4A1) is implicated in mediating chemotherapy-induced apoptosis in cancer cells, its role in ovarian cancer has not been determined. In an ovarian cancer tissue microarray, TR3 protein expression was elevated in stage I tumors, but down-regulated in a significant subset of metastatic tumors. Moreover, TR3 expression was significantly lower in platinum-resistant tumors in patients with metastatic disease, and low TR3 staining was associated with poorer overall and progression-free survival. We have identified a direct role for TR3 in cisplatin-induced apoptosis in ovarian cancer cells. Nucleus-to-cytoplasm translocation of TR3 was observed in cisplatin-sensitive (OVCAR8, OVCAR3, A2780PAR) but not cisplatin-resistant (NCI/ADR-RES, A2780CP20) ovarian cancer cells. Immunofluorescent analyses showed clear overlap between TR3 and mitochondrial Hsp60 in cisplatin-treated cells, which was associated with cytochrome C release. Ovarian cancer cells with stable shRNA- or transient siRNA-mediated TR3 down-regulation displayed substantial reduction in cisplatin effects on apoptotic markers and cell growth in vitro and in vivo. Mechanistic studies demonstrated that the cisplatin-induced cytoplasmic TR3 translocation required for apoptosis induction was regulated by JNK activation and inhibition of Akt. Finally, cisplatin-resistance was partially overcome by ectopic TR3 overexpression, and by treatment with the JNK activator anisomycin and Akt pathway inhibitor, wortmannin. Our results suggest that disruption of TR3 activity, via down-regulation or nuclear sequestration, likely contributes to platinum resistance in ovarian cancer. Moreover, we have described a treatment strategy aimed at overcoming platinum resistance by targeting TR3.
Keywords: Ovarian cancer, TR3, cisplatin, chemoresistance, apoptosis
Introduction
Ovarian cancer is the most lethal gynecologic malignancy and the vast majority of epithelial ovarian malignancies present as biologically aggressive, metastatic disease (1, 2). The high incidence of relapse following standard platinum-based therapy indicates that there is an urgent need for new treatment strategies and novel insight into mechanisms of platinum resistance. Since most high-grade ovarian cancers harbor mutations in p53 (3), identifying anti-tumor effectors that act independently of p53 is an important goal.
TR3 (also known as nur77 and NR4A1) has emerged as a major regulator of cancer cell survival and an attractive therapeutic target (4). TR3 is a member of the NR4A family of nuclear receptors, and mediates apoptosis in various cancer cell types in response to a wide range of chemotherapeutic agents. Known mechanisms of TR3-induced apoptosis include p53-independent nuclear-cytoplasmic translocation, leading to cytochrome C release in response to various pro-apoptotic drugs (5-11), or up-regulation of pro-apoptotic genes and/or down-regulation of anti-apoptotic genes (12-14). Nuclear export of TR3 is known to involve specific changes in its phosphorylation status, such as N-terminal serine phosphorylation by JNK and loss of Akt-mediated phosphorylation on serine 351(15). At the mitochondria, TR3 binds Bcl-2, which induces a conformational change such that Bcl-2 assumes a pro-apoptotic function(9). Apoptosis mediated at least partly through TR3 activity has been reported in an ovarian-derived teratocarcinoma cell line, Pa-1 (5). However, it is unknown whether TR3 mediates apoptotic effects of established chemotherapeutic agents such as cisplatin in ovarian cancer cells of epithelial origin.
Despite the clear pro-apoptotic role for TR3 identified in chemotherapy-treated cancer cells, accumulated evidence indicates that it may play a more complex role in tumorigenesis. TR3 expression is also induced by mitogenic factors in the absence of apoptosis in various cancer cell types and is up-regulated in some solid epithelial tumors (11, 16-19). In contrast, TR3 is down-regulated in some metastatic solid tumors (17, 20) and TR3-mediated suppression of colon cancer tumorigenesis has been recently reported (14, 21). The Cancer Genome Atlas (TCGA) data demonstrate that TR3 mRNA expression is detected in ovarian tumors, although no significant alterations in the TR3 gene, such as mutation, amplification or promoter methylation, are present in these tumors (3). There have been no previous reports measuring TR3 protein expression in epithelial ovarian tumors.
To identify possible roles of TR3 in ovarian cancer, and to relate TR3 protein expression to clinical outcomes, we first determined its expression in a tissue microarray (TMA) generated from tumor samples from 209 ovarian cancer patients. We demonstrated an association between low TR3 expression, resistance to platinum chemotherapy and survival indices. Then, we identified a functional link between TR3 and cisplatin-mediated apoptosis in ovarian cancer cells. Collectively, our results suggest that TR3 is an important regulator of ovarian cancer cell apoptosis and that down-regulation or nuclear sequestration of TR3 contributes to platinum response and resistance. Finally, this study has implications for future treatment strategies to overcome platinum resistance in ovarian cancer by up-regulating TR3 or targeting TR3 for nuclear export.
Materials and Methods
Cell culture, chemicals and plasmids
Growth of the epithelial ovarian cancer cell lines SKOV3, OVCAR3, NCI/ADR-RES, OVCAR5, and OVCAR8, well-characterized as part of the National Cancer Institute (NCI) 60 Cancer Panel (22-24), have been described previously (25)(25). A2780 PAR and A2780 CP20 cells were kind gifts from Professor Anil Sood (MD Anderson Cancer Center, Houston, TX) (26). Growth of normal human ovarian surface epithelium (HOSE) cells has also been described (25). All cell lines were utilized within 6 months of receipt from the aforementioned cell line banks, and all tested negative for mycoplasma. Cells were treated with the DNA-damaging agents, cisplatin and doxorubicin (both from Sigma Chemical Co., St Louis, MO), the histone deacetylase inhibitor SAHA (kind gift from Dr. Edward Holson, Stanley Center for Psychiatric Research; Broad Institute; Cambridge, MA), the nuclear export inhibitor, leptomycin B (Sigma Chemical Co.), the JNK inhibitor, SP600125 (Enzo Life Sciences, Ann Arbor, MI), the PI-3 kinase inhibitor, wortmannin (Enzo Life Sciences), and the JNK activator, anisomycin (Enzo Life Sciences). A 0.01% DMSO solution in cell culture medium was used as the vehicle control for in vitro cell growth and apoptosis experiments described below. A TrueORF® plasmid encoding for DDK (FLAG)-tagged full length TR3, and its corresponding empty vector, were purchased from Origene (Rockville, MD).
