

Abstract
The stressosome is a 1.8 MDa cytoplasmic complex that controls diverse bacterial signaling pathways. Its role is best understood in Bacillus subtilis, where it activates the σB transcription factor in response to a variety of sharp environmental challenges, including acid, ethanol, heat or salt stress. However, details of the signaling mechanism within the stressosome remain uncertain. The core of the complex comprises one or more members of the RsbR co-antagonist family together with the RsbS antagonist protein, which binds the RsbT kinase in the absence of stress. As part of the response, RsbT first phosphorylates the RsbRA co-antagonist on T171 and then RsbS on S59; this latter event correlates with the stress-induced release of RsbT to activate downstream signaling. Here we examine the in vivo consequence of S59 phosphorylation in a model strain whose stressosome core is formed solely with the RsbRA co-antagonist and RsbS. A phosphorylation-deficient S59A substitution in RsbS blocked response to mild stress but had declining impact as stress increased: with strong ethanol challenge response with S59A was 60% as robust as with wild type RsbS. Genetic analysis narrowed this S59-independent activation to the stressosome and established that significant signaling still occurred in a strain bearing both the T171A and S59A substitutions. We infer that S59 phosphorylation increases signaling efficiency but is not essential, and that a second (or underlying) mechanism of signal transduction prevails in its absence. This interpretation nullifies models in which stressosome signaling is solely mediated by control of RsbT kinase activity toward S59.
Introduction
Particular bacterial sensory modules often regulate activity of a variety of output domains to accomplish different signaling tasks. The 1.8 MDa stressosome complex appears widespread among the eubacteria, and its components are encoded in different genome contexts, implying it provides sensory function for diverse signaling pathways [1]–[3]. These components are best defined in Bacillus subtilis, where the stressosome controls activation of the general stress factor σB in response to rapidly increasing physical or chemical signals in the environment [4]–[6]. However, understanding of the mechanism by which the stressosome senses and conveys information remains limited. Here we use a genetic approach to assess the importance of a key phosphorylation event on a core stressosome protein, and as a result provide evidence for another, phosphorylation-independent route of information transfer within the complex.
Genetic and biochemical analysis indicates that the B. subtilis stressosome is formed from three different types of proteins: one or more members of the partially redundant RsbR co-antagonist family, the RsbS antagonist, and the RsbT serine-threonine kinase [7]–[11]. The four RsbR co-antagonists (RsbRA, RB, RC and RD) have different N-terminal, non-heme globin domains and more conserved C-terminal STAS (sulfate transporter/anti-sigma factor antagonist) domains, whereas the smaller RsbS antagonist comprises only a STAS domain [2], [12]. Twenty RsbR and ten RsbS dimers multimerize via these STAS domains to form the pseudo-icosahedral core of the stressosome, and in complexes formed in vitro the RsbT kinase appears to bind the surface of this structure at positions occupied by the RsbS antagonists [2]. The N-terminal domains of the RsbR dimers form outward projections from the stressosome surface and are thought to provide sensory input [2], but with the exception of YtvA – a distinct RsbR family member with an N-terminal, blue-light-sensing LOV domain [13]–[18] – there is no experimental evidence regarding how stress signals are sensed and conveyed to the core [19], [20].
More is known about downstream signaling events. Stressosome output is represented by release of the RsbT kinase, which then binds and activates the RsbU environmental phosphatase by direct protein-protein interaction [7], [21], [22]. As part of this release, the RsbT kinase phosphorylates both RsbRA and RsbS on conserved residues within their STAS domains, both in vitro and in vivo [22]–[25]. The signaling model shown in Fig. 1 is supported by biochemical analysis, the phenotypes of null alleles as well as phosphorylation-deficient and phosphomimetic substitutions, and analysis of the phosphorylation state of key residues during the stress response [5]. Much of this in vitro and in vivo analysis, including structural analysis by hybrid methods [2], has made use of a minimal functional stressosome consisting of only the RsbRA, RsbS and RsbT proteins. Strains encoding such minimal stressosomes more readily reveal the effects of genetic alterations in rsbRA [8], [19], [20]. They also serve as useful models for the numerous bacterial species that lack multiple RsbR paralogs and possess only a single RsbRA-like protein [1], [3]. We therefore used strains encoding minimal stressosomes for the majority of experiments reported here.
Figure 1. Stressosome and model of RsbRA-RsbS-RsbT activation.

(A) Cytoplasmic stressosome complex (RsbR-RsbS-RsbT) controls activation of the environmental phosphatase (RsbU) in response to diverse physical and chemical signals. STAS domains of RsbR and RsbS form the stressosome core (red); N-terminal domains of RsbR are hypothetical sensors (red crosshatch); dissociable RsbT kinase (green) is positive activator of RsbU (yellow). RsbU removes the phosphoryl group (orange) from S56 on the RsbV anti-anti-σ, ultimately activating the σB stress factor. The energy phosphatase (RsbP) and the phosphatase-independent cold stress pathways are shown in dotted outline. Arrowheads indicate activation of protein targets or enzymatic reactions; T-headed lines indicate inhibition. (B) Model of stressosome control of RsbU activity. Stressosome core comprises partially redundant RsbRA, RB, RC, and RD co-antagonists (represented here as RsbRA) and the RsbS antagonist, which binds the RsbT kinase. In unstressed cells RsbT phosphorylates RsbRA on T171, facilitating subsequent activation of RsbT kinase activity toward RsbS (+ arrow). During the stress response RsbT phosphorylates RsbS on S59; RsbT is released to bind and activate RsbU. The RsbX feedback phosphatase (dotted outline) dephosphorylates RsbS-P. (C) RsbS comprises a single STAS domain (red rectangle), whereas the larger RsbRA has an N-terminal non-heme globin domain (red crosshatching) and C-terminal STAS domain (red). Phosphorylated S59 and T171 residues lie in the STAS domains; T205 (light grey) is phosphorylated only under extreme stress (see text). RsbRB, RC and RD are similar to RsbRA, with corresponding phosphorylated residues (not shown); these three paralogs were removed in some strains. YtvA is an RsbR family member that increases stressosome output in response to blue light, sensed by its N-terminal LOV (Light-Oxygen-Voltage) domain (blue). Figure modified from ref [20].
