

Abstract
Located on chromosome 9p21.3, p16INK4a seems lost amongst a cluster of neighboring tumor suppressor genes. While best known for inhibiting cyclin dependent kinase (CDK) activity, p16INK4a is not a one trick pony. Long term p16INK4a expression pushes cells to enter senescence, an irreversible cell cycle arrest that prevents the growth of would-be cancer cells, but also contributes to aging. Loss of p16INK4a is one of the most frequent events in human tumors and allows pre-cancerous lesions to bypass senescence. Therefore, precise regulation of p16INK4a is essential to tissue homeostasis, maintaining a tight balance between tumor suppression and aging. Here, we outline the pathways required for proper p16INK4a regulation and highlight the critical functions of p16INK4a in cancer, aging and human physiology that make this gene special.
Keywords: INK4/ARF, senescence, CDKN2A, polycomb
Introduction
Every day we depend on our cells to make the right decision – to divide or not to divide. Proliferation is essential for tissue homeostasis, but when deregulated can both promote cancer and lead to aging. For this reason, the decision to replicate is tightly controlled by a complex network of cell cycle regulatory proteins. In the early 1990s, it was clear that the catalytic activity of cyclin dependent kinases (CDKs) was required to drive cellular division. Less obvious were the signals that regulate CDK activity and how these became altered in neoplastic disease. In an attempt to address this very question, Beach and colleagues made the observation that CDK4 bound a distinct, 16 kilodalton protein in cells transduced with a viral oncogene (1). Biochemical characterization of this protein, later named p16INK4a, placed it amongst the INK4-class of cell cycle inhibitors, which bind directly to CDK4 and CDK6, blocking phosphorylation of the retinoblastoma tumor suppressor (RB) and subsequent traversal of the G1/S cell cycle checkpoint ((2, 3); Fig. 1A). In the presence of various stressors (e.g. oncogenic signaling, DNA damage), p16INK4a expression blocks inappropriate cellular division, and prolonged induction of p16INK4a leads to an irreversible cell cycle arrest termed ‘cellular senescence’.
Figure 1.

Function, Structure and Polymorphisms of the INK4/ARF Locus. A, p15INK4b and p16INK4a both function in the RB tumor suppressor pathway through inhibition of CDK4/6 activity. Expression of p14ARF inhibits the E3 ubiquitin ligase activity of MDM2, leading to stabilization of p53. The p53 and RB pathways play integral roles in blocking inappropriate cellular proliferation. B, Packed into 35 kilobases of chromosome 9p21.3 are three well-characterized tumor suppressor genes: p14ARF, p15INK4b and p16INK4a. GWAS have implicated 9p21.3 SNPs in cancer, heart disease, glaucoma, type II diabetes, autism and endometriosis. The majority of the SNPs lie outside of transcript and coding regions in a recently discovered long, non-coding RNA, ANRIL. Of the identified SNPs, those that have been shown to correlate with CDKN2A expression in at least one study are filled with grey. Other SNPs that have not been correlated with CDKN2A expression in validation studies, or have yet to be examined are filled with black or white, respectively.
The gene encoding p16INK4a, CDKN2A, lies within the INK4/ARF tumor suppressor locus on human chromosome 9p21.3 (Fig. 1B). CDKN2A encodes two transcripts with alternative transcriptional start sites (4). Both transcripts share exons 2 and 3, but are translated in different open reading frames to yield two distinct proteins: p16INK4a and ARF (p14ARF in humans, p19ARF in mice). In addition to CDKN2A, the INK4/ARF locus encodes a third tumor suppressor protein, p15INK4b, just upstream of the ARF promoter (3). Discovered through homology-based cDNA library screens, p15INK4b functions analogously to p16INK4a, directly blocking the interaction of CDK4/6 with D-type cyclins (2, 3). In contrast to p15INK4b and p16INK4a which function to inhibit RB phosphorylation, ARF expression stabilizes and thereby activates another tumor suppressor, p53 (5, 6). Like the INK family of inhibitors, p53 functions to block inappropriate proliferation and cellular transformation. Through a poorly understood mechanism, likely dependent upon cell type and transcriptional output, p53 activation can trigger either apoptosis or cell cycle arrest (7). A fourth INK4/ARF transcript, ANRIL (Anti-sense Non-coding RNA in the INK4/ARF Locus), was recently discovered in a familial melanoma kindred with neural system tumors (8). The ANRIL transcript runs anti-sense to p15INK4b and encodes a long, non-coding RNA elevated in prostate cancer and leukemia (9, 10). ANRIL is proposed to function as an epigenetic regulator of INK4/ARF gene transcription, targeting histone modifying enzymes to the locus (See discussion below). In summary, the INK4/ARF locus is a relatively small (110kb), but complex locus essential to the proper maintenance of cell cycle control and tumor suppression. In this review, we focus on the founding member of the INK4/ARF locus, p16INK4a, and discuss what is known and unknown about p16INK4a regulation in cancer and aging.
CDK4/6-Independent Roles of p16INK4a
Several lines of evidence suggest that p16INK4a may function both through CDK4/6-dependent and -independent mechanisms to regulate the cell cycle. CYCLIN D-CDK4/6 complexes are stabilized by interactions with the CDK2 inhibitors, p21CIP1, p27KIP1 and p57KIP2, and serve to titrate these proteins away from CDK2 (11–14). Subsequent expression of p16INK4a or p15INK4b causes these complexes to disassociate, releasing sequestered CDK2 inhibitors (15). This process, known as ‘CDK inhibitor reshuffling’, has been documented in a growing list of cell lines, and several lines of evidence support the biological relevance of this model. Mice harboring kinase-dead Cdk4 or Cyclin D1 alleles that retain p27KIP1 binding capacity (Cyclin D1K112E, Cdk4D158N) display heightened CDK2 activity (16–18) and fewer developmental defects than Cyclin D1 knockouts. The same observation holds true for a Cyclin D1 knock-in mutation incapable of binding RB (Cyclin D1ΔLxCxE)(19). As such, it is not surprising that p27KIP1 deletion can rescue the retinal hypoplasia and early mortality phenotypes of Cyclin D1-null mice (20, 21).
More recently the biological relevance of CDK inhibitor re-shuffling has come under scrutiny. Knock-in mice harboring p16INK4a-insensitive Cdk4 and Cdk6 alleles still capable of binding p27KIP1 (Cdk6R31C and Cdk4R21C, respectively) do not display the phenotypes predicted by this model (18, 22). The decreased p16INK4a binding capacity of these mutants should promote p27KIP1 sequestration and enhanced CDK2 activity, but cells from the liver and testes of Cdk4R21C mice show no change in the composition of CDK2-Cyclin complexes, nor do thymocytes harboring the Cdk6R31C allele (18, 22). These data suggest that, in at least a subset of cell types, the kinase activity of CDK4/6 is predominantly responsible for proliferative control. Knockout mice lacking a single CDK4/6 inhibitor (p16INK4a, p15INK4b or p19INK4d) develop normally and are born at expected Medelian ratios ((23–25). In contrast, p18INK4c knockouts are characterized by organomegaly, yet the association of p27KIP1 with CDK2 complexes is unchanged in these animals (26). Work examining combined loss of p15INK4b and p18INK4c (24) or p27KIP1 and p18INK4c (26) in mice suggests that distinct mechanisms are used by each inhibitor to control cellular proliferation. This result is in contrast to the CDK inhibitor re-shuffling model wherein co-deletion would be predicted to concertedly promote CDK2 activity. However, it is important to note that none of these publications contest the fact that CDK2 inhibitors bind CYCLIN D-CDK4/6 complexes and are released upon p16INK4a expression. Moreover, recent findings suggest that p16INK4a may contribute to cell cycle regulation through additional CDK-independent mechanisms. Specifically, expression of p16INK4a has been reported to stabilize p21CIP1, and may inhibit the AUF1-dependent decay of p21CIP1, cyclin D1 and e2f1 mRNA (27, 28). As a whole, these data provide evidence that the cell cycle-related functions of p16INK4a may extend beyond CDK4/6 inhibition to include the regulation of other CDK-CYCLIN targets.
Transcriptional, Translational and Epigenetic Regulation of p16INK4a
To maintain tissue homeostasis and prevent cancer, the ability of p16INK4a to inhibit cellular proliferation must be tightly controlled. In this section, we discuss the role of chromatin, transcriptional co-factors and RNAs in maintaining proper p16INK4a expression. In addition, we highlight the complexity and redundancy of p16INK4a regulatory pathways required for proper proliferative control.
p16INK4a Repression by Polycomb Group Complexes
Chromatin modifications by the polycomb group complexes (PcGs), PRC1 and PRC2, are critical to the homeostatic regulation of INK4/ARF gene expression (Fig. 2A). The PRC2 complex is made up of four core components: EZH1/2, EED, SUZ12 and RBAp46/48. EZH1 or 2 serves as the catalytic subunit of PRC2 and functions only in the presence of EED and SUZ12 to compact chromatin through the di- and tri-methylation of histone H3 lysine 27 (H3K27me2/3) (29, 30). H3K27me3 is recognized by a chromodomain-containing CBX protein family member associated with PRC1. In this manner, PRC1 is recruited to the INK4/ARF locus, where it catalyzes the ubiquitylation of histone H2A lysine 119 (H2AK119ub), resulting in further chromatin compaction and gene silencing (31). Multiple variants of the PRC1 complex have been identified in vivo, each containing homologs of the Drosophila Posterior Sex Comb (Psc; NSPC1/PCGF1, MEL-18/PCGF2, RNF3/PCGF3, BMI1/PCGF4, RNF159/PCGF5, RNF134/PCGF6), Polycomb (Pc; CBX2, CBX4, CBX6, CBX7, CBX8), Sex Combs Extra (RING1, RING2) and Polyhomeotic proteins (Ph; HPH1, HPH2, HPH3) (32). PRC1 complexes contain a single Psc and Pc homolog, yet Maertens et al. observed binding of MEL-18, BMI-1, CBX7 and CBX8 to the repressed INK4/ARF locus (32). Further investigation revealed that multiple PRC1 variants bind to the p16INK4a promoter, working in a concerted manner to control gene expression (32). It remains to be determined whether recently reported non-canonical PRC1 complexes that lack a Pc homolog, contain RYBP/YAF2, and are recruited to chromatin independent of H3K27me3 (33) can also function to regulate INK4/ARF gene expression. However, regardless of their composition, PRC1 and 2 complexes clearly bind throughout the INK4/ARF locus, and repress p16INK4a expression in young, unstressed cells (31). Maintenance of this repression may be partially dependent on the ubiquitin-specific protease, USP11, which was also shown by Maertens et al. to bind and stabilize the PRC1 complex (34). In their work, depletion of USP11 caused polycomb complexes to dissociate from the INK4/ARF locus leading to subsequent de-repression of p16INK4a.