Generation of TR3 knockdown cells
OVCAR-8 cells were transfected (Lipofectamine 2000, Invitrogen Corp., Carlsbad, CA) with pre-designed pGFP-V-RS shRNA HuSH-29 plasmids targeting human TR3 (ShTR3) or control, scrambled shRNA (ShScr) on the same vector background (Origene). Additional details regarding selection, characterization and maintenance of clones are in Supplementary Methods. For transient TR3 knockdown, OVCAR3 cells were transfected with ON-TARGETplus non-targeting (NT) or TR3-targeting siRNA duplexes (Thermo Fisher Scientific, Inc., Waltham, MA) using RNAiMAX transfection reagent (Invitrogen).
Immunofluorescence
Cells were grown, fixed, permeabilized and stained with anti-NR4A1/TR3, anti-Hsp60, anti-cytochrome C, anti-Bcl-2, and anti-DDK (FLAG) primary antibodies as previously described (7). Additional details regarding primary and secondary antibodies, and for cell counts, are provided in Supplementary Methods. Images were acquired and analyzed as previously described (27).
Western Blotting
Whole cell protein isolation, subcellular fractionation, Western Blotting and signal detection were performed as described previously (25, 28) to detect anti-TR3/nur77, anti-Nurr1/NR4A2, anti-NOR1/NR4A3, anti-PARP, anti-caspase-3, anti-β-actin, anti-histone H3, anti-Bcl-2, anti- DDK (FLAG), anti-phospho-JNK (Thr183/Tyr185), anti-phospho-Akt (Ser473) and anti-α-tubulin primary antibodies. Additional details are provided in Supplementary Methods. Co-immunoprecipitation experiments with anti-TR3 on lysates normalized for TR3 expression were performed as previously described (28).
Quantitative real time RT-PCR
RNA isolation and cDNA synthesis were performed as previously described (29). Levels of mRNA expression for TR3 and NR4A2 were determined using TaqMan® gene expression assays. Additional details are provided in Supplementary Methods.
SRB assays
Sulfurhodamine B (SRB) growth assays were performed as previously described (29, 30). Effects on cell growth were measured 72 hours after addition of drugs. Absorbance was measured at 510nm using a Spectramax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA) in the High-Throughput Screening Core of the Vanderbilt Institute of Chemical Biology.
Xenograft Assays
Four to five-week-old female athymic Nude-Foxn1nu mice were purchased from Harlan Laboratories (Indianapolis, IN). For this subcutaneous (SC) xenograft model, 5 × 106 OVCAR8 ShScr or ShTR3 cells in 200 μL of a PBS/Matrigel (BD Biosciences, San Jose, CA) mixture (1:1 v/v) were injected SC into the right flank. After the tumors reached approximately 200 mm3 in volume, mice were randomized and treated with cisplatin (5mg/kg weekly) or vehicle control (PBS) for 3 weeks before euthanasia and necropsy (30). Tumor volume was calculated weekly from caliper measurements of the smallest (SD) and largest diameter (LD) using the formula: volume = [LD × SD2] × π/6 (28). Experiments performed received prior approval from the Vanderbilt University Institutional Animal Use and Care Committee, and all animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care.
Case Selection and TMA generation
Institutional Review Board approval for the tissue studies was obtained at Vanderbilt University Medical Center (VUMC). To compare TR3 expression in normal and neoplastic epithelium, we obtained de-identified tissue from human epithelial ovarian tumors with serous histology and normal human ovarian samples from the Vanderbilt Translational Pathology Shared Resource. For the TMA, 209 patients diagnosed with ovarian cancer at VUMC between 1994 and 2004 had evaluable paraffin tissue blocks. Additional details regarding generation of the TMA, recording of patient clinical data, and for definitions of overall and progression-free survival and platinum sensitivity, are provided in Supplementary Methods. Tumor stage and grade was assigned based on the International Federation of Gynecology and Obstetrics (FIGO) system (31). Clinical data relating to tumor stage, grade, histology and platinum sensitivity of patient tumors are summarized in Table 1.
Table 1. Clinical data for the 209 ovarian cancer patients whose tumors were used to generate a TMA.
| Characteristic | Measure |
|---|---|
|
| |
| Median Age at Diagnoses (years) | 56.76 |
| Tumor stage | |
| IA | 29 (13.9%) |
| IB | 2 (1.0%) |
| IC | 21 (10.0%) |
| IIA | 2 (1.0%) |
| IIB | 1 (0.5%) |
| IIC | 6 (2.9%) |
| IIIA | 5 (2.4%) |
| IIIB | 6 (2.9%) |
| IIIC | 107 (51.2%) |
| IV | 21 (10.0%) |
| Unstaged | 5 (2.4%) |
| Unknown stage | 4 (1.9%) |
| Platinum Sensitive | 86 (41.1%) |
| Platinum Resistant | 43 (20.6%) |
| Grade | |
| 1 | 38 (18.2%) |
| 2 | 50 (23.9%) |
| 3 | 108 (51.7%) |
| Unknown grade | 13 (6.2%) |
| Histology | |
| Epithelial | 202 (96.7%) |
| Serous/Papillary | 140 (67.0%) |
| Endometrioid | 27 (12.9%) |
| Mucinous | 15 (7.2%) |
| Clear Cell | 9 (4.3%) |
| Mixed | 6 (2.9%) |
| Other Epithelial | 5 (2.4%) |
| Other | 7 (3.3%) |
| Progression Free Survival (years) | 2.64 [+/- 2.13] |
| Overall Survival (years) | 3.33 [+/- 2.81] |
Values in round parentheses represent the percentage of the total (209) patients. For Progression Free Survival and Overall Survival, numbers are mean [+/- SD].
Immunohistochemistry
Tissue fixation, processing and sectioning methods have been previously described (32). Hematoxylin and eosin staining for histology and immunostaining for anti-TR3/NR4A1, anti-pan-cytokeratin, anti-cleaved caspase-3 and anti-mib-1/Ki67 primary antibodies were performed as described (32). Additional details for these antibodies, and for cell counts in ovarian xenografts, are provided in Supplementary Methods. For the TMAs, semi-quantitative measurement of TR3 expression in tumors was performed using the automated Ariol® SL-50 Platform (Molecular Devices LLC, Sunnyvale, CA). Additional details are provided in Supplementary Methods.
Statistics
For in vitro experiments, values shown were the mean + SD of 3 independent experiments with p < 0.05 relative to appropriate controls considered to be statistically significant (Student's t test). In our mouse experiments, differences between groups were determined by Mann-Whitney test. For TMA analyses, Kaplan-Meier curves were used to analyze progression-free and overall survival. Survival curves were compared using the Cox proportional hazards model. The likelihood ratio test was used to assess statistical significance. Differences in TR3 expression related to tumor Stage (Stage I versus II/III/IV), grade (grade 1 versus 2/3) and platinum sensitivity or resistance were determined by Mann-Whitney test.