According to the model shown in Fig. 1, T171 of RsbRA is phosphorylated even in unstressed cells; this event is thought to be an important prerequisite but does not by itself trigger the environmental stress response [8], [24]–[26]. Rather, T171 modification appears to facilitate subsequent phosphorylation of residue S59 on RsbS when stress is encountered. A negative charge at the S59 position decreases RsbS affinity for RsbT in pairwise binding experiments [3], [22] and results in RsbT release from the stressosome in vitro [7]. Correspondingly, either S59 phosphorylation or a phosphomimetic S59D substitution correlates with activation of the response in vivo [24], [27]. By contrast, T205 of RsbRA is thought to be phosphorylated only under extreme stress in order to inhibit signaling [25]. RsbX – a feedback phosphatase encoded in the neighborhood of the RsbRST cluster in B. subtilis and other bacteria – resets the system by removing the phosphate from S59 and T205 [1], [22], [25], [26].
Thus the RsbR co-antagonists and the RsbS antagonist have both negative and positive signaling roles, and these roles can be differentially affected by mutations within their structural genes. The known negative role of the RsbR co-antagonists is their required cooperation with RsbS to sequester RsbT in unstressed cells [7], [8]; null mutants missing all four RsbR paralogs or the single RsbS protein result in constitutive signaling in vivo [27], [28]. The presumed positive role of the RsbR co-antagonists is to stimulate RsbT kinase activity toward RsbS during the stress response [23], [26], which is thought to trigger the positive signaling function of RsbS; phosphorylation-deficient substitutions at RsbRA T171 or RsbS S59 cause diminished or nonexistent response in vivo [8], [27], depending on genetic context. In this regard, the S59A substitution in RsbS eliminates detectable phosphorylation in vitro [22], but its original phenotypic characterization suggested that S59A supports at least some signaling activity in an otherwise wild-type strain [27]. This implies that S59 phosphorylation is important but not essential for signaling. In accord with this result, more recent assays of the S59A substitution in model strains whose stressosomes were formed solely with the RsbRA or RsbRC co-antagonists found unexpectedly robust stress responses, indicating the existence of a signaling pathway independent from S59 phosphorylation [8], [19]. However, the nature of this S59-independent pathway has not been further explored, and it remains uncertain whether such signaling is indeed associated with the environmental branch of the regulatory network, or if it instead occurs via the distinct energy- or cold-signaling branches (Fig. 1).
Perhaps due to this uncertainty, prevailing models of stressosome signaling focus on how the phosphorylation state of S59 can be modulated by controlling RsbT kinase activity [2], [4], [24], [29], largely overlooking the observations that environmental stresses can activate σB in an S59A mutant. A further investigation of the influence of S59A on stress signaling therefore seemed warranted. We report here that the S59A substitution decreases signaling efficiency but does not prevent the stress response. Moreover, mutant analysis locates the S59-independent signaling mode to the environmental branch of the network, leading us to propose that it reflects the fundamental mechanism of signal transduction within the stressosome.
Results
Strains Deficient in S59 Phosphorylation Still Respond to Ethanol Stress
For our initial experiments investigating the effect of RsbS S59A on stress signaling, we used three strains whose stressosomes differed in their complement of RsbR family co-antagonists: one had the wild-type set of all four co-antagonists whereas the others had only one of the two principal co-antagonists, either RsbRA alone or RsbRB alone, together with the RsbS antagonist. Such strains activate σB equally well in response to ethanol stress, but their stressosomes exert different degrees of control over the unstressed output of the signaling network, with the wild type complex having the lowest basal output and thus the greatest control, RsbRB slightly less control, and RsbRA less still [8], [10]. As is the case for wild type, strains encoding only RsbRA or RsbRB nonetheless manifest an excess of stressosome core needed to bind the available RsbT, so these differences in basal level appear to reflect a fundamental property of the co-antagonist [10]. That is, when paired with RsbS each co-antagonist species could produce a complex with a characteristic affinity for RsbT or susceptibility to phosphorylation, or perhaps a different sensitivity to the activating signal, and thus would sequester RsbT with different efficiency in unstressed cells [10].