Figure 2.
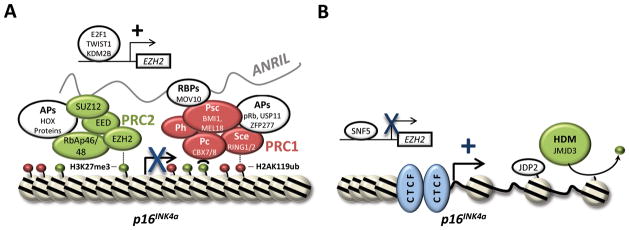
Mechanisms of p16INK4a Regulation by Chromatin Modification. A, p16INK4a is negatively regulated by the histone modifying complexes, PRC1 and PRC2, which combine to lay down repressive marks (e.g. H3K27me3, H2AK119ub) throughout the INK4/ARF locus. Several associated proteins (APs), including RNA binding proteins (RBPs), are reported to facilitate PRC1/2 interaction with the p16INK4a promoter. APs and RBPs discussed in the text are shown. Elevated expression of the PRC2 component, EZH2, is also reported to enhance INK4/ARF gene silencing. Proteins reported to transactivate EZH2, leading to PRC-mediated silencing of p16INK4a are depicted above. B, Activation of p16INK4a is associated with decreases in PRC1/2 levels and the removal of repressive histone marks. JMJD3 demethylates H3K27me3 and subsequent chromatin decondensation promotes access by transcription factors and p16INK4a transcription while JDP2 binds H3K27 and prevents further methylation. CTCF maintains chrosomal boundaries and 3-dimension structure of the chromatin surrounding p16INK4a.
The importance of PRC1 and PRC2 for proper p16INK4a regulation may be best exemplified by the phenotypes of polycomb knockout mice. Deletion of the PRC1 component, Bmi1, results in homeotic skeletal transformations, lymphoid and neurological defects (35). Many of these phenotypes are attributable to the deregulation of homeobox gene expression, however, the lymphoid and neurological defects observed in Bmi1 knockout mice can be almost completely rescued by INK4/ARF deletion (36). Here, INK4/ARF loss reverses the self-renewal defects of Bmi1-null hematopoietic and neuronal progenitors (37, 38). Together, these data show that PRC1 regulation of p16INK4a expression is required for proper development, stem cell maintenance and homeostasis. In contrast to PRC1-null animals which survive gestation, deletion of the PRC2 members, Ezh2 or Suz12, is embryonic lethal (39, 40). For this reason, conditional knockout alleles are required to assess the biological functions of PRC2. Using such alleles to delete Ezh2 in the brain, pancreas and embryonic skin, phenotypic outcomes have been observed. Specifically, loss of Ezh2 in murine pancreatic islets causes a diabetic phenotype associated with increased β-cell expression of both p16INK4a and ARF (41). In contrast, Ezh2 deletion in the brain and skin resulted in only mild differentiation defects (42, 43). The recent observation that EZH1 is expressed in many adult tissues and also catalyzes histone H3 methylation led to the hypothesis that EZH1 may compensate for EZH2 loss in some settings. Indeed, deletion of both Ezh1 and 2 caused severe defects in murine skin morphogenesis associated with a >70-fold increase in p16INK4a/ARF expression (43).
Supporting a role for EZH2 in maintaining proliferative homeostasis, human germline mutations in EZH2 give rise to Weaver syndrome, a congenital disorder characterized by uncontrolled and rapid growth (44). While this phenotype could be attributed to p16INK4a silencing, few reports have attempted to functionally characterize the EZH2 missense mutations commonly associated with Weaver syndrome (44). Instead, work has focused on recurring point mutations reported in B-cell lymphoma. These mutations localize to tyrosine 641 of the EZH2 SET domain, resulting in the production of a neomorphic protein with enhanced H3K27 di- and tri-methylation activity (45). Of importance, not all cancer-associated EZH2 mutants are gain-of-function alleles. In myleloid neoplasms, missense, nonsense and frameshift mutations in EZH2 have been described which lack a catalytic SET domain (46). In addition, the expression of wildtype EZH2 has been reported to cause p16INK4a silencing in SNF5-deficient malignant rhabdoid tumors (MRTs) (47). Deletion or pharmaceutical inhibition of EZH2 activity in cells from these tumors increases p16INK4a expression, resulting in cell cycle inhibition (47, 48). Together, these observations suggest that EZH2 activity must be tightly controlled in order to maintain proliferative homeostasis and proper p16INK4a regulation.
Recruiting Polycomb to CDKN2A
In Drosophila melanogaster, polycomb group proteins are recruited to defined DNA binding sites termed, Polycomb repressive elements (PREs) (Reviewed in: (49)). In contrast, few mammalian PREs have been identified to date, leading many to speculate that other DNA binding proteins or RNAs must guide polycomb complexes to target genes like the INK4/ARF locus. Recent work suggests that long non-coding RNAs (lncRNAs) can serve as scaffolds for polycomb recruitment and epigenetic gene silencing. The discovery of disease-linked polymorphisms within ANRIL prompted investigation into whether this lncRNA could function in a similar manner to regulate INK4/ARF gene transcription. Through RNA binding assays, a study by Yap et al. showed that CBX7 interacts with both ANRIL and H3K27me3 to promote INK4/ARF gene silencing (Fig. 2A and (10)). Subsequently, ANRIL binding to SUZ12, a component of the PRC2 complex, was reported to promote silencing of p15INK4b, but not p16INK4a (50). Together with these data, a report showing that MOV10, a putative RNA helicase and PRC1 binding partner, is required for p16INK4a repression, supports a role for ANRIL in polycomb recruitment to the INK4/ARF locus (51). Whether MOV10 binds ANRIL to facilitate PRC1 interaction with the INK4/ARF locus has yet to be determined. However, together these data make a strong case for the role of ANRIL in epigenetic silencing of p16INK4a.
ANRIL-independent mechanisms are also suggested to recruit PcG complexes to the p16INK4a locus. Recently, H2.0-like homeobox 1 (HLX1), a homeobox (HOX) protein, was shown by Martin et al. to facilitate PRC2 recruitment to the p16INK4a promoter (Fig. 2A and (52)). While the mechanism has yet to be defined, six other homeobox-containing proteins (HOXA9, DLX3, HOXB13, HOXC13, HOXD3 and HOXD8) were similarly reported to participate in p16INK4a silencing (52). Given the role of HOX genes in developmental patterning, it is interesting to speculate that proteins like HLX1 and HOXA9 initiate tissue-specific silencing of p16INK4a. Like HOX proteins, both TWIST1, a basic helix-loop-helix (bHLH) transcription factor, and KDM2B, a histone demethylase, may also facilitate polycomb-mediated silencing of the INK4/ARF locus. Ectopic expression of TWIST1 and KDM2B can cause cellular levels of EZH2 to rise, resulting in an increase in PRC2 activity (53). KDM2B was further reported to function in demethylating H3K36me2/3, a common marker for DNA polymerase II transcription (53). Furthermore, TWIST1 appears to increase BMI1 expression (55), and subsequent recruitment of BMI1 to the p16INK4a promoter has been linked to interactions with phosphorylated RB (pRB) and zinc finger domain-containing protein 277 (ZFP277) (54, 55). In particular, the link between pRB and p16INK4a silencing is intriguing as it suggests the presence of a feedback loop wherein cells entering S-phase repress p16INK4a expression (54). ZFP277-mediated recruitment of BMI1 to the p16INK4a promoter may also be linked to the cell cycle. A study by Negishi et al. showed that reductions in ZFP277 expression caused by oxidative stress could lead to PRC1 dissociation from the p16INK4a promoter and subsequent cell cycle arrest (55). How these mechanisms of PRC recruitment interplay with ANRIL remains to be established, but certainly the complexity of p16INK4a silencing is indicative of the importance of this gene in maintaining tissue homeostasis.
Reversing Polycomb Silencing of the INK4/ARF locus
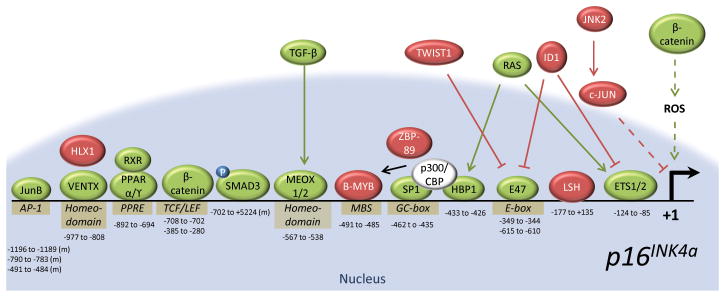
In order for normal cells to traverse the G1/S checkpoint, p16INK4a must be maintained in a repressed state. At the same time, induction of p16INK4a expression in the presence of stress signals is required to prevent inappropriate cell cycle progression. Linking proliferative control to epigenetic regulation of the p16INK4a promoter, Bracken et al. first demonstrated that RB phosphorylation during the G1 to S-phase transition releases E2F1 to transactivate EZH2 and EED (56). Later, these data were confirmed by an independent group who showed that p53 activation represses EZH2 expression through RB-mediated inhibition of E2F1 activity (57). While these results explain how the activity of PRC2 might be curbed in the presence of stress, they do not explain how epigenetic silencing of the p16INK4a promoter is reversed. One mechanism of removing repressive histone marks is via the activity of histone demethylases. In response to oncogenic stressors, levels of the H3K27me3 demethylase, Jmjd3, increase, removing repressive histone marks from the p16INK4a promoter (Fig. 2B; (58, 59)). Following histone demethylation, the Yokoyama lab showed that Jun dimerization protein 2 (JDP2) may help maintain p16INK4a in an active state by binding and sequestering H3K27 away from the actions of PRC2 (60). These findings suggest that the activity of JDP2 and JMJD3 establishes a permissive state for p16INK4a expression. Furthermore, unmethylated H3K27 is no longer recognized by PRC1, and chromatin surrounding the p16INK4a promoter is decondensed. Based upon this activity, it is not surprising that JMJD3, like p16INK4a, serves as a barrier to induced pluripotency (61, 62) (See: “p16INK4a as a Barrier to Pluripotency”). Clearly, interplay between PRC1, PRC2 and histone demethylases is required for homeostatic regulation of the p16INK4a promoter, yet how this crucial balance is maintained is still in question. Moreover, it is possible that other histone demethylases, such as lysine-specific demethylase 6A (KDM6A/UTX), may also play a role in p16INK4a regulation.
