

Abstract
Over the past decade, nine gene therapy clinical trials for Parkinson's disease (PD) have been initiated and completed. Starting with considerable optimism at the initiation of each trial, none of the programs has yet borne sufficiently robust clinical efficacy or found a clear path toward regulatory approval. Despite the immediately disappointing nature of the efficacy outcomes in these trials, the clinical data garnered from the individual studies nonetheless represent tangible and significant progress for the gene therapy field. Collectively, the clinical trials demonstrate that we have overcome the major safety hurdles previously suppressing central nervous system (CNS) gene therapy, for none produced any evidence of untoward risk or harm after administration of various vector-delivery systems. More importantly, these studies also demonstrated controlled, highly persistent generation of biologically active proteins targeted to structures deep in the human brain. Therefore, a renewed, focused emphasis must be placed on advancing clinical efficacy by improving clinical trial design, patient selection and outcome measures, developing more predictive animal models to support clinical testing, carefully performing retrospective analyses, and most importantly moving forward—beyond our past limits.
Background: Impetus and Outcomes with Parkinson's Gene Therapy (GT) Trials
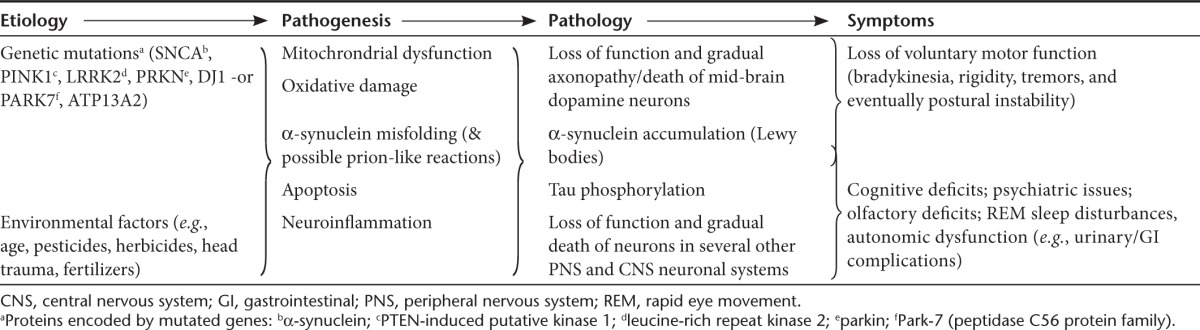
Parkinson's disease (PD) is a chronic, progressive neurodegenerative disease most widely recognized for the profound degeneration of mid-brain dopamine nigrostriatal neurons linked to serious motor symptoms.1 However, PD is far more complex than commonly appreciated, with multiple etiologic variables and pathogenic pathways, complex pathologies, and a wide range of central nervous system (CNS) and non-CNS symptoms (Table 1).2,3 Moreover, wide gaps in our understanding still exist at each disease level (i.e., etiology, pathogenesis, pathology, symptoms), and the cause-effect relationships between them remain especially obscure. Arguably, the most well-characterized relationship exists with regard to nigrostriatal degeneration linked to the key motor symptoms; currrent oral dopaminergic pharmaceuticals are effective in controlling these symptoms at early disease stages. However, the drugs' effectiveness decline with progressive pathology, leading to gradual incapacitation of patients by increased “off” time (i.e., periods of no symptomatic relief) and increasing side effects such as peak-dose dyskinesias.1 Thus, adequate treatment of the nigrostriatal-mediated motor impairments continues to represent a significant unmet medical need, affecting over 4 million people worldwide.4 Though a number of solutions have been conceived to improve the function of the degenerating dopaminergic system, translating these biopharmaceutical concepts to the clinic has been challenging due to obstacles associated with delivering macromolecules to the central nervous system in a persistent and targeted fashion.
Table 1. Complex etiology, pathogenesis, pathology, and symptomatology of Parkinson's disease.