We first tested the effect of the S59A substitution in the strain with a full complement of RsbR family co-antagonists and found it decreased maximal response to ethanol stress about eight-fold (Fig. 2A); in four independent assays the mutant still retained 13% of the activity manifested by the strain with wild type RsbS. This 13% activation was eliminated in a strain bearing a null rsbU allele and thus lacking the RsbU environmental phosphatase: the rsbS-S59A rsbU double mutant had same low activity as the rsbU null control (Fig. 2A). This result indicates that the activation remaining in the S59A mutant originated in the environmental branch of the network and did not involve either the energy stress or cold stress pathways (Fig. 1), which are unaffected by loss of RsbU function [30], [31]. Because previous in vivo assays have shown that RsbU is incapable of transmitting signals of ethanol stress in the absence of input from RsbT [32], we draw the strong inference that the S59A-independent activation signal is conveyed via the recognized environmental signaling pathway upstream from RsbU, and not by RsbU itself or by a hypothetical independent pathway that converges on RsbU.
Figure 2. Strains bearing the S59A substitution retain significant stress activation.
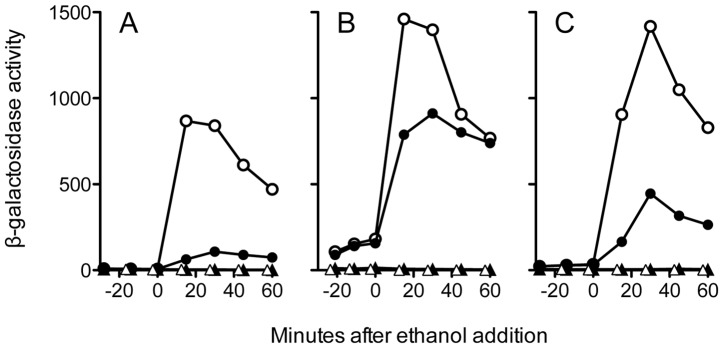
β-galactosidase accumulation from a σB-dependent ctc-lacZ fusion, assayed in logarithmically growing cells either wild type for RsbS (open circles) or with the S59A substitution (closed circles), before and after 4% ethanol addition. In all three panels an RsbU null strain defective for the response served as the negative control (PB495, open triangles). (A) Wild type stressosome with all four co-antagonists and wild type RsbS (PB198, open circles); RsbS-S59A (PB470, closed circles); or RsbU null with RsbS-S59A (PB1274, closed triangles). (B) Minimal stressosome containing RsbRA as sole co-antagonist and wild type RsbS (PB1078, open circles); RsbS-S59A (PB1161, closed circles); or RsbU null with RsbS-S59A (PB1273, closed triangles). (C) Minimal stressosome containing RsbRB as sole co-antagonist and wild type RsbS (PB1255, open circles); RsbS-S59A (PB1256, closed circles); or RsbU null with RsbS-S59A (PB1275, closed triangles). Representative results are shown; in independent experiments S59A supported a response 13% as robust as wild type RsbS in the strain with all four co-antagonists (+/−2.0% SEM, n = 4); 63% in the strain with RsbRA alone (+/−2.1%, n = 7); and 33% in the strain with RsbRB alone (+/−1.3%, n = 3).
We then examined the effects of S59A in strains encoding simplified stressosomes. In the strain with stressosome complexes formed only with RsbRA, the RsbS mutant had 60% of parental activity (Fig. 2B), and with RsbRB about 30% (Fig. 2C). As was the case for the strain encoding all four RsbR co-antagonists, the heightened S59A-independent activation in the RsbRA or RsbRB strains was eliminated by the rsbU null (Fig. 2B and 2C). Moreover, the observation that the magnitude of the S59A-independent activation was significantly influenced by the co-antagonist complement of the stressosome further supports the conclusion that this activation is dependent upon the recognized signaling pathway upstream from RsbU.
Significant Signaling can Occur in the Absence of both S59 and T171 Phosphorylation
The only known signaling element upstream from RsbU is the stressosome itself [5], whose core constituents are the RsbR paralogs and RsbS. To determine if a fully functional RsbRA co-antagonist was required for the S59A-independent activation mechanism, we assayed the effects of the T171A substitution in RsbRA. T171A eliminates the primary site of RsbRA phosphorylation [23], [24], [26] and manifests phenotypes of different severities, depending on the in vivo complement of stressosome components [8]. In a strain encoding all four RsbR co-antagonists and wild type RsbS, T171A effectively prevents signaling, decreasing ethanol stress response by 100 fold. By contrast, in a strain encoding only RsbRA and wild type RsbS, T171A decreases the response by 10–12 fold [8].
In agreement with these previous results, we found that the T171A substitution eliminated signaling in an otherwise wild type strain encoding all four RsbR paralogs (Fig. 3A). T171A also blocked signaling in the RsbS S59A mutant, indicating that in the wild type stressosome the S59A-independent activity requires full RsbRA co-antagonist function. This result provides independent evidence that the activity originates in the environmental signaling pathway and does not involve either the energy or cold stress pathways. However, in this genetic background the strong T171A phenotype potentially obscured any epistatic interaction with S59A.
Figure 3. Positive epistasis between S59A and T171A in a model stressosome.