In Drosophila, the SWI/SNF chromatin remodeling complex serves as a trithorax group activator, opposing the actions of polycomb-mediated silencing. Similarly, SWI/SNF functions to counteract PcG silencing of p16INK4a in mammals. Cancer cell lines deficient in the SWI/SNF component, SNF5, induce high levels of p16INK4a upon the restoration of SNF5 expression (63). In untransformed cell lines, SNF5 binds and directly inhibits the transcription of EZH2, resulting in decreased polycomb occupancy at the p16INK4a promoter (47). Tumor cells lacking SNF5 overexpress EZH2 and are dependent upon the activity of PRC2 to silence p16INK4a expression and drive proliferation (47, 64).
Chromatin Regulation of p16INK4a by Non-Polycomb Proteins
Epigenetic modification of the INK4/ARF locus extends beyond polycomb-mediated silencing. The well-conserved genomic insulator, CCCTC-motif binding factor (CTCF), binds throughout the INK4/ARF locus (65, 66) and functions to regulate both chromatin compaction and gene expression (Fig. 2B). Witcher et al. first reported interaction of CTCF with a chromosomal boundary ~2kb upstream of the p16INK4a promoter (65). In their studies of cancer cell lines, knockdown of CTCF resulted in the spread of heterochromatin DNA into the INK4/ARF locus leading to epigenetic silencing of p16INK4a (65). In contrast to this model, Hirosue et al. recently reported that decreases in CTCF expression associated with oncogene induced senescence promote decondensation of the INK4/ARF locus and lead to heightened levels of p16INK4a mRNA (66). Reconciliation of these two results is possible as rapid p16INK4a induction following CTCF knockdown would place enormous pressure on would-be cancer cells to epigenetically silence the INK4/ARF locus. In fact, the observation that epigenetic silencing of p16INK4a in breast cancers is accompanied by CTCF disassociation from the locus (65) is consistent with a role for CTCF in proper epigenetic regulation of INK4/ARF.
Transcriptional Activators and Repressors of p16INK4a
Opposing Transcriptional Regulators
The presence of a permissive chromatin state alone is insufficient for p16INK4a expression. Binding of activation factors and the subsequent recruitment of RNA polymerase is required to initiate p16INK4a transcription (Fig. 3). Similar to the interplay between PRC2 and JMJD3, transcriptional regulation of the p16INK4a promoter is tightly controlled by antagonistic pathways. Serving as a classic example, induction of p16INK4a by the E-box binding transcription factors E-26 transformation-specific 1 (ETS1), E-26 transformation-specific 2 (ETS2) and E47, is directly opposed by Inhibitor of DNA Binding 1 (ID1) (67). In response to oncogenic and senescent signaling, ETS1, ETS2 and/or E47 bind to E-box motifs (CANNTG) within the p16INK4a promoter to stimulate gene expression (67, 68). This action is directly antagonized by ID1 which prevents interaction of ETS1, ETS2 and E47 with the p16INK4a promoter (67, 68). As such, it is not surprising that Id1 null MEFs undergo premature senescence in culture (69), and that age-related increases in p16INK4a expression correlate directly with ets-1 levels in mice and rats (70).
Figure 3.
Transcriptional Regulation of p16INK4a. Expression of p16INK4a requires the action of transcription factors that recruit and/or facilitate RNA polymerase association with the promoter (shown in green). Opposing this action are transcriptional repressors (shown in red). Direct interactions with the p16INK4a promoter are depicted by a solid line, indirect interactions with a dotted line. The numbers below each binding site indicate the position of protein interaction relative to the p16INK4a transcriptional start site. All locations correspond to the human genome unless designated by an ‘(m)’, which signifies the mouse genome. Proteins predicted to share a common binding site are depicted over top of one another.
Similar to ID1, TWIST1 is reported to oppose transcriptional activation of p16INK4a. The relationship between ID1 and TWIST1 was initially suggested in studies examining the progression of benign human nevi to melanoma. In general, nevi are non-proliferative and express elevated levels of p16INK4a; yet, upon progression to melanoma these lesions frequently silence p16INK4a (71). Analysis of TWIST1 and p16INK4a expression in nevi and melanomas revealed an inverse correlation between these two proteins, putting forth the hypothesis that they function in antagonistic pathways (72). Preliminary work suggested that this antagonism might be mediated through direct interaction of TWIST1 with ETS2(72), however, in a recent publication by Cakouros et al., TWIST1 was reported to inhibit p16INK4a transcription by decreasing E47 expression (73). Clearly, further work is required to fully understand the physiological relationship between p16INK4a and TWIST1. In addition, it will be of interest to assess the potential role of p16INK4a in classical TWIST1 pathways including epithelial to mesenchymal transition, stem cell maintenance and tumor metastasis.
In line with the relationship between TWIST, ID1 and E-box binding transcription factors, antagonistic interplay has also been described between members of the Activator Protein-1 (AP-1) family of transcription factors, c-JUN and JUNB. While JUNB serves to activate p16INK4a transcription by binding to three identified AP1-like sites within the p16INK4a promoter (74), c-JUN limits p16INK4a expression (75). Certainly, a delicate balance between inhibitory (ID1, TWIST, c-JUN) and stimulatory (JUNB, ETS, E47) pathways is required for proper regulation of p16INK4a. Upsetting this balance through the overexpression of inhibitors or repression of p16INK4a activators promotes the bypass of senescence and can lead to cancer (72, 73, 76–78).
The HOX family of proteins could also be viewed as antagonistic p16INK4a regulators. Above, we discussed the potential role of HLX1, HOXA9, DLX3, HOXB13, HOXC13, HOXD3 and HOXD8 in polycomb-mediated repression of the INK4/ARF locus. In contrast, the HOX proteins, VENTX, MEOX1 and MEOX2 have each been reported to bind the p16INK4a promoter and activate gene transcription (79–81). This proposed interplay between HOX genes and p16INK4a regulation is suggestive of a role for CDK4/6 inhibition in embryonic development. In spite of this, developmental defects are not observed in p16INK4a knockout mice or melanoma-prone kindreds harboring germline p16INK4a deficiencies (25, 82). Whether compensatory mechanisms are required to combat the functional loss of p16INK4a during development is still to be determined.
Role of Acetyltransferases and Deacetylases in p16INK4a Regulation
Histone acetylation facilitates chromatin decondensation and subsequent gene transactivation. As such, transcriptional coactivators often harbor or recruit histone acetyltransferase (HAT) activity to target gene promoters. In a pair of recent publications, Wang et al. describe how the transcription factors, SP1 and HMG box-containing protein 1 (HBP1), recruit p300, a well-known HAT, to the p16INK4a promoter (83, 84). In their studies, acetylation of local chromatin as well as HBP1, promoted decondensation of the p16INK4a promoter and subsequent gene transactivation. However, numerous targets of p300 acetylation have been identified to date, including B-MYB, a putative repressor of p16INK4a transcription (83, 85, 86). Therefore, the p300-p16INK4a relationship is likely complex and may be dependent upon the available pool of transcriptional co-factors within a given cell type.
HAT activity is opposed by histone deacetylases (HDACs) which promote transcriptional silencing. In human cell lines, HDACs 1–4 have all been reported to bind and repress transcription from the p16INK4a promoter (52, 87–89). Most of these interactions have been linked to bridging transcription factors such as Lymphoid Specific Helicase (LSH), HLX1 and ZBP-89 (87, 89); albeit one report suggested that HDAC2 may directly bind the p16INK4a promoter (88). While loss of Hdac1, 2, 3 or 4 causes developmental abnormalities and lethality in mice, none of these phenotypes have been attributed to defects in p16INK4a regulation (90).
Age-related Signaling Pathways Influence p16INK4a Expression
The observation that p16INK4a levels are high in senescent cells has prompted several investigations into the connection between pro-senescent signaling and p16INK4a regulation. For example, age-related metabolic pathologies have long been associated with activation of the peroxisome proliferator-activated receptors (PPARs). It is now known that these nuclear receptors directly bind and activate the p16INK4a promoter leading to subsequent cell cycle arrest (91, 92). Similarly, alterations in TGF-β signaling have been linked to a variety of age-related diseases including cancer, osteoarthritis, cardiovascular disease and Alzheimer’s (93). Work by the Conboy lab has demonstrated that elevated TGF-β signaling reduces the capacity of muscle stem cells to regenerate (94). Here, phospho-SMAD3 has been shown to directly bind the p16INK4a promoter, stimulating gene transcription and cell cycle arrest (95). Due to crosstalk between the TGF-β and β-CATENIN/WNT signaling pathways, it is not surprising that aberrant WNT signaling is also associated with age-related disease (95). β-CATENIN can directly bind and activate the p16INK4a promoter in both human and murine cells (96–98), and recent evidence links the induction of p16INK4a by reactive oxygen species (ROS) to β-CATENIN/WNT signaling (99). Together these findings suggest a strong association between age-promoting, ‘gerontogenic’ signals and p16INK4a expression.