Results
TR3 expression is heterogeneous in ovarian cancer cells
While TR3 exerts diverse effects in cancer cells, such as mediating apoptosis induction by multiple chemotherapeutic agents (5-11) or promoting cell growth in response to mitogenic stimuli (11), its role in ovarian cancer cell biology is unknown. Data extracted from publically available databases confirmed that TR3 mRNA expression is detected at variable levels in primary ovarian tumor samples (3, 33) and the NCI60 panel of ovarian cancer cell lines (22-24).
To determine protein expression levels of TR3, we examined 5 ovarian cancer cell lines represented in the NCI60 panel. As shown in Figure 1A, TR3 expression was heterogeneous, with relatively high expression levels in NCI/ADR-RES, OVCAR8 and OVCAR3 cells. Low TR3 expression was observed in SKOV3 and OVCAR5 cells. We then confirmed that TR3 was detected by immunohistochemical staining in ovarian tumors (Supplementary Figure 1). Prominent nuclear TR3 expression was observed in areas of tumor tissue with correspondingly high levels of the epithelial marker, pan-cytokeratin, and the proliferation marker, Ki67. TR3 expression was also detected in the single layer of cytokeratin-positive epithelial cells in normal ovary (Supplementary Figure 1), consistent with its abundant expression in cells derived from normal human ovarian surface epithelium (HOSE) (Figure 1A).
Figure 1. Expression of TR3 in ovarian tumors.

(A) TR3 protein expression in 5 ovarian cancer cell lines and in normal human ovarian surface epithelial cells (HOSE). (B) Immunohistochemical analysis of TR3 expression in tumor cells in early-stage (I) and late-stage (II-IV) serous tumors. High power photomicrographs are shown in the bottom panel. (C) Box-and-whiskers plot showing association between TR3 expression and serous tumor Stage, grade and platinum resistance. (D) Overall survival (OS) and (E) progression-free survival (PFS) in patients with serous ovarian cancer with high (>70% TR3-positive cells) and low (<70% TR3-positive cells) expression.
Low TR3 expression in a subset of metastatic human ovarian cancers is associated with platinum resistance and reduced survival
To investigate the relationship between TR3 expression in ovarian tumors and clinical outcomes, we determined the percentage of TR3-positive tumor cells in each section of our TMA. Since all cells positive for TR3 within the tumor displayed nuclear staining, with only a small subset of tumors (10/209) containing cells displaying both nuclear and cytoplasmic staining, counts were based on nuclear TR3 expression. TR3 expression for each tumor and associated clinical data are shown in Supplementary Table 1. The median TR3-positivity in these tumors was 72.1%. No significant differences were observed in TR3 expression in the epithelial tumor subtypes (Supplementary Figure 2). Because the majority of epithelial tumors were of papillary serous histology (140/202), and only relatively small numbers of other histological classes were represented, we chose to restrict subsequent analyses to serous tumors.
As shown in Figure 1B&C, a high percentage of epithelial cells in early stage (I) serous tumors displayed TR3 expression. In contrast, a large subset of late stage, metastatic serous tumors showed significantly lower TR3 expression. Lower TR3 nuclear staining was also associated with higher grade tumors (Figure 1B), with reduced overall and disease-free survival (Figure 1C) and, strikingly, with platinum resistance (Figure 1B). We then compared our data to results extracted from publically available ovarian cancer microarray databases, TCGA (3) and Yoshihara et al (33). TCGA data indicate that 70% (186/268) of ovarian tumors show reduced TR3 mRNA expression levels compared to normal tissue, consistent with our results. However, the TCGA data displayed no significant association between TR3 mRNA expression and platinum resistance or survival (Supplementary Figure 3A&B). In contrast, a significant association between low TR3 mRNA expression and reduced progression-free survival, but not overall survival, was demonstrated in the Yoshihara data set at Top/Bottom 20%, 35% and 50% cutoffs (Supplementary Figure 3C&D).
Cisplatin induces TR3 expression in ovarian cancer cells
Our TMA data showed an association between low TR3 expression and reduced platinum response and survival. However, no direct functional link between endogenous TR3 and platinum response in ovarian cancer cells has been previously demonstrated. To determine the role of endogenous TR3 in cisplatin-induced growth inhibition and apoptosis, we selected 3 ovarian cancer cell lines with relatively high levels of basal TR3 expression (OVCAR8, NCI/ADR-RES and OVCAR3) for further study (Figure 1A). SRB cell growth experiments showed that NCI/ADR-RES cells were resistant to cisplatin (Figure 2A), consistent with previous observations in these multi-drug resistant cells (27). NCI/ADR-RES cells are a derivative of OVCAR8 cells (23), which were more sensitive to cisplatin than NCI/ADR-RES and OVCAR3 cells (Figure 2A). A similar pattern of effect across the cell lines was observed when cisplatin-induced apoptosis was measured by PARP cleavage (Figure 2B). Interestingly, TR3 expression was induced by cisplatin in all 3 cell lines, including the cisplatin resistant NCI/ADR-RES cells following 24h treatment (Figure 2B). These results suggest that stimulation of TR3 expression alone is not sufficient for growth inhibition or apoptosis and other mechanisms play a role.
Figure 2. Effects of cisplatin in ovarian cancer cells.

(A) Effects of 72 hour exposure to 5 μM cisplatin in OVCAR8, NCI/ADR-RES and OVCAR3 cells in SRB assays. (B) Protein levels ofTR3 and cleaved PARP (Cl PARP) in cells treated with 5 μM cisplatin (24h). (C) High power photomicrographs showing subcellular localization of TR3 (green) relative to DAPI-stained nuclei (blue) and mitochondrial Hsp60 (red) in control (Con) and cisplatin (Cisp)-treated cells (5μM, 24h). Overlap between TR3 and Hsp60 is shown in yellow. Counts showing percentage of cells displaying (D) cytoplasmic TR3 expression and (E) significant TR3 and Hsp60 overlap. (F) TR3 associates with the mitochondrial Bcl-2 in cisplatin-treated OVCAR8 cells. Control and cisplatin-treated IP lysates were normalized to ensure equivalent amounts of TR3. Inset: Overlap of Bcl-2 (red) and a TR3- DDK (FLAG) (green) expression vector. (G) High power photomicrographs of OVCAR8 and OVCAR3 cells showing punctate staining of cytochrome C (red) in control, untreated cells and diffuse staining in cisplatin-treated cells (5μM, 24h). (H) Percentage of OVCAR8 and OVCAR3 cells displaying diffuse cytochrome C staining and TR3 cytoplasmic localization. All values are mean + SD of 3 independent experiments. * p < 0.01 relative to control, Student's t test.