Progress achieved in the realm of gene therapy (GT) over the past decade has offered solutions to many of the delivery constraints,5 and several aspects of PD present it overtly as an ideal clinical indication to target using GT: (i) the well-defined, localizable, and targetable neuronal systems involved with major motor symptoms, (ii) the need for relatively small titer and volume of vector targeted to those sites, which avoids the systemic circulation of immunogenic materials, and (iii) the large and increasing demand for improved therapeutics with an aging population,4 which in whole bolsters impact and financial support for research and development. Given this rationale, PD has, for better or worse, become a key exemplar for CNS GT. To date, the results of completed PD GT trials have supported the safety of GT targeting in the brain and many have further confirmed the successfully targeted expression of bioactive proteins in specific brain sites. However, none of the programs has yet produced sufficiently robust or reliable efficacy data to enable initiation of a pivotal phase 3 trial required for regulatory approval. We attempt to integrate the many successes and formidable challenges of GT treatment of PD, in an effort to seek the best path forward for PD and CNS GT as a whole.
The Look-See Approach
As compared with conventional small molecule drug testing, GT in the CNS is limited with respect to establishing initial dosing, quantifying targeting success, and accessing a comprehensive gauge of transgene production and localization, all of which provide the basis for iterative improvement with traditional drug development. While we expect that such limitations are attributable to a short-lasting gap between demand and current technological capability, the ramifications of such limitations include poor predictive power and a loss of continuity and progress between successive trials.
Whereas continuing progress is being made in the visualization of viral vectors as they are delivered to the brain (e.g., the use of gadolinium-containing liposomes coupled with magnetic resonance imaging),6,7 there is currently no direct means to determine the distribution or magnitude of transgene expression, since protein expression with CNS GT trials is both generated and produced entirely within the confines of the human brain. Therefore, it is not yet possible to confirm proper dose selection or monitor dosing levels, as would normally be done using standard pharmacokinetic (i.e., plasma level) methods. This severely limits the predictive and correlative strength of treatment endpoint measurements. One surrogate means by which transgene expression and function has been assessed is positron emission tomography (PET) imaging coupled with radiolabeled L-Dopa.8 This approach has enabled the live imaging of the dopamine precursor's uptake, which when compared to pretreatment levels can be directly informative to aromatic L-amino acid decarboxylase (AADC) and tyrosine hydroxylase (TH) transgene production and function. One problem with this approach is the inadequacy of using such technology as a dosing criterion: a sufficient signal is not expected until weeks or months after vector is administered, making adjustments to dosing inefficient at best, even within the flexibility of a phase 1 trial.
Due to the relative blindness of the researcher/clinician to targeting and dosing data, the process of translating and validating clinical results from prior animal data in GT programs has been characterized a “look-see” or trial and error approach. While this approach can still enable iterative improvement and therapeutic success, as demonstrated by the continued improvements to hemophilia GT treatment (discussed further below), it is not a paradigm easily applied to all drug development endeavors. Moreover, the repeated efforts can widen the so-called “valley of death” one confronts when attempting to move a program forward from initial, small open-label trials to the multi-center, double-blind, sham-surgery controlled trials required to establish efficacy. As we will point out in the next section, each of the PD approaches described below offer vivid examples of the look-see approach, often to the program's detriment.
Parkinson's GT Clinical Trials to Date
To date, five divergent GT approaches have been developed to treat the major motor symptoms of PD, all with the use of AAV or lentivirus vector platforms (Table 2). Each of the clinical approaches has focused on aspects of the basal ganglia, a group of subcortical neural sites located near the base of the forebrain that communicate intimately with the cerebral cortex and other brain areas. Most GT approaches directly target the terminals of the degenerating nigrostriatal neurons for gene delivery, while one approach indirectly attempts to resolve striatal neurochemical imbalance by increasing inhibitory control from the subthalamic nucleus.
Table 2. Summary of gene therapy clinical programs for Parkinson's disease.