Cellular Structure and p16INK4a Expression
An emerging theme in p16INK4a regulation is the potential for cytoskeletal rearrangements to influence INK4/ARF gene transcription. In an siRNA screen of over 20,000 genes, Bishop and colleagues recently identified GLI2, a member of the Hedgehog signaling pathway, as an activator of p16INK4a expression (100). Interestingly, GLI2 partially localizes to a non-motile cytoskeletal protrusion called the primary cilium, and cultured human mammary epithelial cells with a primary cilium expressed lower levels of p16INK4a than those without (100). Upon ablation of p16INK4a, the number of cells with a primary cilium increased, suggesting a link between cellular structure and p16INK4a expression (100). Supporting this observation, the ACTIN nucleating enzymes ARP2 and 3 have been implicated in a second connection between cytoskeletal structure and Ink4/Arf gene transcription. Here, the generation of stable ARP2/3 knockdown cells required concomitant Ink4/Arf deletion (101). These data suggested that lamellipodial dysfunction triggers growth inhibitory Ink4/Arf gene transcription; however the specific role of p16INK4a in this process has yet to be examined.
The Role of microRNAs and RNA Binding Proteins in p16INK4a Regulation
While literature defining transcriptional and epigenetic regulators of p16INK4a is extensive, far fewer studies have examined post-transcriptional mechanisms of p16INK4a regulation. Discovered in an unbiased search for miRNAs silenced during senescence (102), two independent groups have reported that miR-24 binds and inhibits p16INK4a translation (102, 103). Consistent with this observation, antagonists of miR-24 cause a moderate decrease in the proliferation of normal human keratinocytes (103). In a similar manner, knockdown of miR-31 has been proposed to regulate cell cycle progression in murine embryo fibroblasts with altered nuclear structure (104). Here, loss of Lamin B1 was associated with increased miR-31 expression and p16INK4a instability (104). This work suggests another possible connection between cellular structure and p16INK4a regulation, linking changes in nuclear integrity to p16INK4a expression. Together, the associations between miR-31, miR-24 and p16INK4a suggest that miRNAs function to control proliferation via interactions with p16INK4a mRNA; however, it is important to note that other cell cycle regulators have been identified as miR-24 targets (e.g. cdk4, cyclin A2, cyclin B1, myc, e2f2, p14ARF and p27KIP1; (103, 105, 106)) and miR-31 (e.g. ets1, cdk1; (107)). Therefore, cell cycle defects caused by perturbations in miRNA expression likely reflect the outcome of both p16INK4a-dependent and -independent pathways.
The let-7 family of microRNAs has also been implicated in p16INK4a regulation, proliferative control and stem cell aging. The Morrison group first reported that murine let-7b expression increased with age in neuronal stem cells (108). Although let-7b did not bind p16INK4a mRNA directly, overexpression of let-7b reduced the expression of High-Mobility Group AT-Hook 2 (HMGA2), causing p16INK4a levels to increase (108). In addition to hmga2, a growing number of RNAs involved in growth and proliferative control have been identified as let-7b targets, and therefore, it is not surprising that the levels of most let-7 family members decrease during tumor progression (109).
In addition to miRNAs, RNA binding proteins can also regulate p16INK4a translation. Interaction of the Hu RNA binding protein, HuR, with the p16INK4a 3′UTR have been reported to destabilize the transcript in an miRNA-independent fashion (110). Work by Zhang et al. suggests that this action is opposed by the tRNA methyltransferase, NSUN2, which methylates the 3′UTR of p16INK4a to prevent HuR binding and subsequent mRNA degradation (111). In this way, interplay between HuR and NSUN2 may tightly control p16INK4a translation. For example, in the presence of oxidative stress, NSUN2 levels appear to increase, tipping the balance towards mRNA stability, p16INK4a expression and subsequent cell cycle arrest (111).
The Function and Significance of p16INK4a Expression in Cancer
In the absence of p16INK4a, both mice and humans are predisposed to cancer (25, 82). Loss of p16INK4a function can occur through gene deletion, methylation or mutation, and therefore, comprehensive genetic analyses are required to determine the frequency of p16INK4a silencing in cancer. Adding to the challenge of assessing p16INK4a status in human tumors, few studies distinguish between the two CDKN2A transcripts: p16INK4a and ARF. Here, we compiled data from the Cancer Genome Atlas (TCGA) to reveal that functional inactivation of CDKN2A is a frequent event in most tumor types ((112–118); Fig. 4). We probed 16 tumor types to determine the percent of cases wherein mutational and/or copy number changes disrupted genes critical to the p16INK4a tumor suppressor pathway (i.e. RB1, CDKN2A, CCND1 (CYCLIN D1), CCND2 (CYCLIN D2), CCND3 (CYCLIN D3), CCNE1 (CYCLIN E1), and CCNE2 (CYCLIN E2)). Tumor types displaying a high percentage of p16INK4a tumor suppressor pathway alterations included urothelial carcinoma (BLUCA; 77%), glioblastoma multiforme (GBM; 77%), head and neck squamous cell carcinoma (HNSC; 63%), lung squamous cell carcinoma (LUSC; 58%) and cutaneous melanoma (SKCM; 58%) (Fig. 4A). Examination of individual tumor profiles revealed infrequent overlap between CDKN2A and RB1 deletion, implying that these are often mutually exclusive tumorigenic events (Fig. 4B). In support of these data, an inverse correlation between p16INK4a and RB inactivation was previously described in lung cancer cell lines (119) and human tumors (120). In contrast to RB, we frequently noted alterations within other components of the p16INK4a tumor suppressor pathway (i.e. Cyclin amplification) in CDKN2A mutant tumors from the TCGA dataset (Fig. 4B). However, in cases where the p16INK4a tumor suppressor pathway appeared genetically intact, high levels of RB1 or CDKN2A methylation were observed (e.g. kidney cancers (KIRP, KIRC), colorectal adenocarcinomas (COAD/READ), low grade glioma (LGG), and acute myeloid leukemia (AML); Fig. 4C). Combining these genetic and epigenetic data suggests that functional inactivation of the p16INK4a-RB axis approaches 100% in many cancer types. A prior study by Schutte et al. supports this claim, demonstrating inactivation of the p16INK4a/RB-axis in 49 of 50 pancreatic carcinomas (121). However, further comprehensive assessment of p16INK4a functionality in a wide variety of tumors will require thorough experimental and bioinformatic analyses.
Figure 4.

Alterations in the p16INK4a Tumor Suppressor Pathway are Frequent in Human Cancer. A, Data from TCGA was obtained and analyzed using cBioPortal (112, 113). The tumors analyzed are as follows: Bladder Urothelial Carcinoma (BLUCA), Glioblastoma Multiforme (GBM), Head and Neck Squamous Cell Carcinoma (HNSC), Lung Squamous Cell Carcinoma (LUSC), Skin Cutaneous Melanoma (SKCM), Ovarian Serous Cystadenocarcinoma (OV), Lung Adenocarcinoma (LUAD), Stomach Adenocarcinoma (STAD), Breast Invasive Cancer (BRCA), Uterine Corpus Endometrial Carcinoma (UCEC), Brain Lower Grade Glioma (LGG), Colon and Rectum Adenocarcinoma (COAD/READ), Prostate Adenocarcinoma (PRAD), Kidney Renal Papillary Cell Carcinoma (KIRP), Kidney Renal Clear Cell Carcinoma (KIRC), Acute Myeloid Leukemia (AML). The percent of tumors with mutations or copy number changes in the p16INK4a tumor suppressor pathway are shown. For the purposes of this analysis the p16INK4a tumor suppressor pathway was defined to contain: CDKN2A (p16INK4a), RB1 (RB), CCND1 (CYCLIN D1), CCND2 (CYCLIN D2), CCND3 (CYCLIN D3), CCNE1 (CYCLIN E1), and CCNE2 (CYCLIN E2). B, OncoPrints from cBioPortal show aberrations in individual tumors across the X-axis. C, Methylation of CDKN2A (black bars) and RB1 (grey bars) was quantified using HM27 or HM450 TCGA data. Genes were considered methylated if β-values exceeded 0.2.
p16INK4a Expression in Tumors
In most tumor types, p16INK4a inactivation occurs early in tumorigenesis. For example, pancreatic intraepithelial neoplasias frequently inactivate p16INK4a upon progression to invasive disease (122). Pressure to silence p16INK4a presumably stems from oncogenic engagement of the RB tumor suppressor pathway. Therefore, tumors with early defects in RB signaling continue to express p16INK4a independent of progression (Reviewed in: (123)). Intense activation of the p16INK4a promoter is believed to represent a futile attempt by the cell to curb oncogenic proliferation in the absence of a functional G1/S checkpoint. In the clinic, immunohisotchemical p16INK4a staining is used to identify cervical and head and neck tumors driven by oncogenic human papilloma virus (HPV) infection. Here, the HPV viral oncoprotein, E7, functionally inactivates RB leading to increases in p16INK4a expression (124). Although it is believed that RB inactivation is requisite for the elevation of p16INK4a expression in cancer, aberrations in the RB pathway are not obvious in every tumor. The RB checkpoint is deregulated by multiple mechanisms independent of RB1 mutation, deletion or methylation. Viral oncogene expression and cyclin gene amplification represent just two possible forms of alternative RB inactivation. Unfortunately, there is a paucity of studies that comprehensively examine the status of the RB pathway in multiple human tumor types. Therefore, the critical role of this p16INK4a-induced checkpoint may be underestimated.
Limited analyses of the RB pathway in human tumors have revealed that p16INK4a expression can be predictive of both tumor subtype and therapeutic response. Elevated p16INK4a levels discern small cell lung cancer from lung adenocarcinoma (119, 125) and characterize the basal-like breast cancer subtype (126). Given that tumor subtypes often show distinct therapeutic response profiles, it is not surprising that p16INK4a expression can predict therapeutic efficacy. For example, elevated p16INK4a levels predict a higher initial response to radiation therapy in prostatic adenocarcinoma (127). However, these same p16INK4a positive tumors are the most likely to fail androgen-deprivation therapy (128). In oropharyngeal cancer, p16INK4a serves as a marker of oncogenic HPV infection and is predictive of improved therapeutic response and patient survival (129). While p16INK4a expression clearly serves as a relevant clinical marker, combined analysis of multiple RB pathway members may be of additional value. Likewise, recent data suggests that the localization of p16INK4a staining within a tumor may provide further clinical insight (see below).