Mitochondrial targeting of TR3, leading to cytochrome C release, is associated with cisplatin-induced apoptosis in ovarian cancer cells
Cytotoxic drug-induced changes in the subcellular localization of TR3 have emerged as a primary mechanism for apoptosis induction. Previous studies have shown that various pro-apoptotic stimuli promote translocation of TR3 from the nucleus to the cytoplasm (5-11), often with direct mitochondrial targeting and alters function of Bcl-2 to become pro-apoptotic (9). Therefore, we evaluated subcellular localization of TR3 before and after cisplatin treatment by immunofluorescence analyses. Consistent with previous reports (5-7), TR3 expression was predominantly localized to DAPI-stained nuclei in untreated ovarian cancer cells (Figure 2C). Cytoplasmic translocation of TR3 was significantly induced by cisplatin in OVCAR8 and OVCAR3 cells, but not in cisplatin-resistant NCI/ADR-RES cells (Figure 2C&D). To determine mitochondrial localization, we demonstrated that the punctate TR3 cytoplasmic staining pattern in cisplatin-treated cells co-localized with the well-established mitochondrial marker, Hsp60 (Figure 2C&E). To investigate the interaction between TR3 and Bcl-2, we performed co-immunoprecipitation and immunofluorescence experiments and demonstrated an association between TR3 and Bcl-2 in cisplatin-treated OVCAR8 cells (Figure 2F).
To validate the possible link between cisplatin resistance and a lack of cisplatin-induced cytoplasmic targeting of TR3 in ovarian cancer cells, we used another well-characterized isogenic cell line model of cisplatin resistance in ovarian cancer, A2780 PAR and CP20 cells (26). As shown in Supplementary Figure 5, there was a similar lack of TR3 mitochondrial targeting in cisplatin-treated CP20 cells compared to PAR cells. To link cytoplasmic TR3 translocation with apoptosis induction, we performed immunofluorescence analyses of cytochrome C release. Cytochrome C is localized to the inner mitochondrial membrane in untreated cells and following stimulation with pro-apoptotic agents is released into the cytosol to form a diffuse staining pattern (6, 7). As shown in Figure 2G-H, cisplatin-treated OVCAR8 and OVCAR3 cells displayed diffuse cytochrome C staining. The majority of the cells (>80%) simultaneously displayed cytoplasmic TR3 expression, implicating mitochondrial targeting of TR3 as a key factor in cytochrome C release and apoptosis. A small proportion of cells displayed cytochrome C release without mitochondrial TR3 targeting. Therefore, other mechanisms are likely to mediate cisplatin-induced apoptosis in these ovarian cancer cells.
To investigate TR3 in normal cells treated with cisplatin, we used HOSE ovarian epithelial cells as a model. We demonstrated that in cisplatin-treated HOSE cells TR3 expression is not only upregulated, but found in the cytoplasm, co-localized with mitochondrial Hsp60 (Supplementary Figure 4).
The nuclear export inhibitor leptomycin B reduces cisplatin-induced apoptosis and cytoplasmic targeting of TR3
To better understand the link between mitochondrial targeting of TR3and apoptosis in ovarian cancer cells, we performed experiments with the well-established nuclear export inhibitor, leptomycin B (LMB) (7). First, we confirmed that treatment of OVCAR8 cells with LMB prior to addition of cisplatin significantly reduced the number of cells showing cytoplasmic localization in immunofluorescence experiments (Figure 3A). We validated these results with subcellular fractionation experiments (Figure 3B). In cisplatin-treated cells where TR3 cytoplasmic translocation was inhibited, we observed a significant reduction in the number of cells showing cytochrome C release (Supplementary Figure 6) and reduced levels of cleaved PARP and cleaved caspase-3 (Figure 3B). Similar results were observed in OVCAR3 cells (data not shown). We then tested the possibility that LMB-mediated inhibition of apoptosis was due to reduced TR3 expression. However, cisplatin induced TR3 to a similar degree with and without LMB pre-treatment (Figure 3D). These studies suggest that cytoplasmic TR3 translocation from the nucleus to the cytoplasm was essential for cisplatin-induced apoptosis in these ovarian cancer cells.
Figure 3. Cisplatin-induced apoptosis requires nuclear export of TR3.

Following treatment of OVCAR8 cells with 5 μM cisplatin (24h), with and without 2h pre-treatment with leptomycin B (1 ng/ml): (A) Immunofluorescence analysis of subcellular TR3 localization; (B) TR3 expression in nuclear and cytoplasmic fractions extracted from OVCAR8 cells treated with cisplatin and/or LMB. Fractionation efficiency was determined by probing for total histone H3 and a-tubulin in nuclear and cytoplasmic fractions, respectively; (C) Immunofluorescence analysis of cytochrome C release. (D) Protein levels of TR3, cleaved PARP and cleaved caspase-3. Values are mean + SD of 3 independent experiments. # p < 0.01 relative to cisplatin effect, Student's t test.
Ovarian cancer cells with TR3 down-regulation are resistant to cisplatin-induced apoptosis in vitro and in vivo
Having established a functional link between TR3 and cisplatin-induced apoptosis, we next aimed to model the TR3 down-regulation observed in ovarian tumors. To this end, we generated OVCAR8 cell clones stably expressing a GFP-tagged shRNA plasmid targeting TR3 (ShTR3). We identified three GFP-positive clones displaying substantial TR3 down-regulation at the protein level compared to GFP-positive clones expressing control, scrambled ShRNA (Figure 4A&B). The highest levels of TR3 expression were observed in ShScr#1 cells, comparable to TR3 levels in parental OVCAR8 cells (data not shown). Demonstrating the specificity of TR3 down-regulation, protein expression of the structurally related NR4A family members, NR4A2/Nurr1 and NR4A3/Nor1, was not altered in these TR3 knockdown clones (Figure 4A). A similar pattern of effect was observed at the mRNA level (Figure 4C).
Figure 4. Cisplatin resistance in ovarian cancer cells with TR3 knockdown in vitro.

(A) Protein levels of TR3 (NR4A1), NR4A2/Nurr1 and NR4A3/Nor1, and p21 in OVCAR-8 clones stably transfected with ShRNA targeting TR3 (ShTR3) or control scrambled ShRNA (ShScr). (B) Immunofluorescence analysis of GFP expression (green) and TR3 expression (red) in ShTR3#3 and ShScr#1 cells. DAPI-stained nuclei are in blue. (C) Quantitative RT-PCR analysis of TR3 and NR4A2 mRNA expression relative to corresponding GAPDH levels in ShTR3#3 cells compared to ShScr#1 cells. * p < 0.01 relative to expression in ShScr control, Student's t test. (D) Effects of increasing concentrations of cisplatin in SRB assays (72h treatment). Values are the percentage growth inhibition by cisplatin in ShScr#1 and ShTR3#3 clones at each concentration tested. # p < 0.01 relative to cisplatin effect in ShScr cells, Student's t test). Protein levels of TR3, cleaved PARP and cleaved caspase-3 in (E) ShTR3 and ShScr cells, and (F) OVCAR3 cells transiently transfected with non-targeting (NT) or two distinct siRNAs targeting TR3 (siTR3), treated with 5 μM cisplatin for 24h. All values are mean + SD of 3 independent experiments.