β-galactosidase accumulation from a ctc-lacZ fusion in logarithmically growing cells, before and after 4% ethanol addition. (A) Wild type stressosome with all four co-antagonists and wild type RsbS and RsbRA (PB198, open circles); RsbS-S59A (PB470, closed circles); RsbRA-T171A (PB830, open squares); or RsbS-S59A together with RsbRA-T171A (PB1219, closed squares). (B) Minimal stressosome containing RsbRA as sole co-antagonist and wild type RsbS and RsbRA (PB1078, open circles); RsbS-S59A (PB1161, closed circles); RsbRA-T171A (PB1205, open squares); or RsbS-S59A with RsbRA-T171A (PB1190, closed squares). Representative results are shown; in independent experiments the T171A-S59A mutant manifested a response 0.2% that of wild type RsbRA and RsbS in the strain with all four co-antagonists (+/−0.03% SEM, n = 3); and 21% in the strain with RsbRA alone (+/−5.8%, n = 3).
We therefore turned to the strain encoding the model stressosome formed from the RsbRA co-antagonist and RsbS. In this strain the T171A substitution only attenuated signaling about 12 fold, and produced an unexpected result when paired with S59A: the two substitutions manifested positive epistasis, with S59A in RsbS partially suppressing the phenotype of T171A in RsbRA (Fig. 3B). Positive (or alleviating) epistasis often results when two gene products act in concert within the same pathway [33]–[35]. In light of the extensive structural and biochemical data regarding interaction between RsbRA and RsbS [2], [7], [8], [10], [32], this additional genetic support is not surprising. However, if T171 and S59 phosphorylation were in fact key to the signaling mechanism [2], [4], [24], [29], the substantial stress activation evident in the T171A-S59A double mutant is unexpected (Fig. 3B). A structural basis for this effect is unlikely. When RsbRA and RsbS are incorporated into the pseudoatomic structure of the stressosome, both T171 and S59 lie on the exterior, solvent-exposed faces of their respective STAS domains, and alanine substitution at these residues would not impact surfaces thought to be important for STAS interaction within the core [2]. We therefore interpret our results to indicate that neither T171 nor S59 phosphorylation is required for signaling in the model stressosome.
The S59-independent Route of Signal Transduction does not Involve the YtvA Light Sensor
We wished to further explore the nature of the S59-independent route of signaling within the stressosome. In this regard, Purcell et al. [36] proposed that some blue-light-sensing LOV domains may also have redox sensing capability within the bacterial cytoplasm. Notably, another protein associated with the B. subtilis stressosome is the blue-light-sensing YtvA regulator, an RsbR family member that carries an N-terminal LOV domain (Fig. 1). In contrast to the related RsbR co-antagonists, YtvA by itself does not appear capable of forming a stressosome with RsbS in vitro [37] and is only known to have a positive signaling role [11], [14], [15], [38]. Nonetheless, if its LOV domain were capable of redox as well as blue-light sensing, YtvA is a candidate for an S59-independent route of signaling, possibly detecting the secondary oxidative stress elicited by ethanol challenge [39].
To test the involvement of YtvA in ethanol stress signaling, we introduced a null ytvA allele into the parental and S59A mutant strains with RsbRA as the only co-antagonist. As shown in Fig. 4, loss of YtvA function reduced ethanol response of both the parent and the S59A mutant by a similar amount (20–25%), as expected for loss of a common positive regulator. However, the S59A mutant manifested the same significant fraction of the parental ethanol response in either the presence or absence of YtvA. We conclude that the S59A-independent route of signal transmission does not depend on YtvA.
Figure 4. S59-independent activation does not require the YtvA blue-light sensor.
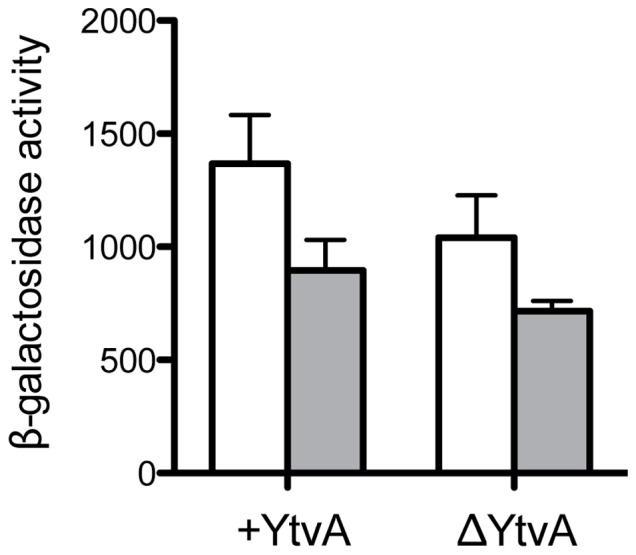
Peak β-galactosidase accumulation from a ctc-lacZ fusion after 4% ethanol stress, in the presence or absence of YtvA (+YtvA or ΔYtvA). Strains encoded a minimal stressosome containing RsbRA as sole co-antagonist together with either wild type RsbS (PB1078 for +YtvA or PB1085 for ΔYtvA; open bars) or RsbS bearing the S59A substitution (PB1161 or PB1272; shaded bars). Error bars represent range in two independent assays.
RsbS Phosphorylation Increases Response Efficiency
The results shown in Figures 2–4 were obtained in response to 4% ethanol stress. We next examined the effect of S59A on response to ethanol challenge over a range of lesser concentrations, using the strain encoding only the prototype co-antagonist, RsbRA [32]. As shown in Fig. 5A, S59A almost completely blocked response to a mild 0.75% ethanol stress, but its influence decreased significantly as ethanol challenge increased. This effect is readily apparent in Fig. 5B, which plots the data from Fig. 5A as the fraction of parental activity retained by the S59A mutant relative to the peak stress response of the parental strain. We infer from these results that S59 phosphorylation is critical for the transmission of low amplitude signals but becomes less significant as signal strength grows.