The Significance of Stromal p16INK4a Expression
While a myriad of signals are linked to p16INK4a regulation, most of these are triggered by intrinsic, cell-autonomous events. Employing a recently developed luciferase reporter mouse, p16LUC, we have observed a second, cell-non-autonomous mechanism of p16INK4a activation (130). The p16LUC allele expresses firefly luciferase from the endogenous p16INK4a promoter, allowing investigators to dynamically track p16INK4a transcription in live animals. When the p16LUC allele was crossed with genetically engineered mouse models (GEMMs) of human cancer, a luminescent signal appeared only in the location of future tumor formation (130). These luminescent foci were visible weeks before tumors could be visualized or palpated, providing a significant detection advantage over traditional monitoring methods. This incredible sensitivity, along with the observation that tumors maintained luciferase activity during progression, led to the hypothesis that p16INK4a transcription is activated in the surrounding tumor stroma. Indeed, this was the case as syngeneic transplantation of six p16LUC-negative tumor cell lines harboring a wide variety of oncogenic drivers caused stromal induction of p16LUC (Fig. 5A and (130)). Although the specific stromal cell types responsible for this observation have yet to be identified, bone marrow transplantation studies suggest that the p16LUC signal originates in part from bone marrow-derived cells. Therefore, it appears that alterations in the milieu surrounding a growing neoplasm can promote the expression of p16INK4a in a cell-non-autonomous fashion (Fig. 5B).
Figure 5.

Extrinsic Versus Intrinsic Activation of p16INK4a. A, Injection of Lkb1 null endometrial cancer cells into a syngenic mouse heterozygous for the p16LUC reporter causes stromal luciferase expression upon tumor growth. Injection of Matrigel vehicle on the opposite flank does not alter p16INK4a expression. See also: (130) B, Intrinsic signals induces p16INK4a expression in damaged, senescent or transformed cells. Alterations in surrounding the cellular milieu can trigger the induction of p16INK4a in nearby undamaged cells through an unknown pathway.
Mounting evidence supports a role for stromal p16INK4a expression in tumor initiation and progression. Fibroblasts ectopically expressing p16INK4a have altered cellular metabolism and overproduce high-energy mitochondrial fuels such as L-lactate (131). In xenograft studies, co-injection of these fibroblasts with MDA-MB-231 breast cancer cells increased tumor size 2 fold, suggesting that altered stromal metabolism caused by p16INK4a expression promotes tumor growth (131). In support of this observation, elevated p16INK4a levels in the stroma of human mammary ductal carcinoma in situ (DCIS) lesions are predictive of disease recurrence independent of other histopathological markers such as ER positivity (132). It remains to be determined whether the promotion of tumorigenesis by p16INK4a-positive stromal cells is solely a reflection of altered local metabolism. Studies from the p16LUC mouse model suggest that p16INK4a induction in tumor infiltrating immune cells may also promote cancer progression by dampening the anti-tumor response. Regardless of the mechanism, these data make it clear that p16INK4a expression in the tumor stroma should not be ignored.
Therapeutic Mimicry of p16INK4a
Pharmaceutical efforts to mimic the function of p16INK4a arose after the anti-tumor effects of flavopiridol were linked to CDK inhibition (Reviewed in: (133)). Flavopiridol (alvocidib) was originally touted as an inhibitor of epidermal growth factor receptor (EGFR), but later shown to inhibit the growth of a wide range of cancer cells at an IC50 much lower than that required to block EGFR activation. Broad inhibition of cyclin dependent kinases (CDKs), including CDK4 (IC50 <120nM), was later identified as the mechanism of flavopiridol action (134). After several promising phase I trials, flavopiridol failed in phase II, showing significant activity only in cases of relapsed chronic lymphocytic leukemia (135). Based upon these disappointing results, Sanofi-Aventis halted production of flavopiridol in 2010. For other non-selective CDK inhibitors, multiple toxicities and poor solubility prevented successful transition to the clinic. However, recent efforts to generate potent and selective CDK inhibitors appear more fruitful.
Of particular interest to the pharmaceutical industry, selective CDK4/6 inhibitors have shown promise in early clinical trials (Reviewed in: (136)). Knowledge of p16INK4a regulation and function has bolstered these efforts by providing biomarkers of therapeutic efficacy and identifying patient populations wherein response is likely. Often clinical trials of CDK4/6 inhibitors now exclude RB null tumors, which are naturally resistant to the effects of ectopic p16INK4a expression. In addition, the observation that p16INK4a is markedly upregulated in response to RB-inactivating viral oncoproteins (e.g. E7, T-Antigen) has prompted the exclusion of tumors expressing high levels of p16INK4a. Finally, studies of CDK knockout mice are assisting these efforts, identifying cell and tumor types which are more reliant upon the activity of CDK4/6 to drive proliferation than that of CDK2. Employing this knowledge, CDK4/6 inhibitors are showing efficacy, and a number of drugs have now entered phase II and III clinical trials (Phase 2- LEE011(Novartis) and Ly2835219(Lilly); Phase 3- MK-7965/dinaciclib (Merck) and PD-0332991/palbociclib (Pfizer)) (137).
p16INK4a selectively binds CDK4 and 6 in vitro (2), however achieving such therapeutic specificity is more challenging. In fact, Merck attributes the success of dinaciclib in chronic lymphocytic leukemia to inhibition of CDK9, not CDK4/6 (137). Moreover, the observation that p16INK4a may contribute to CDK4/6-independent cell cycle regulation (i.e. through CDK inhibitor reshuffling) suggests that pharmaceutical CDK4/6 inhibitors may not fully recapitulate the potency of p16INK4a. While adverse effects associated with CDK4/6-selective inhibitors are mild (i.e. limited bone marrow suppression) (136), concerns about the long-term efficacy of such therapeutics is rising. In particular, mechanisms to bypass the requirement for CDK4/6 are already known including: RB loss and increased CDK2 activity. How quickly tumors will exploit these pathways to subvert therapeutic treatment is unknown. Initial trials of PD-0332991 in mantle cell lymphomas speak to this concern. While 89% of study participants showed reduced phospho-RB staining at 3 weeks, only 18% of tumors responded to therapy, suggesting that resistance was rapidly acquired (138). In addition to concerns about resistance, the role of p16INK4a expression in the tumor stroma remains undefined and inhibition of the local immune response or altered cellular metabolism may function to promote growth and metastasis. In fact, fibroblasts exposed to PD-0332991 adapt a tumor promoting, metabolic phenotype similar to that of fibroblasts overexpressing p16INK4a (131). Therefore, therapeutic mimetics could have similar, detrimental consequences on the tumor microenvironment. A final concern is the long-term effects of CDK4/6 inhibition on cellular senescence. Prolonged expression of p16INK4a promotes senescence and decreases regenerative capacity (See below). In a similar manner, CKD4/6 inhibitors may influence tissue aging. However, under normal physiological conditions, stem cell populations are by and large quiescent and thus unlikely altered by CDK4/6 inhibitors. It may be that stress and/or mitogenic signaling, which often accompany p16INK4a induction in vitro, is required to elicit senescence, and therefore the effects of CDK4/6 inhibition on aging will be minimal. As more than half of all cancers are diagnosed in people 65 and older, determining whether CDK4/6 therapeutics exacerbate age-related disease will be of significant interest.
The Role of p16INK4a in Senescence and Aging
p16INK4a Marks Biological Senescence
As noted first by Sherr and colleagues (139), p16INK4a levels increase with aging. In fact, detailed quantification has demonstrated a direct increase in p16INK4a expression with chronological age in all mammalian species tested to date (140). Such increases in p16INK4a are exponential, rising ~16 fold during the average human lifespan and making p16INK4a one of the most robust aging biomarkers characterized to date (141). The induction of senescence and p16INK4a expression is traditionally associated with a wide variety of intrinsic cellular stressors including: DNA damage, telomere erosion, reactive oxygen species, and stalled replication forks (Reviewed in: (142)). However, use of the p16LUC reporter mouse has provided evidence that undefined, extrinsic signals can also trigger p16INK4a transcription in a cell non-autonomous fashion (130). Using this same reporter to compare the dynamics of p16INK4a expression in mice with that observed in humans showed a direct correlation between the rate of p16INK4a accumulation and lifespan (130). Furthermore, data from progeroid and calorically restricted rodents suggest that p16INK4a serves as a marker of biological rather than chronological aging (70, 143, 144). Supporting this observation in humans, smoking and chemotherapy are associated with elevated p16INK4a expression in the human population (141, 145). Moreover, skin biopsies from long-lived nonagenarian cohorts have fewer p16INK4a positive cells than their age-matched partners (146). Clinically, use of p16INK4a as a marker of biological aging could provide quantitative measures of patient fitness including immune function and chemotherapeutic tolerance (145, 147). Assessment of p16INK4a levels prior to organ transplantation may also aid in the identification of biologically ‘younger’ donor tissues with increased potential for success (148–150).