In SRB assays, ShTR3#3 cells showed significantly reduced basal growth compared to all three ShScr-expressing clones (Supplementary Figure 6). Growth inhibition following TR3 knockdown was accompanied by up-regulation of basal levels of the cyclin-dependent kinase inhibitor p21 (Figure 4A). Notably, ShTR3#3 cells showed the highest p21 levels among the TR3 knockdown clones, consistent with the highest level of basal growth inhibition in these cells. As shown in Supplementary Figure 7, the inhibitory effects of 5 μM cisplatin in ShScr clones in SRB assays were reduced in all three TR3 knockdown clones, with greatest inhibition observed in ShTR3#3 cells. Therefore, we selected the combination of ShScr#1 and ShTR3#3 cells for further analysis. We confirmed that cisplatin resistance in TR3 knockdown cells was observed over a range of cisplatin concentrations (2-10 μM) (Figure 4D). Moreover, a major contribution to this effect in ShTR3 cells was inhibition of cisplatin-induced apoptosis, as evidenced by reduced levels of cleaved PARP and cleaved caspase-3 (Figure 4E). We also confirmed that TR3 down-regulation conferred resistance to cisplatin-induced apoptosis in OVCAR3 cells, using two distinct TR3-targeting siRNAs (Figure 4F). Reduced cytoplasmic translocation of TR3 in ShTR3 and siTR3-transfected cells was confirmed by subcellular fractionation and immunofluorescence (data not shown).
The inhibitory effects of mechanistically distinct cytotoxic drugs, the DNA damaging agent, doxorubicin and the histone deacetylase inhibitor, SAHA, were also inhibited by TR3 knockdown in SRB assays (Supplementary Figure 7A). We confirmed that doxorubicin and SAHA both induced mitochondrial targeting of TR3 in OVCAR8 cells (Supplementary Figure 7B). These findings indicate that the reduced growth inhibitory and apoptotic effects following TR3 knockdown were not specific to cisplatin.
We next examined the response of ShScr#1 and ShTR3#3 cells to cisplatin in vivo. As shown in Figure 5A-C, 3 weeks' treatment with cisplatin via IP injection induced pronounced reduction in size of tumors derived from ShScr cells, an effect abrogated by approximately 52 ± 8% by TR3 knockdown. Basal growth of ShTR3 xenografts was reduced by 14 ± 5% compared to ShScr tumors, although this difference failed to reach statistical significance (p = 0.081, Mann-Whitney test). Immunohistochemical analyses were performed to quantify the number of apoptotic and proliferating cells by staining for cleaved caspase-3 and Ki67, respectively. As shown in Figure 5D&F, the cisplatin-induced increase in cleaved caspase-3 was significantly abrogated in ShTR3 cells. In contrast, the cisplatin-induced reduction in cells expressing Ki67 was similar between ShTR3 and control ShScr cells (Figure 5E&F). These results suggest that reduced cisplatin-induced apoptosis in tumors with TR3 down-regulation likely contributed more to cisplatin resistance than reduced growth inhibitory effects.
Figure 5. Cisplatin resistance in TR3 knockdown cells in vivo.

(A) Time course of growth of ShScr#1 and ShTR3#3 tumors in nude mice treated with vehicle (PBS) or cisplatin (5 mg/kg weekly) for 3 weeks. (B) Waterfall plot showing the percentage change in tumor volume from Time 0 (when drug injections were initiated) for individual tumors. Tumors are shown in (C). Immunohistochemical analyses of expression of (D) cleaved caspase-3 and (E) Ki67. Values are mean + SD; # p < 0.01 relative to cisplatin effect in ShScr tumors, Student's t test). (F) High power images for TR3, cleaved caspase-3 and Ki67 staining in vehicle and cisplatin-treated tumors.
TR3 overexpression sensitizes ovarian cancer cells to cisplatin-induced apoptosis in vitro
Since a large proportion of ovarian tumors retain high TR3 expression which is associated with relative sensitivity to platinum chemotherapy, we next determined whether increasing TR3 expression increased response to cisplatin. We selected SKOV3 cells for these studies because of their relatively low endogenous TR3 protein expression (see Figure 1A), which we confirmed in immunofluorescence experiments (data not shown). As shown in Figure 6A, SKOV3 cells transiently transfected with a TR3-myc/DDK (FLAG)-tagged expression vector were sensitized to cisplatin-induced apoptosis. We confirmed that the TR3-FLAG vector was targeted to the mitochondria in cisplatin-treated cells by immunofluorescence staining (Figure 6B).
Figure 6. Sensitization of ovarian cancer cells to cisplatin-induced apoptosis.
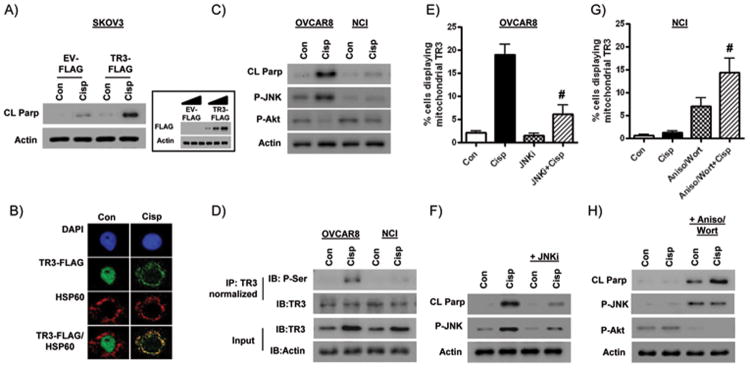
(A) Protein levels of cleaved PARP in SKOV3 cells transiently transfected with a DDK (FLAG)-tagged TR3 expression vector (1μg) or an equivalent amount of empty vector (EV) and treated with 5 μM cisplatin for 24h. Inset: Specific expression of the FLAG tag in cells transfected with increasing amounts of the TR3-FLAG vector. (B) Mitochondrial translocation of the TR3-FLAG vector following cisplatin treatment in SKOV3 cells. Effects of cisplatin treatment (5μM; 24h) on (C) protein levels of phospho-JNK (Thr183/Tyr185) and phospho-Akt (Ser473), and (D) serine phosphorylation levels of TR3, in OVCAR8 and NCI/ADR-RES cells. Control and cisplatin-treated IP lysates were normalized to ensure equivalent amounts of TR3 in (D). Effects of 2h pre-treatment and then co-treatment with the JNK inhibitor SP600125 (20 μM) on (E) mitochondrial localization of TR3 and (F) protein levels of cleaved PARP and phospho-JNK (Thr183/Tyr185) in cisplatin-treated OVCAR8 cells (5μM; 24h). Effects of 2h pre-treatment and co-treatment (3 hourly pulses) with the JNK activator anisomycin (25 ng/ml) and Akt inhibitor wortmannin (5 μM) on (G) mitochondrial localization of TR3 and (H) protein levels of cleaved PARP, phospho-JNK (Thr183/Tyr185) and phospho-Akt (Ser473) in cisplatin-treated NCI/ADR-RES cells (5μM; 12h). All values are mean + SD of 3 independent experiments. # p < 0.01 relative to cisplatin alone, Student's t test.