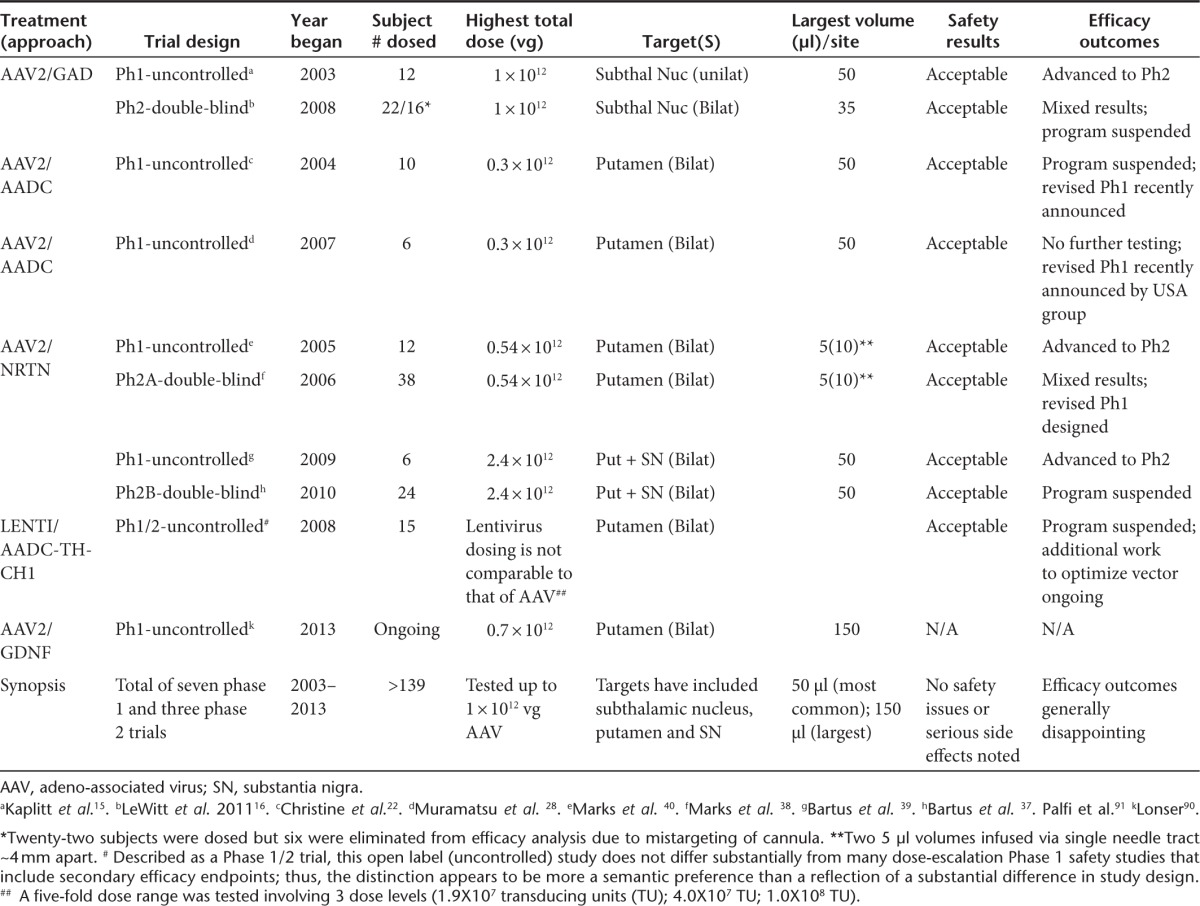
Three of the five PD GT approaches have attempted to ameliorate motor symptoms by altering the neurochemical conductivity of neurons mediating the motor behavior. This general strategy has involved delivering neurotransmitter-producing enzymes. In a first clinical study, AAV2/GAD (adeno-associated viral vector serotype 2/glutamic acid decarboxylase (GABA), a combination of GAD-65 and GAD-67) was delivered to the subthalamic nucleus9,10 (Figure 1b). GAD catalyzes the synthesis of gamma-aminobutyric acid, the major inhibitory neurotransmitter in the CNS, potentially providing lost inhibitory control in the basal ganglion motor system, thus restoring appropriate transynaptic balance. A second approach has attempted to improve the ability of the putamen to synthesize dopamine from exogenous L-Dopa (the immediate precursor for dopamine and the mainstay therapeutic for early-stage PD) by expressing the major L-DOPA-converting enzyme AADC in the putamen. Whereas one clinical investigation used AAV to deliver this enzyme,8 a second study used a trisictronic (delivery of three transgenes from a single vector cassette) lenti-vector expressing AADC as well as TH and GTP-CH1 (guanosine 5′-triphosphate cyclohydrolase1), with the latter two enzymes also significantly contributing to dopamine's synthesis11 (Figure 1c).