Figure 5. Ability to phosphorylate S59 increases stressosome sensitivity.
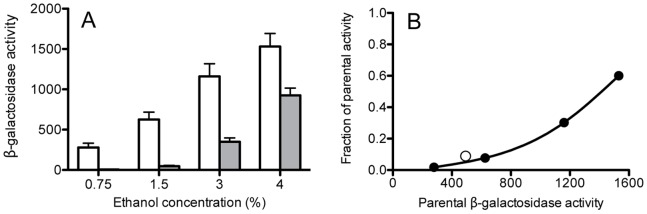
(A) Peak β-galactosidase accumulation from a ctc-lacZ fusion after addition of different concentrations of ethanol. Strains encoded a minimal stressosome containing RsbRA as sole co-antagonist and either wild type RsbS (PB1078; open bars) or RsbS bearing the S59A substitution (PB1161; shaded bars). Error bars represent range in two independent assays. (B) Closed symbols indicate fraction of parental activity manifested by S59A mutant at each ethanol concentration (data from A); open symbol indicates fraction at 0.3 M NaCl (see text).
We attempted a parallel experiment with increasing salt stress. However, concentrations above 0.3 M NaCl had secondary effects that interfered with cell growth (data not shown). We were therefore unable to achieve responses of the magnitude necessary for full comparison to the ethanol data. However, the 0.3 M NaCl experiment did yield a potentially interesting result: the maximum response of the parental strain was 472 units (+/−SEM of 38; n = 3) whereas that of the S59A mutant was 28 units (+/−SEM of 9; n = 3), for a fractional response of 0.06. When this additional point was plotted as the open symbol in Fig. 5B, it fell near the line representing the ethanol data. This suggests that the effect of S59 phosphorylation is independent from the primary stress that elicits the response.
Discussion
Most existing models of stressosome operation have focused on phosphorylation of RsbS S59 as the key signaling event within the stressosome complex [2], [4], [24], [29]. A presumed conformational change in the RsbR-RsbS core subunits has been suggested to influence activity of the RsbT kinase toward RsbS, either by allosteric control of kinase activity or by unmasking substrate residues within the core [2]. Along these lines, a recent computational model of stressosome operation was said to support the allosteric control of RsbT activity by phosphorylated RsbRA [29]. An acknowledged shortcoming of this model was its inability to fully replicate the phosphorylation kinetics of the in vivo study on which it was partly based. The in vivo study found that a rapid, post-stress increase in S59-phosphorylated RsbS occurred prior to a modest increase in T171-phosphorylated RsbRA [24], whereas in the computational model the increase in phosphorylated RsbS necessarily lagged the increase in phosphorylated RsbRA [29]. An unacknowledged shortcoming of the computational model was that another in vivo study, using a different growth medium, found no post-stress increase in T171-phosphorylated RsbRA at all [25]. These anomalies suggest that stress-dependent modulation of T171 phosphorylation does not well explain signal transmission within the stressosome, and that other mechanisms must play an important role.
In strong support of this view, the genetic results presented here speak against the prevailing phosphorylation models in their simplest forms. Stressosomes composed solely of mutant RsbRA and RsbS proteins, with phosphorylation-deficient T171A and S59A substitutions, were nonetheless capable of transmitting environmental stress signals in vivo (Fig. 3B). In vitro no other phosphorylation sites were detectable in the T171A-T205A mutant form of RsbRA or the S59A mutant form of RsbS [22], [23], and in vivo there is likewise no evidence for any additional sites on either protein [24], [25], [40]. Because in vivo phosphorylation of T205 primarily occurs under stress conditions more severe than we used here [24], [25], and this modification appears to suppress rather than activate signaling, for the purposes of our study its effects can be ignored. We therefore infer that significant in vivo activation of the stressosome can occur in the absence of both RsbRA and RsbS phosphorylation.
Our demonstration of a phosphorylation-independent signaling mechanism chiefly relied on a strain encoding a widely-used model stressosome in which RsbRA was the sole co-antagonist. This strain serves as an archetype for those bacterial species whose genomes encode only a single RsbR co-antagonist [1], the best characterized of which most closely resembles RsbRA [3]. However, we also extended these findings to show that RsbS phosphorylation was partly dispensable in two other strains (Fig. 2), one of which had wild-type stressosomes comprising all four co-antagonists [8], [9]. Our results should therefore be broadly applicable to stressosome operation in other bacterial signaling pathways, whether these stressosomes have but a single RsbR co-antagonist, as is the norm, or multiple co-antagonists, as is the case for B. subtilis and several other bacteria [1].
The existence of an RsbS-independent signaling role for RsbRA was initially suggested by the genetic epistasis test of Kim et al. [8], who found that the loss-of-signaling phenotype caused by the T171A substitution in RsbRA could not be fully reversed by the phosphomimetic S59D substitution in RsbS. This result points to a signaling role for RsbRA independent from its recognized ability to influence the rate of RsbS phosphorylation by RsbT [23], [26]. Here we confirm an RsbS-independent role by another means: a phosphorylation-deficient S59A substitution in RsbS decreased but did not completely block signaling, having the effect of altering the dose-response curve to require greater input for a comparable output (Fig. 5). This raises the intriguing possibility that S59 phosphorylation is an evolutionary addition that overlays a primordial signaling mechanism, increasing its sensitivity by increasing the dissociation rate of RsbT.