JNK and Akt mediate TR3 phosphorylation and nuclear export in association with cisplatin sensitivity in ovarian cancer cells
To investigate mechanisms linking nuclear export of TR3 to cisplatin sensitivity in ovarian cancer cells, we examined TR3 phosphorylation by JNK activation and Akt inhibition. It is known that N-terminal serine phosphorylation by JNK and loss of Akt-mediated phosphorylation on serine 351 are essential for nuclear export of TR3 (15). Furthermore, a less sustained JNK activation in cisplatin-resistant A2780 CP20 cells compared to PAR cells is observed following cisplatin treatment (34).
First, we demonstrated JNK activation and AKT inhibition in OVCAR8 cells compared to cisplatin-resistant NCI/ADR-RES cells after 24 hours cisplatin treatment (Figure 6C). Then we showed in co-immunoprecipitation experiments that OVCAR8 cells display increased levels of serine phosphorylation of TR3 following cisplatin treatment, an effect not observed in NCI/ADR-RES cells (Figure 6D). To directly test the role of JNK activation in cisplatin sensitivity, we treated the cells with the JNK-selective inhibitor SP600125 (35). When JNK was inhibited, cytoplasmic translocation and mitochondrial targeting of TR3 was reduced (Figure 6E&F); effects were associated with reduced levels of cleaved PARP. Similar results were observed in cisplatin-treated OVCAR3 cells (Supplementary Figure 8A&B).
Having established a role for JNK activation in cisplatin sensitivity, we asked if cisplatin resistance could be overcome by the well-characterized JNK activator, anisomycin (36). Since inhibition of Akt is also necessary for TR3 nuclear export (15), we tested the effects of the Akt pathway inhibitor wortmannin (37). Treatment with anisomycin or wortmannin alone did not alter TR3 localization or apoptosis in cisplatin-resistant NCI/ADR-RES cells (Supplementary Figure 8). However, simultaneous treatment with wortmannin, anisomycin and cisplatin in NCI/ADR-RES cells as shown in Figure 6G&H, increased the number of cells with mitochondrial TR3 localization in association with apoptosis, and sensitized the cells to cisplatin-induced apoptosis. Similar results were seen in A2780 CP20 cells (Supplementary Figure C&D).
Discussion
A clinical challenge in the treatment of metastatic ovarian cancer is resistance to platinum-based chemotherapy. In this study, we demonstrate a role for TR3 in contributing to platinum-resistance in ovarian cancer. First, we show that low nuclear expression of TR3 is associated with platinum resistance and decreased survival in a large subset of metastatic serous ovarian cancers represented on a TMA we designed. Then, we demonstrate a role for TR3 in mediating chemotherapy-induced apoptosis in epithelial ovarian cancer cells. Finally, we identify two plausible mechanisms by which TR3 contributes to cisplatin resistance in ovarian cancer by: 1) down-regulation of expression; and, 2) aberrant nuclear sequestration.
To date, no previous reports have related protein expression of the TR3 nuclear orphan receptor in ovarian tumors to clinical outcomes. In our TMA, a large subset of metastatic ovarian serous tumors had low TR3 protein levels, which were associated with reduced responses to platinum chemotherapy, and decreased overall and progression-free survival. TCGA data indicate that mRNA expression is not significantly different in platinum resistant and sensitive tumors, and demonstrate no significant association between TR3 expression and survival indices (3). Although limited by fewer cases, Yoshihara data set reveals reduced progression-free survival with low TR3 mRNA levels (33). Moreover, our results are consistent with previous reports of down-regulated TR3 expression in other types of metastatic solid tumors (17, 20).
Targeting the pro-apoptotic effects of TR3 as a therapeutic tool is emerging as an attractive strategy for cancer treatment (4). In response to pro-apoptotic factors or direct agonists such as derivatives of cytosporone B and C-DIM (8, 16, 38), the TR3 orphan nuclear receptor is implicated in mediating apoptosis in cancer cells by at least three distinct mechanisms: (i) nucleus-to-cytoplasmic translocation, resulting in cytochrome C release through direct mitochondrial targeting of TR3 (5, 6, 8-11); (ii) mechanisms independent of mitochondrial association (7); and, (iii) activation of TR3-mediated transcription (12-14). Although TR3 has been associated with vitamin K2-induced apoptosis in ovarian-derived teratocarcinoma Pa-1 cells (5), a role for TR3 in mediating chemotherapy-induced apoptosis in human ovarian cancer cells of epithelial origin has not been previously described. Here, we have identified nuclear export to the mitochondria as a major factor in determining cisplatin response in epithelial ovarian cancer cells.
Cytoplasmic translocation of TR3 was observed in relatively cisplatin-sensitive cells (OVCAR8, OVCAR3 and A2780 PAR), but not in cisplatin-resistant NCI/ADR-RES or A2780 CP20 cells. We show that TR3 is directly targeted to the mitochondria in ovarian cancer cells treated with cisplatin and other pro-apoptotic drugs, doxorubicin and the histone deacetylase inhibitor, SAHA. Moreover, several lines of evidence indicate that TR3 localized to the mitochondria is directly implicated in apoptosis induction. First, cytochrome C release was observed in approximately 80% of cells expressing cytoplasmic TR3. Second, inhibition of nuclear export of TR3 with leptomycin B significantly reduced the extent of cisplatin-induced apoptosis. Third, shRNA- and siRNA-mediated down-regulation of TR3 expression resulted in significant resistance to the anti-tumor effects of cisplatin and other chemotherapeutic drugs, accompanied by reduced levels of TR3 in the cytoplasm. While these studies indicate a role for direct TR3-mediated activation of the intrinsic apoptotic pathway, the fact that leptomycin B did not completely inhibit cisplatin-induced apoptosis suggests that TR3 may also be exerting pro-apoptotic effects through in the nucleus. This result is consistent with the observed pro-apoptotic effects of the methylene-substituted diindolylmethane (C-DIM) family of TR3 agonists in cancer cells in the absence of cytoplasmic TR3 translocation(13, 38). To fully understand the role of TR3 in ovarian cancer apoptosis, we will explore nuclear TR3 pro-apoptotic effects in future studies.