Figure 1.

Illustration of Parkinson's disease gene therapy strategies to date.(a) In a sagittal slice of the human brain, dopaminergic pathways are depicted projecting within various deep-brain basal ganglia structures particularly affected in Parkinson's disease (PD), such as the substantia nigra (SN) projecting to the striatum (STR). (b) In the first gene therapy (GT) clinical trial for PD, adeno-associated virus (AAV) carrying genes encoding for glutamic acid decarboxylase (GAD), a gamma-aminobutyric acid (GABA)-producing enzyme, were delivered to the subthalamic nucleus (STN), to neurons projecting to, and ostensibly inhibiting hyperactive putaminal (PUT) neurons of the striatum. (c,d) The majority of PD GT clinical trials to date, including those incorporating neurotrophic factors (NRTN, GDNF) and dopamine-producing enzymes (AADC, TH) have targeted the therapeutic vector to striatal neurons, relying on retrograde transport to the SN cell bodies to achieve maximal therapeutic response. Delivery of the therapeutic vector directly to cell bodies of the SN has been used as an alternative or complementary strategy to overcome the transport deficiences in this pathological brain tissue in an attempt enhance therapeutic response. (e) One of the two major STR/SN gene therapy approaches involves expression and secretion of neurotrophic factors glial-derived neurotrophic factor (GDNF) or neurturin (NRTN) in SN neurons projecting to the striatum, and thereby attempting to repair the SN-STR dopamine pathway and restore its function. The other GT approach in this system relies on expression of enzymes aromatic L-amino acid decarboxylase (AADC) or tyrosine hydroxylase (TH), each of which is involved in the synthesis of dopamine from neurochemical substrates found in in striatum-projecting neurons.
The final two of five PD GT approaches have employed a neurotrophic factor delivery approach, which rather than directly attempting to modulate neuronal activity, instead confer “trophic” support to the dopaminergic and surrounding neuronal populations by inducing repair genes in activated cells12,13 (Figure 1c,e). This offers potential improvement in symptoms as well as delay in disease progression.5,14 Both neurotrophic GT investigations to date have used AAV2 as the delivery vector; the studies diverge merely in the choice of which of two structural and functional analog proteins (neurturin (NRTN) or glia cell–derived neurotrophic factor (GDNF)) is packaged and expressed. Below we offer perspective on the outcomes of each of the PD GT clinical approaches to date.
AAV/GAD
Following publication of nonhuman efficacy and safety data10 and the successful completion of the first safety and tolerability trial for PD GT,15 a single controlled phase 2 trial with AAV/GAD was initiated. The trial involved 45 total subjects (roughly 50:50, treated:sham) and was completed without serious incident.16 Several interesting protocol innovations were employed, including prescreening all subjects with 18F-fluorodeoxyglucose PET to help assure a definitive diagnosis of PD. While this investigation is unique in that it achieved statistical significance on the primary efficacy endpoint (i.e., UPDRS motor-off), which represents a significant accomplishment in its own right, the clinical effect was described as “modest”.17 Indeed, even the significant primary endpoint produced only a 3.4-point difference on the UPDRS, part 3 scale (clinician-scored motor evaluation), compared to sham control. Moreover, the slim statistical difference between groups (P = 0.04) likely positively influenced by the use of a reasonable, protocol-prescribed data analysis approach that would not meet the more rigorous “intent to treat” standards traditionally held by the US Food and Drug Administration (FDA). Specifically, five subjects, four of whom showed no benefit on the primary endpoint, were eliminated after the trial was completed because an magnetic resonance imaging-based review of the targeting identified their injections as being off-target.16 While two other efficacy measurements (both clinical impressions rating scales) also showed statistical significance (P < 0.05), these effects were also quite modest (i.e., < ½ point difference), while 21 other efficacy endpoints, including those commonly considered important for confirming clinical benefit (e.g., PDQ-39; multiple motor diary scores), showed no difference between groups. Thus, despite technically meeting the primary endpoint, the efficacy data from this trial were modest at best, which likely was responsible for the trial's sponsor (Neurologix, Fort Lee, NJ) discontinuing the AAV/GAD program.