Based on the results reported here, we conclude that the stressosome manifests two mechanisms of signal transduction in response to acute environmental stress. We further suggest that these two mechanisms share an underlying commonality: a hypothetical signal-induced shift in conformational equilibria within the stressosome core [2]. One mechanism is dependent on RsbS phosphorylation and likely reflects RsbRA enhancement of RsbT kinase activity toward RsbS. This enhancement may involve active stimulation of the kinase or an unmasking of the RsbS substrate within the core; either could be elicited by the hypothetical conformational shift [2]. The other mechanism is independent of RsbS phosphorylation and its molecular basis is presently unknown. However, we propose that the same conformational shift can trigger RsbT release even in the absence of RsbS phosphorylation, albeit less efficiently. In this view, the signal-induced conformational shift is the fundamental signaling response, which then secondarily elicits activation of the RsbT kinase. Our genetic experiments with minimal stressosomes have uncovered this fundamental response, and offer a potential avenue for its exploration.
Materials and Methods
Bacterial Strains and Genetic Methods
B. subtilis strains shown in Table 1 are derivatives of PB2, a 168 Marburg strain originally obtained from Patrick Piggot [41]. Plasmids are shown in Table 2. Strain constructions employed two-step allele replacement [44], standard recombinant methods [45], and natural transformation [46]; all were confirmed by sequencing the coding regions of interest. A single-copy, ctc-lacZ transcriptional fusion provided an indirect measure of σB activity [42].
Table 1. Bacillus subtilis strains.
| Name | Genotype or description | Constructiona |
| PB2 | trpC2 | Marburg strain [41] |
| PB198 | amyE::ctc-lacZ trpC2 | [42] |
| PB470 | rsbS S59A amyE::ctc-lacZ trpC2 | [27] |
| PB491 | rsbRAΔ1 amyE::ctc-lacZ trpC2 | [32] |
| PB495 | rsbUΔ2 amyE::ctc-lacZ trpC2 | [31] |
| PB830 | rsbRA T171A amyE::ctc-lacZ trpC2 | [8] |
| PB1078 | rsbRBΔ2 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | [19] |
| PB1085 | rsbRBΔ2 rsbRCΔ1::spc rsbRDΔ2 ytvAΔ1::ery amyE::ctc-lacZ trpC2 | [19] |
| PB1161 | rsbS S59A rsbRBΔ2 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | [19] |
| PB1190 | rsbS S59A rsbRA T171A rsbRBΔ2 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | pTG6027→PB1161b |
| PB1205 | rsbRA T171A rsbRBΔ2 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | pTG6027→PB1078b |
| PB1219 | rsbRA T171A rsbS S59A amyE::ctc-lacZ trpC2 | pTG6027→PB470b |
| PB1254 | rsbRAΔ1 rsbRDΔ2 amyE::ctc-lacZ trpC2 | pTG5943→PB491b |
| PB1255 | rsbRAΔ1 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | pSA82c→PB1254 |
| PB1256 | rsbS S59A rsbRAΔ1 rsbRCΔ1::ery rsbRDΔ2 amyE::ctc-lacZ trpC2 | pTG6009→PB1255b |
| PB1271 | rsbS S59A rsbRBΔ2 rsbRCΔ1::spc rsbRDΔ2 amyE::ctc-lacZ trpC2 | pEr::Spc→PB1161 |
| PB1272 | rsbS S59A rsbRBΔ2 rsbRCΔ1::spc rsbRDΔ2 ytvAΔ1::ery amyE::ctc-lacZ trpC2 | pSA68c→PB1271 |
| PB1273 | rsbS S59A rsbRBΔ2 rsbRCΔ1::ery rsbRDΔ2 rsbUΔ3 amyE::ctc-lacZ trpC2 | pTG6110→PB1161b |
| PB1274 | rsbS S59A rsbUΔ3 amyE::ctc-lacZ trpC2 | pTG6110→PB470b |
| PB1275 | rsbS S59A rsbRAΔ1 rsbRCΔ1::ery rsbRDΔ2 rsbUΔ3 amyE::ctc-lacZ trpC2 | pTG6110→PB1256b |
Arrow indicates transformation from donor to recipient.
Two-step allele replacement.
Linearized plasmid.
Table 2. Plasmids for strain construction.
| Plasmid | Relevant feature | Reference |
| pEr::Sp | Converts ery to spc | [43] |
| pSA68 | ytvAΔ1::ery in pUC19 integrative plasmid | [28] |
| pSA82 | rsbRCΔ1::ery in pUC19 integrative plasmid | [28] |
| pTG5916 | NdeI site converted to I-SceI in pUS19 integrative plasmid | [19] |
| pSS4332 | Expresses I-SceI for two-step allele replacement (pTG5916 vectors) | [44] |
| pTG5943 | rsbRDΔ2 in pTG5916 | [19] |
| pTG6009 | rsbS S59A in pTG5916 | [19] |
| pTG6027 | rsbRA T171A in pTG5916 (ACC→GCC) | This work |
| pTG6110 | rsbUΔ3 in pTG5916 (codons 12–331 deleted) | This work |
β-galactosidase Accumulation Assays
Assays were conducted as described previously [19]. Cultures were grown at 37 C in shake flasks containing buffered Luria broth lacking salt, and with moderate white light illumination (3 to 4 µmol m−2 s−1). This illumination saturated the blue light-sensing positive regulator YtvA to establish a constant effect on assay results [47]. Unstressed samples were taken during early exponential growth up to a cell density of 20 absorbance units (Klett-Summerson colorimeter equipped with a number 66 transmission filter); ethanol stress was then imposed at the final concentrations indicated in the figures. Samples were treated as described by Miller [48], but with activity defined as ΔA 420×1,000 min−1 mg protein−1 (Protein Assay Reagent; Bio-Rad Laboratories, Hercules, CA). Stress activation was calculated as maximal post-stress activity minus basal activity just prior to stress addition.