We acknowledge that multiple alternative mechanisms of cisplatin-induced apoptosis and platinum resistance have been reported in cancer cells (39-44) that are TR3-independent. However, in this study, we have identified two mechanisms by which deregulation of TR3 function contributes to cisplatin resistance: down-regulation of expression and aberrant nuclear sequestration. We also provide evidence that these effects may at least be partially overcome by TR3 overexpression or by stimulating TR3 phosphorylation and nuclear export by JNK activation and inhibition of Akt.
While reduced TR3 levels in advanced cancer may impair response to chemotherapy as suggested by our data, a study in breast cancer showed that down-regulation of TR3 promotes cell invasion and migration(17). Therefore, TR3 may play a more complex role in ovarian cancer than simply as a putative target of therapy. Determining whether TR3 down-regulation in ovarian cancer cells promotes other critical pro-tumorigenic cellular processes, such as tumor cell invasion, and/or activation of oncogenic signaling pathways (19), will be the focus of future studies. Adding to the complexity of the possible roles of TR3 in ovarian cancer, we also showed that there was reduced growth in TR3 knockdown cells. These findings suggest a pro-proliferative role for TR3, which may have significance in some metastatic ovarian cancer cells and normal ovarian epithelial cells expressing high levels of TR3. Such a cell-type and context-dependent pro-proliferative role for TR3 would be consistent with 1) the ability of mitogens to induce TR3 in the absence of apoptosis (7) and 2) observations in various solid tumors, where TR3 is over-expressed in cancer cells compared to normal epithelium (16-19).
In conclusion, we have shown that TR3 is an important regulator of apoptosis and plays a role in mediating response to cytotoxic chemotherapy such as cisplatin in subtypes of ovarian cancer. Further investigation into cell-type and context-dependent mechanisms is planned. Nevertheless, our results suggest that targeting TR3 by activating its expression and promoting its nuclear export are rational therapeutic strategies for overcoming cisplatin resistance in ovarian cancer.
Supplementary Material
Acknowledgments
The Vanderbilt University High-Throughput Screening Core, the Vanderbilt Immunohistochemistry Core, the Vanderbilt Genome Shared Resources Core, the Vanderbilt Epithelial Biology Center Core. Ms. Lynne Black for administrative and editorial support.
Grant Support: The project described was supported by the National Center for Research Resources, Grant UL1 RR024975-01, and is now at the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06. AJW was also supported by the Marsha Rivkin Ovarian Cancer Foundation (Seattle, WA). DK was supported by NIH grants 5P30 CA068485, CA091408 5 U54, 1UL1 RR024975 and K08CA148887-01.
Footnotes
Conflict of interest: The authors disclose no potential conflicts of interest
Author' Contributions: Conceptions and design: A.J. Wilson, D. Khabele
Development of methodology: A.J. Wilson, S.A. Fletcher, J.C. Slaughter, O. Fadare, D. Khabele
Acquisition of data: A.J. Wilson, A.Y. Liu, O.B. Adebayo, S. James, O. Fadare, D. Khabele
Analysis and interpretation of data (e.g. statistical analysis, biostatistics, computational analysis): A.J. Wilson, A. Liu, J. Roland, O.B. Adebayo, S.A. Fletcher, J.C. Slaughter, O. Fadare, D. Khabele
Writing, review and/or revision of the manuscript: A.J. Wilson, A.Y. Liu, J. Roland, O.B. Adebayo, S.A. Fletcher, J. C. Slaughter, J. Saskowski, M.A. Crispens, H.W. Jones, III, S. James, O. Fadare, D. Khabele
Administrative, technical or material support: A.J. Wilson, O.B. Adebayo, J. Saskowski, M.A. Crispens, H.W. Jones, III, S. James, O. Fadare, D. Khabele
Study supervision: A.J. Wilson, D. Khabele
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Landen CN, Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol. 2008;26:995–1005. doi: 10.1200/JCO.2006.07.9970. [DOI] [PubMed] [Google Scholar]
- 3.Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SO, Li X, Khan S, Safe S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin Ther Targets. 2011;15:195–206. doi: 10.1517/14728222.2011.547481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sibayama-Imazu T, Fujisawa Y, Masuda Y, Aiuchi T, Nakajo S, Itabe H, et al. Induction of apoptosis in PA-1 ovarian cancer cells by vitamin K2 is associated with an increase in the level of TR3/Nur77 and its accumulation in mitochondria and nuclei. J Cancer Res Clin Oncol. 2008;134:803–12. doi: 10.1007/s00432-007-0349-z. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–64. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 7.Wilson AJ, Arango D, Mariadason JM, Heerdt BG, Augenlicht LH. TR3/Nur77 in colon cancer cell apoptosis. Cancer Res. 2003;63:5401–7. [PubMed] [Google Scholar]
- 8.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–56. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 9.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–40. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 10.Yu H, Kumar SM, Fang D, Acs G, Xu X. Nuclear orphan receptor TR3/Nur77 mediates melanoma cell apoptosis. Cancer Biol Ther. 2007;6:405–12. doi: 10.4161/cbt.6.3.3755. [DOI] [PubMed] [Google Scholar]
- 11.Wu Q, Liu S, Ye XF, Huang ZW, Su WJ. Dual roles of Nur77 in selective regulation of apoptosis and cell cycle by TPA and ATRA in gastric cancer cells. Carcinogenesis. 2002;23:1583–92. doi: 10.1093/carcin/23.10.1583. [DOI] [PubMed] [Google Scholar]
- 12.Shin HJ, Lee BH, Yeo MG, Oh SH, Park JD, Park KK, et al. Induction of orphan nuclear receptor Nur77 gene expression and its role in cadmium-induced apoptosis in lung. Carcinogenesis. 2004;25:1467–75. doi: 10.1093/carcin/bgh135. [DOI] [PubMed] [Google Scholar]
- 13.Yoon K, Lee SO, Cho SD, Kim K, Khan S, Safe S. Activation of nuclear TR3 (NR4A1) by a diindolylmethane analog induces apoptosis and proapoptotic genes in pancreatic cancer cells and tumors. Carcinogenesis. 2011;32:836–42. doi: 10.1093/carcin/bgr040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao LM, He JP, Chen HZ, Wang Y, Wang WJ, Wu R, et al. Orphan receptor TR3 participates in cisplatin-induced apoptosis via Chk2 phosphorylation to repress intestinal tumorigenesis. Carcinogenesis. 2012;33:301–11. doi: 10.1093/carcin/bgr287. [DOI] [PubMed] [Google Scholar]