In retrospect, the AAV/GAD clinical trial highlighted several key early concerns for the PD GT field to consider: (i) whether modest nonhuman primate efficacy data (e.g., a 1-point improvement versus control on a 32-point behavioral scale) justified moving into clinical testing, (ii) the disappointingly modest efficacy data generated in the clinic, and (iii) the need for such high exclusion rates of treated subjects. The latter concern hints at potential challenges in performing consistent, accurate intracranial targeting—a concern that continues to be addressed and potentially resolved,18 albeit using real-time imaging instrumentation and methods not yet available in most stereotactic surgical operating rooms. Perhaps more importantly, even the hint of exclusionary data, regardless of the specific circumstances, can mar the perceived integrity of the results, and discount the apparent maturity of the field at large. Here, the modest efficacy data may be best ascribed to modest non-human primate findings. However, as we will examine, such modest clinical efficacy reported in this initial trial foreshadowed that of each subsequent clinical trial, even where preclinical studies showed more robust results. This clinical disappointment, therefore, can best be examined in the broader context of PD GT.
AAV/AADC
Another early PD GT approach utilized AAV/AADC, which after showing preclinical efficacy in MPTP monkeys,8 moved into clinical testing for 15 moderately advanced subjects in an open-label phase 1 clinical trial. The protocol for this trial was publicly discussed at the recombinant DNA advisory committee in late 2003,19 but subjects were not treated until 1 year later, during which time Genzyme, Boston, MA acquired the program from Avigen (Alameda, CA).20 First, it is worth noting the success of this trial, which like the preceding AAV/GAD study, included preliminary evidence for safety.21,22 However, despite a reasonable scientific rationale and several animal studies demonstrating reasonably robust enhancement in nigrostriatal dopamine function with AAV2-AADC,8,23,24,25 the phase 1 trial found only very modest efficacy,17,26 and was even described as neither “clear cut” nor what “we needed” by a Genzyme spokesperson.27 A second phase 1 study was performed in Japan using the identical vector (provided by Genzyme) and dosing paradigm, as well as a similar clinical protocol.28 Not surprisingly, the open-label efficacy results were not markedly different from the trial conducted in the USA. Recently, Genzyme agreed to allow the program's academic originators, in collaboration with Michael J. Fox Foundation for Parkinson's Research (MJFF), to resurrect a modified version of the clinical program, admitting that without the MJFF financial support, the program would not likely have moved forward.27
The reinitiated program offers an example of the look-see approach currently necessary for CNS GT work, which given the 10-year separation between studies, might allow for at least marginal redesign and advancement of the forthcoming study. First, we should reexamine the overriding approach: the use of AADC as the lynchpin of the clinical trial. Some researchers have questioned whether elevating levels of AADC is necessary or sufficient to enhance dopamine, citing either the redundancy of other decarboxylases in the CNS, or the maintained functionality of the decarboxylase activity in patients with advanced PD, who can often still benefit from low doses of L-Dopa.29 However, the preclinical data are strikingly clear in terms of efficacy expectations,8 and recent clinical successes with AAV/AADC in a compassionate use study of AADC deficiency30 reaffirm the potential for this transgene to restore L-Dopa conversion to dopamine in humans (to the extent that the monogenic disease may be comparable to the complex etiology of PD). Thus, an alternative explanation for the lack of efficacy might be due to delivery—a more manageable hurdle. The originators of the AADC trial acknowledge that the dosing parameters used in their initial clinical effort were suboptimal. Specifically, the researchers noted a somewhat lacking transduction pattern in the putamen: whereas the prior dosing parameters led to 35–40% coverage of this region as a whole, this corresponded with only 5–6% of AADC-expressing neurons within the delivery area.25 Thus, a clear priority for the reinitiated trial will be to extend the putaminal coverage as well as the transduced cell density, which the group expects to accomplish by using increased vector volumes and doses.21 This solution, though abbreviated in their discussion, merits attention: certainly using magnetic