A Causal Role for p16INK4a in Aging
Several lines of evidence suggest that p16INK4a is not only a biomarker of aging, but also causes aging in many cell types. Using p16INK4a transgenic and knockout mice, the cell-autonomous role of p16INK4a in aging has been investigated. In murine hematopoietic stem cells, T-cells, pancreatic β-cells and neural progenitors of the subventricular zone, age-related p16INK4a expression causes a decline in regenerative capacity (151–154). A caveat to these initial studies was the use of germline knockout mice; however strategies employing conditional p16INK4a loss or siRNAs have since confirmed these findings in T-cells and pancreatic β-cells (154, 155). Supporting the idea that tissues expressing less p16INK4a are more biologically fit, kidney transplant success is higher in p16INK4a-low donor organs (148–150). These data put forth the hypothesis that p16INK4a expression drives cellular senescence, resulting in decreased regenerative potential. As a proof of principal, work from the Van Deursen lab showed that deletion of p16INK4a-expressing cells from a BubR1 deficient, progeroid mouse model reduced many aging phenotypes (e.g. sarcopenia, cataracts, loss of adiposity) (144). Further work by this group showed that both muscle and adipocyte progenitors from these mice express high levels of p16INK4a, suggesting that even the deletion of senescent stem cells can promote improved regenerative capacity (156). Unfortunately, these animals did not live longer, owing to the development of cardiac pathologies, and a similar experiment has yet to be reported in wild type mice (144). One explanation for why these animals were not long-lived is that the gerontogenic effects of p16INK4a expression are tissue-specific. Indeed, p16INK4a levels do not seem to influence the regenerative capacity of murine melanocyte stem cells or neuronal progenitors of the dentate gyrus (unpublished observation and (153, 157)). Other mechanisms of curbing age-related p16INK4a induction have been reported in mice. For example, activation of platelet-derived growth factor receptor (PDGFR) signaling in elderly murine pancreatic β-cells increased the regenerative capacity of these cells via repression of p16INK4a expression (158). Likewise, administration of fibroblast growth factor 7 (FGF7) reduced p16INK4a levels and increased the number of early T-cell progenitors in 15–18 month-old mice, thereby partially rescuing the known decline in thymopoiesis with age (159). Apart from these studies, analyses of p16INK4a-mediated regenerative decline are limited to a small number of tissues; therefore the potential outcome of p16INK4a-directed therapies remains uncertain. Clearly, further characterization of the relationship between p16INK4a expression, senescence and regenerative capacity in vivo would have broad implications for the therapeutic treatment of age-related disease.
The Role of p16INK4a in Intrinsic vs. Extrinsic Aging
Senescence is typically viewed as a response to detrimental, intrinsic cellular events; however, recent evidence suggests that extrinsic signals also contribute to tissue aging. Above, we discussed the potential for neoplastic transformation to initiate cell non-autonomous p16INK4a expression. Although the extracellular mediators of stromal p16INK4a induction are undefined, several signaling pathways have been linked to the expression of p16INK4a in aging tissues. In muscle satellite cells, age-associated increases in local TGF-β production activate SMAD3, which in turn binds to the p16INK4a promoter to initiate gene transcription (94). Notch signaling antagonizes TGF-β, and in doing so can alleviate age-related declines in satellite cell function (94). Similar to TGF-β signaling, changes in thymic and bone marrow structure are reported to promote aging in local progenitor cell populations (142). Together, these findings put forth the model of “niche aging” wherein stromal changes influence the regenerative capacity of local progenitors. However, parsing the role of the senescent cell versus the niche in aging biology becomes somewhat of a chicken and egg question. After all, senescent cells themselves secrete a large number of pro-inflammatory cytokines associated with age-related disease (Reviewed in: (160)). Future studies aimed at identifying cell non-autonomous p16INK4a activation signals are clearly needed to better understand the induction of senescence during physiological aging.
p16INK4a as a Barrier to Pluripotency
Work with induced pluripotent stem cells (iPS) also implicates p16INK4a as a modulator of regenerative capacity. During iPS generation, senescence induced by the four factor cocktail of OCT4, SOX2, KLF4 and c-MYC serves as a barrier to efficient reprogramming (161). In cells where reprogramming is effective, silencing of INK4/ARF gene transcription is observed concomitant with the induction of molecular markers indicative of stem cell phenotypes (61). Therefore, it is not surprising that iPS production efficiency is increased by cellular immortalization (162), or shRNA knockdown of INK4/ARF genes (61, 161). Currently, efficient generation of iPS from older patients represents a major hurdle for regenerative medicine. Therefore, novel reprogramming approaches aimed at curbing the senescent phenotype, may improve iPS technology for the future (163).
Genome-Wide Association Studies (GWAS) Linking p16INK4a to Age-Related Disease
Meta-analysis of genome-wide association studies (GWAS) suggests that we have only begun to scratch the surface in understanding the role of p16INK4a in age-related disease. Single nucleotide polymorphisms (SNPs) in chromosome 9p21.3 have been linked to cancer, atherosclerosis, diabetes, frailty, cataracts and late onset Alzheimer’s disease (Fig. 1B and (164)). In many cases, expression of p16INK4a correlates directly with SNP genotype, suggesting a causal role for p16INK4a in diverse, age-associated diseases (164). Several mechanistic models have been suggested to explain how SNPs, some of which are more than 100 kb from the gene promoter, influence p16INK4a expression. One model proposes that the activity of distal enhancer elements is modified by SNP genotype (165). Another provides evidence that ANRIL expression and splicing is directly influenced by 9p21 SNPs (166). We believe that these models are not mutually exclusive.
While mechanisms by which 9p21.3 SNPs influence p16NK4a transcription have been proposed, the link between p16INK4a expression and age-related diseases is not always apparent. For example, atherosclerosis is frequently associated with lipid metabolism, yet 9p21.3 SNPs have emerged as robust indicators of atherosclerotic risk in some of the most widely replicated GWASs conducted to date (167). Evidence from animal models suggests that 9p21.3 SNPs may reduce INK4/ARF gene transcription, leading to altered cellular proliferation and apoptosis which exacerbate disease progression (168–170). However, transgenic mice carrying multiple copies of the INK4/ARF locus are equally susceptible to atherosclerosis (171). Therefore, the mechanisms by which 9p21.3 SNPs influence a large number of age-related human disease remain a subject of ongoing investigation.
Conclusions
Since the discovery of p16INK4a more than 20 years ago, numerous advances have led to an increasingly complex view of p16INK4a regulation and function. The role of p16INK4a clearly extends beyond cancer and aging. Dynamic induction of p16INK4a is observed during mammary involution, wound healing, nerve regeneration and infection (unpublished data and (130, 172–174)). It has been proposed that the induction of p16INK4a during these highly proliferative events is critical to the maintenance of proper tissue homeostasis. Whether the same signals that trigger p16INK4a expression under physiological conditions play a role in tumorigenesis or aging is still unclear. However, p16INK4a expression is only transient during processes like mammary involution and wound healing (130, 172, 173). Are p16INK4a positive cells cleared by the immune system or can they revert to a phenotype conducive to proliferation? Understanding the role of p16INK4a in normal physiology will be critical to the development of ‘senolytic’ therapies, which aim to lengthen our healthspan by eliminating senescent cells in the body.
p16INK4a is different than the other INK4/ARF family members. The dynamics of p16INK4a expression during senescence make it a robust biomarker of mammalian aging. Human tumors silence p16INK4a with greater frequency than ARF or p15INK4b (140), suggesting that the tumor suppressor function of p16INK4a is somehow more critical than the other INK4/ARF family members. As such, the therapeutic restoration of p16INK4a activity appears to be a promising avenue for anti-neoplastic development. Ironically, while drug development teams in the field of oncology work fervidly to move CKD4/6 inhibitors into the clinic; aging biologists aim to block the accumulation of p16INK4a-positive cells. Oddly, the key to longevity likely lies in the hands of both groups, as a careful balance of p16INK4a expression is required to stave off cancer and prevent aging.
Acknowledgments
The authors thank M. Waqas, J. Gillahan, S.T. Nguyen and Drs. D. Beach (Barts, UK), C. J. Burd (OSU) and N. Sharpless (UNC) for critical reading of the manuscript. Dr. K. Hoadley (UNC) provided advice and guidance regarding the analysis of TCGA data. This work was supported by NIH R00AG036817 (C.E.B) and American Cancer Society IRG-67-003-50 (C.E.B.).
Footnotes
Conflict of Interest Disclosure: The authors declare no conflicts of interest.