- 15.Han YH, Cao X, Lin B, Lin F, Kolluri SK, Stebbins J, et al. Regulation of Nur77 nuclear export by c-Jun N-terminal kinase and Akt. Oncogene. 2006;25:2974–86. doi: 10.1038/sj.onc.1209358. [DOI] [PubMed] [Google Scholar]
- 16.Cho SD, Yoon K, Chintharlapalli S, Abdelrahim M, Lei P, Hamilton S, et al. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Res. 2007;67:674–83. doi: 10.1158/0008-5472.CAN-06-2907. [DOI] [PubMed] [Google Scholar]
- 17.Alexopoulou AN, Leao M, Caballero OL, Da Silva L, Reid L, Lakhani SR, et al. Dissecting the transcriptional networks underlying breast cancer: NR4A1 reduces the migration of normal and breast cancer cell lines. Breast Cancer Res. 2010;12:R51. doi: 10.1186/bcr2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moll UM, Marchenko N, Zhang XK. p53 and Nur77/TR3 - transcription factors that directly target mitochondria for cell death induction. Oncogene. 2006;25:4725–43. doi: 10.1038/sj.onc.1209601. [DOI] [PubMed] [Google Scholar]
- 19.Fahrner TJ, Carroll SL, Milbrandt J. The NGFI-B protein, an inducible member of the thyroid/steroid receptor family, is rapidly modified posttranslationally. Mol Cell Biol. 1990;10:6454–9. doi: 10.1128/mcb.10.12.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 21.Chen HZ, Liu QF, Li L, Wang WJ, Yao LM, Yang M, et al. The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating Wnt signalling. Gut. 2012;61:714–24. doi: 10.1136/gutjnl-2011-300783. [DOI] [PubMed] [Google Scholar]
- 22.Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y, Chanock SJ, et al. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8:713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scudiero DA, Monks A, Sausville EA. Cell line designation change: multidrug-resistant cell line in the NCI anticancer screen. J Natl Cancer Inst. 1998;90:862. doi: 10.1093/jnci/90.11.862. [DOI] [PubMed] [Google Scholar]
- 24.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther. 2007;6:795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 26.Spannuth WA, Mangala LS, Stone RL, Carroll AR, Nishimura M, Shahzad MM, et al. Converging evidence for efficacy from parallel EphB4-targeted approaches in ovarian carcinoma. Mol Cancer Ther. 2010;9:2377–88. doi: 10.1158/1535-7163.MCT-10-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson AJ, Holson E, Wagner F, Zhang YL, Fass DM, Haggarty SJ, et al. The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol Ther. 2011;12:484–93. doi: 10.4161/cbt.12.6.15956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariadason JM, Arango D, Shi Q, Wilson AJ, Corner GA, Nicholas C, et al. Gene expression profiling-based prediction of response of colon carcinoma cells to 5-fluorouracil and camptothecin. Cancer Res. 2003;63:8791–812. [PubMed] [Google Scholar]
- 30.Wilson AJ, Lalani AS, Wass E, Saskowski J, Khabele D. Romidepsin (FK228) combined with cisplatin stimulates DNA damage-induced cell death in ovarian cancer. Gynecol Oncol. 2012;127:579–86. doi: 10.1016/j.ygyno.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denny L, Quinn M, Hacker N. FIGO Cancer Report 2012. Int J Gynaecol Obstet. 2012;119(2):S89. doi: 10.1016/S0020-7292(12)00458-4. [DOI] [PubMed] [Google Scholar]
- 32.Khabele D, Fadare O, Liu AY, Wilson AJ, Wass E, Osteen K, et al. An orthotopic model of platinum-sensitive high grade serous fallopian tube carcinoma. Int J Clin Exp Pathol. 2012;5:37–45. [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshihara K, Tajima A, Yahata T, Kodama S, Fujiwara H, Suzuki M, et al. Gene expression profile for predicting survival in advanced-stage serous ovarian cancer across two independent datasets. PLoS One. 2010;5:e9615. doi: 10.1371/journal.pone.0009615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li F, Meng L, Zhou J, Xing H, Wang S, Xu G, et al. Reversing chemoresistance in cisplatin-resistant human ovarian cancer cells: a role of c-Jun NH2-terminal kinase 1. Biochem Biophys Res Commun. 2005;335:1070–7. doi: 10.1016/j.bbrc.2005.07.169. [DOI] [PubMed] [Google Scholar]
- 35.Shin M, Yan C, Boyd D. An inhibitor of c-jun aminoterminal kinase (SP600125) represses c-Jun activation, DNA-binding and PMA-inducible 92-kDa type IV collagenase expression. Biochim Biophys Acta. 2002;1589:311–6. doi: 10.1016/s0167-4889(02)00195-7. [DOI] [PubMed] [Google Scholar]
- 36.Hazzalin CA, Le Panse R, Cano E, Mahadevan LC. Anisomycin selectively desensitizes signalling components involved in stress kinase activation and fos and jun induction. Mol Cell Biol. 1998;18:1844–54. doi: 10.1128/mcb.18.4.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitley BR, Beaulieu LM, Carter JC, Church FC. Phosphatidylinositol 3-kinase/Akt regulates the balance between plasminogen activator inhibitor-1 and urokinase to promote migration of SKOV-3 ovarian cancer cells. Gynecol Oncol. 2007;104:470–9. doi: 10.1016/j.ygyno.2006.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei P, Abdelrahim M, Cho SD, Liu X, Safe S. Structure-dependent activation of endoplasmic reticulum stress-mediated apoptosis in pancreatic cancer by 1,1-bis(3′-indoly)-1-(p-substituted phenyl)methanes. Mol Cancer Ther. 2008;7:3363–72. doi: 10.1158/1535-7163.MCT-08-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chock KL, Allison JM, Shimizu Y, ElShamy WM. BRCA1-IRIS overexpression promotes cisplatin resistance in ovarian cancer cells. Cancer Res. 2010;70:8782–91. doi: 10.1158/0008-5472.CAN-10-1352. [DOI] [PubMed] [Google Scholar]
- 41.Ali AY, Abedini MR, Tsang BK. The oncogenic phosphatase PPM1D confers cisplatin resistance in ovarian carcinoma cells by attenuating checkpoint kinase 1 and p53 activation. Oncogene. 2012;31:2175–86. doi: 10.1038/onc.2011.399. [DOI] [PubMed] [Google Scholar]
- 42.Marchion DC, Cottrill HM, Xiong Y, Chen N, Bicaku E, Fulp WJ, et al. BAD phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin Cancer Res. 2011;17:6356–66. doi: 10.1158/1078-0432.CCR-11-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Mao H, Hao Q, Yang Y, Shen L, Huang S, et al. Association of expression of XIAP-associated factor 1 (XAF1) with clinicopathologic factors, overall survival, microvessel density and cisplatin-resistance in ovarian cancer. Regul Pept. 2012;178:36–42. doi: 10.1016/j.regpep.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Zhang P, Liu SS, Ngan HY. TAp73-mediated the activation of c-Jun N-terminal kinase enhances cellular chemosensitivity to cisplatin in ovarian cancer cells. PLoS One. 2012;7:e42985. doi: 10.1371/journal.pone.0042985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.