Funding Statement
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number RO1 GM42077. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pané-Farré J, Lewis RJ, Stülke J (2005) The RsbRST stress module in bacteria: a signalling system that may interact with different output modules. J Mol Microbiol Biotechnol 9: 65–76. [DOI] [PubMed] [Google Scholar]
- 2. Marles-Wright J, Grant T, Delumeau O, van Duinen G, Firbank SJ, et al. (2008) Molecular architecture of the “stressosome,” a signal integration and transduction hub. Science 322: 92–96. [DOI] [PubMed] [Google Scholar]
- 3. Quin MB, Berrisford JM, Newman JA, Baslé A, Lewis RJ, et al. (2012) The bacterial stressosome: a modular system that has been adapted to control secondary messenger signaling. Structure 20: 350–363. [DOI] [PubMed] [Google Scholar]
- 4. Hecker M, Pané-Farré J, Völker U (2007) SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol 61: 215–236. [DOI] [PubMed] [Google Scholar]
- 5.Price CW (2010) General stress response in Bacillus subtilis and related Gram positive bacteria. In: Storz G, Hengge R, editors. Bacterial stress responses. 2nd ed. Washington, DC: ASM Press. 301–318.
- 6. Young JW, Locke JC, Elowitz MB (2013) Rate of environmental change determines stress response specificity. Proc Natl Acad Sci U S A 110: 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen CC, Lewis RJ, Harris R, Yudkin MD, Delumeau O (2003) A supramolecular complex in the environmental stress signalling pathway of Bacillus subtilis . Mol Microbiol 49: 1657–1669. [DOI] [PubMed] [Google Scholar]
- 8. Kim TJ, Gaidenko TA, Price CW (2004) A multicomponent protein complex mediates environmental stress signaling in Bacillus subtilis . J Mol Biol 341: 135–150. [DOI] [PubMed] [Google Scholar]
- 9. Delumeau O, Chen CC, Murray JW, Yudkin MD, Lewis RJ (2006) High-molecular-weight complexes of RsbR and paralogues in the environmental signaling pathway of Bacillus subtilis . J Bacteriol 188: 7885–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reeves A, Martinez L, Haldenwang W (2010) Expression of, and in vivo stressosome formation by, single members of the RsbR protein family in Bacillus subtilis . Microbiology 156: 990–998. [DOI] [PubMed] [Google Scholar]
- 11. van der Steen JB, Ávila-Pérez M, Knippert D, Vreugdenhil A, van Alphen P, et al. (2012) Differentiation of function among the RsbR paralogs in the general stress response of Bacillus subtilis with regard to light perception. J Bacteriol 194: 1708–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murray JW, Delumeau O, Lewis RJ (2005) Structure of a nonheme globin in environmental stress signaling. Proc Natl Acad Sci U S A 102: 17320–17325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Losi A, Polverini E, Quest B, Gärtner W (2002) First evidence for phototropin-related blue-light receptors in prokaryotes. Biophys J 82: 2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaidenko TA, Kim TJ, Weigel AL, Brody MS, Price CW (2006) The blue-light receptor YtvA acts in the environmental stress signaling pathway of Bacillus subtilis . J Bacteriol 188: 6387–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ávila-Pérez M, Hellingwerf KJ, Kort R (2006) Blue light activates the σB-dependent stress response of Bacillus subtilis via YtvA. J Bacteriol 188: 6411–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Möglich A, Moffat K (2007) Structural basis for light-dependent signaling in the dimeric LOV domain of the photosensor YtvA. J Mol Biol 373: 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Möglich A, Ayers RA, Moffat K (2009) Design and signaling mechanism of light-regulated histidine kinases. J Mol Biol 385: 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diensthuber RP, Bommer M, Gleichmann T, Möglich A (2013) Full-length structure of a sensor histidine kinase pinpoints coaxial coiled coils as signal transducers and modulators. Structure 21: 1127–1136. [DOI] [PubMed] [Google Scholar]
- 19. Gaidenko TA, Bie X, Baldwin EP, Price CW (2011) Substitutions in the presumed sensing domain of the Bacillus subtilis stressosome affect its basal output but not response to environmental signals. J Bacteriol 193: 3588–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaidenko TA, Bie X, Baldwin EP, Price CW (2012) Two surfaces of a conserved interdomain linker differentially affect output from the RST sensing module of the Bacillus subtilis stressosome. J Bacteriol 194: 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Delumeau O, Dutta S, Brigulla M, Kuhnke G, Hardwick SW, et al. (2004) Functional and structural characterization of RsbU, a stress signaling protein phosphatase 2C. J Biol Chem 279: 40927–40937. [DOI] [PubMed] [Google Scholar]
- 22. Yang X, Kang CM, Brody MS, Price CW (1996) Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev 10: 2265–2275. [DOI] [PubMed] [Google Scholar]
- 23. Gaidenko TA, Yang X, Lee YM, Price CW (1999) Threonine phosphorylation of modulator protein RsbR governs its ability to regulate a serine kinase in the environmental stress signaling pathway of Bacillus subtilis . J Mol Biol 288: 29–39. [DOI] [PubMed] [Google Scholar]