References
- 1.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes & development. 1993;7:1572–83. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 2.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 3.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–61. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 4.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 5.Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–23. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–34. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 7.Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell death and differentiation. 2013;20:576–88. doi: 10.1038/cdd.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer research. 2007;67:3963–9. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 9.Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–6. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Molecular cell. 2010;38:662–74. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes & development. 1995;9:1831–45. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 12.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, et al. New functional activities for the p21 family of CDK inhibitors. Genes & development. 1997;11:847–62. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 13.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. The EMBO journal. 1999;18:1571–83. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang H, Chou HS, Zhu L. Requirement of cyclin E-Cdk2 inhibition in p16(INK4a)-mediated growth suppression. Molecular and cellular biology. 1998;18:5284–90. doi: 10.1128/mcb.18.9.5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 16.Macias E, Miliani de Marval PL, De Siervi A, Conti CJ, Senderowicz AM, Rodriguez-Puebla ML. CDK2 activation in mouse epidermis induces keratinocyte proliferation but does not affect skin tumor development. The American journal of pathology. 2008;173:526–35. doi: 10.2353/ajpath.2008.071124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 18.Hu MG, Deshpande A, Schlichting N, Hinds EA, Mao C, Dose M, et al. CDK6 kinase activity is required for thymocyte development. Blood. 2011;117:6120–31. doi: 10.1182/blood-2010-08-300517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landis MW, Brown NE, Baker GL, Shifrin A, Das M, Geng Y, et al. The LxCxE pRb interaction domain of cyclin D1 is dispensable for murine development. Cancer research. 2007;67:7613–20. doi: 10.1158/0008-5472.CAN-07-1207. [DOI] [PubMed] [Google Scholar]
- 20.Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:194–9. doi: 10.1073/pnas.011522998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong W, Pollard JW. Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Molecular and cellular biology. 2001;21:1319–28. doi: 10.1128/MCB.21.4.1319-1328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sotillo R, Dubus P, Martin J, de la Cueva E, Ortega S, Malumbres M, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. The EMBO journal. 2001;20:6637–47. doi: 10.1093/emboj/20.23.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zindy F, van Deursen J, Grosveld G, Sherr CJ, Roussel MF. INK4d-deficient mice are fertile despite testicular atrophy. Molecular and cellular biology. 2000;20:372–8. doi: 10.1128/mcb.20.1.372-378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. The EMBO journal. 2000;19:3496–506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 26.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes & development. 1998;12:2899–911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Khalaf HH, Aboussekhra A. p16(INK4A) Positively Regulates p21(WAF1) Expression by suppressing AUF1-dependent mRNA decay. PloS one. 2013;8:e70133. doi: 10.1371/journal.pone.0070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Khalaf HH, Colak D, Al-Saif M, Al-Bakheet A, Hendrayani SF, Al-Yousef N, et al. p16( INK4a) positively regulates cyclin D1 and E2F1 through negative control of AUF1. PloS one. 2011;6:e21111. doi: 10.1371/journal.pone.0021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Molecular cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 31.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes & development. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maertens GN, El Messaoudi-Aubert S, Racek T, Stock JK, Nicholls J, Rodriguez-Niedenfuhr M, et al. Several distinct polycomb complexes regulate and co-localize on the INK4a tumor suppressor locus. PloS one. 2009;4:e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner SA, Bracken AP. A “complex” issue: deciphering the role of variant PRC1 in ESCs. Cell stem cell. 2013;12:145–6. doi: 10.1016/j.stem.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 34.Maertens GN, El Messaoudi-Aubert S, Elderkin S, Hiom K, Peters G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. The EMBO journal. 2010;29:2553–65. doi: 10.1038/emboj.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes & development. 1994;8:757–69. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 36.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 37.Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. The Journal of experimental medicine. 2006;203:2247–53. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–7. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Molecular and cellular biology. 2001;21:4330–6. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. The EMBO journal. 2004;23:4061–71. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes & development. 2009;23:975–85. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereira JD, Sansom SN, Smith J, Dobenecker MW, Tarakhovsky A, Livesey FJ. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15957–62. doi: 10.1073/pnas.1002530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes & development. 2011;25:485–98. doi: 10.1101/gad.2019811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatton-Brown K, Rahman N. The NSD1 and EZH2 overgrowth genes, similarities and differences. American journal of medical genetics Part C, Seminars in medical genetics. 2013;163:86–91. doi: 10.1002/ajmg.c.31359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20980–5. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics. 2010;42:665–7. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 47.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer cell. 2010;18:316–28. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7922–7. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature reviews Molecular cell biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 50.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–62. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El Messaoudi-Aubert S, Nicholls J, Maertens GN, Brookes S, Bernstein E, Peters G. Role for the MOV10 RNA helicase in polycomb-mediated repression of the INK4a tumor suppressor. Nature structural & molecular biology. 2010;17:862–8. doi: 10.1038/nsmb.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin N, Popov N, Aguilo F, O’Loghlen A, Raguz S, Snijders AP, et al. Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK(4)a regulation. The EMBO journal. 2013;32:982–95. doi: 10.1038/emboj.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2641–6. doi: 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes & development. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Negishi M, Saraya A, Mochizuki S, Helin K, Koseki H, Iwama A. A novel zinc finger protein Zfp277 mediates transcriptional repression of the Ink4a/arf locus through polycomb repressive complex 1. PloS one. 2010;5:e12373. doi: 10.1371/journal.pone.0012373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. The EMBO journal. 2003;22:5323–35. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23:5759–69. doi: 10.1038/sj.onc.1207706. [DOI] [PubMed] [Google Scholar]
- 58.Agger K, Cloos PA, Rudkjaer L, Williams K, Andersen G, Christensen J, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes & development. 2009;23:1171–6. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenfuhr M, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes & development. 2009;23:1177–82. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakade K, Pan J, Yamasaki T, Murata T, Wasylyk B, Yokoyama KK. JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. The Journal of biological chemistry. 2009;284:10808–17. doi: 10.1074/jbc.M808333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Collado M, Villasante A, Strati K, Ortega S, Canamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–9. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao W, Li Q, Ayers S, Gu Y, Shi Z, Zhu Q, et al. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell. 2013;152:1037–50. doi: 10.1016/j.cell.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LM, Mohd-Sarip A, et al. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. The Journal of biological chemistry. 2004;279:3807–16. doi: 10.1074/jbc.M309333200. [DOI] [PubMed] [Google Scholar]
- 64.Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Molecular and cellular biology. 2008;28:3457–64. doi: 10.1128/MCB.02019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Molecular cell. 2009;34:271–84. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirosue A, Ishihara K, Tokunaga K, Watanabe T, Saitoh N, Nakamoto M, et al. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging cell. 2012;11:553–6. doi: 10.1111/j.1474-9726.2012.00809.x. [DOI] [PubMed] [Google Scholar]
- 67.Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, et al. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–70. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 68.Zheng W, Wang H, Xue L, Zhang Z, Tong T. Regulation of cellular senescence and p16(INK4a) expression by Id1 and E47 proteins in human diploid fibroblast. The Journal of biological chemistry. 2004;279:31524–32. doi: 10.1074/jbc.M400365200. [DOI] [PubMed] [Google Scholar]
- 69.Alani RM, Young AZ, Shifflett CB. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7812–6. doi: 10.1073/pnas.141235398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. The Journal of clinical investigation. 2004;114:1299–307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 72.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 73.Cakouros D, Isenmann S, Cooper L, Zannettino A, Anderson P, Glackin C, et al. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Molecular and cellular biology. 2012;32:1433–41. doi: 10.1128/MCB.06315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. The EMBO journal. 2000;19:2969–79. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ke H, Harris R, Coloff JL, Jin JY, Leshin B, Miliani de Marval P, et al. The c-Jun NH2-terminal kinase 2 plays a dominant role in human epidermal neoplasia. Cancer research. 2010;70:3080–8. doi: 10.1158/0008-5472.CAN-09-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee TK, Man K, Ling MT, Wang XH, Wong YC, Lo CM, et al. Over-expression of Id-1 induces cell proliferation in hepatocellular carcinoma through inactivation of p16INK4a/RB pathway. Carcinogenesis. 2003;24:1729–36. doi: 10.1093/carcin/bgg145. [DOI] [PubMed] [Google Scholar]
- 77.Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, et al. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Developmental cell. 2003;4:879–89. doi: 10.1016/s1534-5807(03)00161-8. [DOI] [PubMed] [Google Scholar]
- 78.Polsky D, Young AZ, Busam KJ, Alani RM. The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer research. 2001;61:6008–11. [PubMed] [Google Scholar]
- 79.Wu X, Gao H, Ke W, Hager M, Xiao S, Freeman MR, et al. VentX trans-activates p53 and p16ink4a to regulate cellular senescence. The Journal of biological chemistry. 2011;286:12693–701. doi: 10.1074/jbc.M110.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Irelan JT, Gutierrez Del Arroyo A, Gutierrez A, Peters G, Quon KC, Miraglia L, et al. A functional screen for regulators of CKDN2A reveals MEOX2 as a transcriptional activator of INK4a. PloS one. 2009;4:e5067. doi: 10.1371/journal.pone.0005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Douville JM, Cheung DY, Herbert KL, Moffatt T, Wigle JT. Mechanisms of MEOX1 and MEOX2 regulation of the cyclin dependent kinase inhibitors p21 and p16 in vascular endothelial cells. PloS one. 2011;6:e29099. doi: 10.1371/journal.pone.0029099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 83.Wang W, Pan K, Chen Y, Huang C, Zhang X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic acids research. 2012;40:981–95. doi: 10.1093/nar/gkr818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L, et al. P300 plays a role in p16(INK4a) expression and cell cycle arrest. Oncogene. 2008;27:1894–904. doi: 10.1038/sj.onc.1210821. [DOI] [PubMed] [Google Scholar]
- 85.Huang Y, Wu J, Li R, Wang P, Han L, Zhang Z, et al. B-MYB delays cell aging by repressing p16 (INK4alpha) transcription. Cellular and molecular life sciences : CMLS. 2011;68:893–901. doi: 10.1007/s00018-010-0501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson LR, Johnson TK, Desler M, Luster TA, Nowling T, Lewis RE, et al. Effects of B-Myb on gene transcription: phosphorylation-dependent activity ans acetylation by p300. The Journal of biological chemistry. 2002;277:4088–97. doi: 10.1074/jbc.M105112200. [DOI] [PubMed] [Google Scholar]
- 87.Feng Y, Wang X, Xu L, Pan H, Zhu S, Liang Q, et al. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. The FEBS journal. 2009;276:4197–206. doi: 10.1111/j.1742-4658.2009.07128.x. [DOI] [PubMed] [Google Scholar]
- 88.Kim JK, Noh JH, Eun JW, Jung KH, Bae HJ, Shen Q, et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Molecular cancer research : MCR. 2013;11:62–73. doi: 10.1158/1541-7786.MCR-12-0332. [DOI] [PubMed] [Google Scholar]