- 24. Kim TJ, Gaidenko TA, Price CW (2004) In vivo phosphorylation of partner switching regulators correlates with stress transmission in the environmental signaling pathway of Bacillus subtilis . J Bacteriol 186: 6124–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eymann C, Schulz S, Gronau K, Becher D, Hecker M, et al. (2011) In vivo phosphophorylation patterns of key stressosome proteins define a second feedback loop that limits activation of Bacillus subtilis σB . Mol Microbiol 80: 798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen CC, Yudkin MD, Delumeau O (2004) Phosphorylation and RsbX-dependent dephosphorylation of RsbR in the RsbR-RsbS complex of Bacillus subtilis . J Bacteriol 186: 6830–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang CM, Brody MS, Akbar S, Yang X, Price CW (1996) Homologous pairs of regulatory proteins control activity of Bacillus subtilis transcription factor σB in response to environmental stress. J Bacteriol 178: 3846–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Akbar S, Gaidenko TA, Kang CM, O’Reilly M, Devine KM, et al. (2001) New family of regulators in the environmental signaling pathway which activates the general stress transcription factor σB of Bacillus subtilis . J Bacteriol 183: 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liebal UW, Millat T, Marles-Wright J, Lewis RJ, Wolkenhauer O (2013) Simulations of stressosome activation emphasize allosteric interactions between RsbR and RsbT. BMC Syst Biol 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brigulla M, Hoffmann T, Krisp A, Völker A, Bremer E, et al. (2003) Chill induction of the SigB-dependent general stress response in Bacillus subtilis and its contribution to low-temperature adaptation. J Bacteriol 185: 4305–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vijay K, Brody MS, Fredlund E, Price CW (2000) A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the σB transcription factor of Bacillus subtilis . Mol Microbiol 35: 180–188. [DOI] [PubMed] [Google Scholar]
- 32. Akbar S, Kang CM, Gaidenko TA, Price CW (1997) Modulator protein RsbR regulates environmental signalling in the general stress pathway of Bacillus subtilis . Mol Microbiol 24: 567–578. [DOI] [PubMed] [Google Scholar]
- 33. St Onge RP, Mani R, Oh J, Proctor M, Fung E, et al. (2007) Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat Genet 39: 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mani R, St Onge RP, Hartman JL, Giaever G, Roth FP (2008) Defining genetic interaction. Proc Natl Acad Sci U S A 105: 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phillips PC (2008) Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Purcell EB, McDonald CA, Palfey BA, Crosson S (2010) An analysis of the solution structure and signaling mechanism of LovK, a sensor histidine kinase integrating light and redox signals. Biochemistry 49: 6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jurk M, Schramm P, Schmieder P (2013) The blue-light receptor YtvA from Bacillus subtilis is permanently incorporated into the stressosome independent of the illumination state. Biochem Biophys Res Commun 432: 499–503. [DOI] [PubMed] [Google Scholar]
- 38. Ávila-Pérez M, Vreede J, Tang Y, Bende O, Losi A, et al. (2009) In vivo mutational analysis of YtvA from Bacillus subtilis: mechanism of light activation of the general stress response. J Biol Chem 284: 24958–24964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Höper D, Völker U, Hecker M (2005) Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis . J Bacteriol 187: 2810–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, et al. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis . Mol Cell Proteomics 6: 697–707. [DOI] [PubMed] [Google Scholar]
- 41. Piggot PJ (1973) Mapping of asporogenous mutations of Bacillus subtilis: a minimum estimate of the number of sporeulation operons. J Bacteriol 114: 1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boylan SA, Rutherford A, Thomas SM, Price CW (1992) Activation of Bacillus subtilis transcription factor σB by a regulatory pathway responsive to stationary-phase signals. J Bacteriol 174: 3695–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steinmetz M, Richter R (1994) Plasmids designed to alter the antibiotic resistance expressed by insertion mutations in Bacillus subtilis, through in vivo recombination. Gene 142: 79–83. [DOI] [PubMed] [Google Scholar]
- 44. Cybulski RJ Jr, Sanz P, Alem F, Stibitz S, Bull RL, et al. (2009) Four superoxide dismutases contribute to Bacillus anthracis virulence and provide spores with redundant protection from oxidative stress. Infect Immun 77: 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- 46. Dubnau D, Davidoff-Abelson R (1971) Fate of transforming DNA following uptake by competent Bacillus subtilis. I. Formation and properties of the donor-recipient complex. J Mol Biol 56: 209–221. [DOI] [PubMed] [Google Scholar]
- 47. Ávila-Pérez M, van der Steen JB, Kort R, Hellingwerf KJ (2010) Red light activates the σB-mediated general stress response of Bacillus subtilis via the energy branch of the upstream signaling cascade. J Bacteriol 192: 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller JH (1972) Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.