- 89.Zhou R, Han L, Li G, Tong T. Senescence delay and repression of p16INK4a by Lsh via recruitment of histone deacetylases in human diploid fibroblasts. Nucleic acids research. 2009;37:5183–96. doi: 10.1093/nar/gkp533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews Genetics. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gan Q, Huang J, Zhou R, Niu J, Zhu X, Wang J, et al. PPAR{gamma} accelerates cellular senescence by inducing p16INK4{alpha} expression in human diploid fibroblasts. Journal of cell science. 2008;121:2235–45. doi: 10.1242/jcs.026633. [DOI] [PubMed] [Google Scholar]
- 92.Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. The Journal of clinical investigation. 2005;115:3228–38. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krieglstein K, Miyazono K, ten Dijke P, Unsicker K. TGF-beta in aging and disease. Cell and tissue research. 2012;347:5–9. doi: 10.1007/s00441-011-1278-3. [DOI] [PubMed] [Google Scholar]
- 94.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–32. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carlson ME, Silva HS, Conboy IM. Aging of signal transduction pathways, and pathology. Experimental cell research. 2008;314:1951–61. doi: 10.1016/j.yexcr.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes & development. 2007;21:2923–35. doi: 10.1101/gad.450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Induction of p16INK4A mediated by beta-catenin in a TCF4-independent manner: implications for alterations in p16INK4A and pRb expression during trans-differentiation of endometrial carcinoma cells. International journal of cancer Journal international du cancer. 2006;119:2294–303. doi: 10.1002/ijc.22112. [DOI] [PubMed] [Google Scholar]
- 98.Wassermann S, Scheel SK, Hiendlmeyer E, Palmqvist R, Horst D, Hlubek F, et al. p16INK4a is a beta-catenin target gene and indicates low survival in human colorectal tumors. Gastroenterology. 2009;136:196–205. e2. doi: 10.1053/j.gastro.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 99.Zhang DY, Pan Y, Zhang C, Yan BX, Yu SS, Wu DL, et al. Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through promoting the ROS production. Molecular and cellular biochemistry. 2013;374:13–20. doi: 10.1007/s11010-012-1498-1. [DOI] [PubMed] [Google Scholar]
- 100.Bishop CL, Bergin AM, Fessart D, Borgdorff V, Hatzimasoura E, Garbe JC, et al. Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A) Molecular cell. 2010;40:533–47. doi: 10.1016/j.molcel.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 101.Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–87. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R, Jr, Srikantan S, et al. p16(INK4a) translation suppressed by miR-24. PloS one. 2008;3:e1864. doi: 10.1371/journal.pone.0001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Giglio S, Cirombella R, Amodeo R, Portaro L, Lavra L, Vecchione A. MicroRNA miR-24 promotes cell proliferation by targeting the CDKs inhibitors p27 and p16. Journal of cellular physiology. 2013 doi: 10.1002/jcp.24368. [DOI] [PubMed] [Google Scholar]
- 104.Malhas A, Saunders NJ, Vaux DJ. The nuclear envelope can control gene expression and cell cycle progression via miRNA regulation. Cell cycle. 2010;9:531–9. doi: 10.4161/cc.9.3.10511. [DOI] [PubMed] [Google Scholar]
- 105.Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O’Day E, et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Molecular cell. 2009;35:610–25. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.To KH, Pajovic S, Gallie BL, Theriault BL. Regulation of p14ARF expression by miR-24: a potential mechanism compromising the p53 response during retinoblastoma development. BMC cancer. 2012;12:69. doi: 10.1186/1471-2407-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–46. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–39. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell cycle. 2009;8:843–52. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang N, Yi J, Guo G, Liu X, Shang Y, Tong T, et al. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Molecular and cellular biology. 2010;30:3875–86. doi: 10.1128/MCB.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang X, Liu Z, Yi J, Tang H, Xing J, Yu M, et al. The tRNA methyltransferase NSun2 stabilizes p16INK(4) mRNA by methylating the 3′-untranslated region of p16. Nature communications. 2012;3:712. doi: 10.1038/ncomms1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Otterson GA, Kratzke RA, Coxon A, Kim YW, Kaye FJ. Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wildtype RB. Oncogene. 1994;9:3375–8. [PubMed] [Google Scholar]
- 120.Meyerson M, Franklin WA, Kelley MJ. Molecular classification and molecular genetics of human lung cancers. Seminars in oncology. 2004;31:4–19. doi: 10.1053/j.seminoncol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 121.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer research. 1997;57:3126–30. [PubMed] [Google Scholar]
- 122.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:2969–72. [PubMed] [Google Scholar]
- 123.Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell cycle. 2011;10:2497–503. doi: 10.4161/cc.10.15.16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuan J, Knorr J, Altmannsberger M, Goeckenjan G, Ahr A, Scharl A, et al. Expression of p16 and lack of pRB in primary small cell lung cancer. The Journal of pathology. 1999;189:358–62. doi: 10.1002/(SICI)1096-9896(199911)189:3<358::AID-PATH452>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 126.Subhawong AP, Subhawong T, Nassar H, Kouprina N, Begum S, Vang R, et al. Most basal-like breast carcinomas demonstrate the same Rb−/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. The American journal of surgical pathology. 2009;33:163–75. doi: 10.1097/PAS.0b013e31817f9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chakravarti A, DeSilvio M, Zhang M, Grignon D, Rosenthal S, Asbell SO, et al. Prognostic value of p16 in locally advanced prostate cancer: a study based on Radiation Therapy Oncology Group Protocol 9202. J Clin Oncol. 2007;25:3082–9. doi: 10.1200/JCO.2006.08.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee CT, Capodieci P, Osman I, Fazzari M, Ferrara J, Scher HI, et al. Overexpression of the cyclin-dependent kinase inhibitor p16 is associated with tumor recurrence in human prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:977–83. [PubMed] [Google Scholar]
- 129.Rischin D, Young RJ, Fisher R, Fox SB, Le QT, Peters LJ, et al. Prognostic significance of p16INK4A and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol. 2010;28:4142–8. doi: 10.1200/JCO.2010.29.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–51. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Witkiewicz AK, Rivadeneira DB, Ertel A, Kline J, Hyslop T, Schwartz GF, et al. Association of RB/p16-pathway perturbations with DCIS recurrence: dependence on tumor versus tissue microenvironment. The American journal of pathology. 2011;179:1171–8. doi: 10.1016/j.ajpath.2011.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. Journal of cancer research and clinical oncology. 2011;137:1409–18. doi: 10.1007/s00432-011-1039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer research. 1996;56:2973–8. [PubMed] [Google Scholar]
- 135.Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, Andritsos LA, et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J Clin Oncol. 2009;27:6012–8. doi: 10.1200/JCO.2009.22.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guha M. Cyclin-dependent kinase inhibitors move into Phase III. Nature reviews Drug discovery. 2012;11:892–4. doi: 10.1038/nrd3908. [DOI] [PubMed] [Google Scholar]
- 137.Guha M. Blockbuster dreams for Pfizer’s CDK inhibitor. Nat Biotech. 2013;31:187. doi: 10.1038/nbt0313-187a. [DOI] [PubMed] [Google Scholar]
- 138.Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597–607. doi: 10.1182/blood-2011-10-388298. [DOI] [PubMed] [Google Scholar]
- 139.Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–11. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- 140.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 141.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging cell. 2009;8:439–48. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu Y, Sharpless NE. Tumor suppressor mechanisms in immune aging. Current opinion in immunology. 2009;21:431–9. doi: 10.1016/j.coi.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, Prolla TA. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC genomics. 2007;8:80. doi: 10.1186/1471-2164-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Muss HBJK, Alston SM, Lacy AC, Jolly TA, Williams G, Carey LA, Dees EC, Anders CK, Irvin WJ, Sharpless NE. p16INK4a expression after chemotherapy in older women with early-stage breast cancer. J Clin Oncol. 2011:29. [Google Scholar]
- 146.Waaijer ME, Parish WE, Strongitharm BH, van Heemst D, Slagboom PE, de Craen AJ, et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging cell. 2012;11:722–5. doi: 10.1111/j.1474-9726.2012.00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nelson JA, Krishnamurthy J, Menezes P, Liu Y, Hudgens MG, Sharpless NE, et al. Expression of p16(INK4a) as a biomarker of T-cell aging in HIV-infected patients prior to and during antiretroviral therapy. Aging cell. 2012;11:916–8. doi: 10.1111/j.1474-9726.2012.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Melk A, Schmidt BM, Braun H, Vongwiwatana A, Urmson J, Zhu LF, et al. Effects of donor age and cell senescence on kidney allograft survival. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2009;9:114–23. doi: 10.1111/j.1600-6143.2008.02500.x. [DOI] [PubMed] [Google Scholar]
- 149.McGlynn LM, Stevenson K, Lamb K, Zino S, Brown M, Prina A, et al. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging cell. 2009;8:45–51. doi: 10.1111/j.1474-9726.2008.00447.x. [DOI] [PubMed] [Google Scholar]
- 150.Koppelstaetter C, Schratzberger G, Perco P, Hofer J, Mark W, Ollinger R, et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging cell. 2008;7:491–7. doi: 10.1111/j.1474-9726.2008.00398.x. [DOI] [PubMed] [Google Scholar]
- 151.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–6. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 152.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–7. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 153.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–52. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, et al. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117:3257–67. doi: 10.1182/blood-2010-09-304402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes & development. 2009;23:906–11. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell reports. 2013;3:1164–74. doi: 10.1016/j.celrep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Inomata K, Aoto T, Binh NT, Okamoto N, Tanimura S, Wakayama T, et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137:1088–99. doi: 10.1016/j.cell.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 158.Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, et al. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–55. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Berent-Maoz B, Montecino-Rodriguez E, Signer RA, Dorshkind K. Fibroblast growth factor-7 partially reverses murine thymocyte progenitor aging by repression of Ink4a. Blood. 2012;119:5715–21. doi: 10.1182/blood-2011-12-400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of clinical investigation. 2013;123:966–72. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes & development. 2009;23:2134–9. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–8. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Ait-Hamou N, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes & development. 2011;25:2248–53. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging cell. 2012;11:727–31. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1671–7. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- 166.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS genetics. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cunnington MS, Keavney B. Genetic mechanisms mediating atherosclerosis susceptibility at the chromosome 9p21 locus. Current atherosclerosis reports. 2011;13:193–201. doi: 10.1007/s11883-011-0178-z. [DOI] [PubMed] [Google Scholar]
- 168.Kuo CL, Murphy AJ, Sayers S, Li R, Yvan-Charvet L, Davis JZ, et al. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2483–92. doi: 10.1161/ATVBAHA.111.234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–12. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gonzalez-Navarro H, Abu Nabah YN, Vinue A, Andres-Manzano MJ, Collado M, Serrano M, et al. p19(ARF) deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. Journal of the American College of Cardiology. 2010;55:2258–68. doi: 10.1016/j.jacc.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 171.Fuster JJ, Molina-Sanchez P, Jovani D, Vinue A, Serrano M, Andres V. Increased gene dosage of the Ink4/Arf locus does not attenuate atherosclerosis development in hypercholesterolaemic mice. Atherosclerosis. 2012;221:98–105. doi: 10.1016/j.atherosclerosis.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 172.Natarajan E, Omobono JD, 2nd, Guo Z, Hopkinson S, Lazar AJ, Brenn T, et al. A keratinocyte hypermotility/growth-arrest response involving laminin 5 and p16INK4A activated in wound healing and senescence. The American journal of pathology. 2006;168:1821–37. doi: 10.2353/ajpath.2006.051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gadd M, Pisc C, Branda J, Ionescu-Tiba V, Nikolic Z, Yang C, et al. Regulation of cyclin D1 and p16(INK4A) is critical for growth arrest during mammary involution. Cancer research. 2001;61:8811–9. [PubMed] [Google Scholar]
- 174.Gomez-Sanchez JA, Gomis-Coloma C, Morenilla-Palao C, Peiro G, Serra E, Serrano M, et al. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain : a journal of neurology. 2013 doi: 10.1093/brain/awt130. [DOI] [PubMed] [Google Scholar]