resonance imaging-guided CED (convection-enhanced delivery) and an increased dose should increase putaminal coverage (even to the proposed 60% goal). Further, it is conceivable that the two separate concerns of transduction volume and density can be resolved with CED, and increased vector concentration per volume (perhaps a far greater dose). At the same time, however, efforts to increase volume and also increase density might represent competing goals, for as volume is increased, density will decrease (all else being equal) and vice versa. With the steady advances in vector development, we argue that the vector itself should also be considered a variable that can have major consequences on transduction success. For instance, AAV1 and AAV5 are more efficacious than AAV2 in transduction of the primate substantia nigra (SN) and caudate nucleus,31,32 and several other serotypes have shown far superior transduction efficiency than AAV2 in rodent models, with respect to both volume and density.33,34 Should this improved efficiency translate to the Parkinsonian brain, it might allow for reduction in viral dose to achieve the same or greater volume and density of therapeutic gene expression. To the other extreme, assuming far higher AADC levels could be achieved through any or all of the above alternatives, one needs to consider the possibility of increased dyskinesias, as observed in monkeys with high focal doses of AAV-AADC.35 While similar side effects have not been reported in other nonhuman primate studies or in any of the 15 human subjects dosed a decade ago, the correlation between bioactivity in animal models and human studies remains uncertain and a potential problem might reemerge with the proposed changes in CED parameters and dose. The side effects reported when AAV-GDNF was delivered to the SN (and beyond) using aggressive CED parameters in nonhuman primates18 provides a clear, empirical example for why one should be cautious about unexpected outcomes when dosing parameters are adjusted to significantly increase spread of vector or protein. Importantly, neither the spread of protein far outside the targeted SN, nor the side effects reported with aggressive CED were seen in animal studies36 or clinical tests in PD patients37 when somewhat more moderate dosing parameters were employed, involving more modest CED in conjunction with multiple, distributed injection sites.
So what is the greater context of the original and pending follow-up study? The degree by which the reiterated trial digresses and improves upon the original study will be vitally significant for the CNS GT field as a whole, where with each clinical setback the stakes are raised ever higher for this still-fledgling field. As with many emerging technologies, simultaneous advances are likely required, such as improved vectors, more representative animal models, more pointed clinical methodology that can detect negative outcomes, and a backdrop of supportive funding mechanisms.
Lenti-AADC/TH/GTP-CH1
Following publication of efficacy results in nonhuman primates with the trisictronic lenti-vector expressing three enzymes involved in dopamine production,11 an uncontrolled, open label, dose-escalation study was conducted in 15 PD subjects. Three ascending doses were tested, involving a 5-fold dose range. Like the ‘look-see' approach applied by others, significant adjustments were made to the dosing parameters (e.g., needle tracts per hemisphere were reduced from 5 to 3; infusions per needle tract were reduced from several to one; infusion rate was increased from 1 µl/min to 3 µl /min) in the middle of the protocol. The safety profile looked favorable, with most adverse events being mild and deemed unrelated to treatment. Dose-related increases in dyskinesias reportedly resolved when the L-dopa dose was lowered. Modest improvements (from baseline) in UPDRS-motor off scores were seen at 6 and 12 months, roughly comparable to that seen in prior GT phase 1 studies. The authors concluded that they applied an iterative process in this trial to try to optimize delivery and that a more definitive double-blind placebo controlled trial will not be conducted until they are able to achieve an optimal dose and delivery mode, presumably requiring further dosing iterations.
AAV/NRTN (CERE-120)
This first trophic factor approach to PD GT has seen two phase 2 trials conducted to date,38,39,41 each preceded by the completion of an appropriate open-label phase 1 trial,37,40 as well as an extensive nonclinical program before that.5,36,41,42 The efficacy data for both phase 2 trials were mixed and disappointing, in that neither met the primary endpoint (UPDRS motor-off) within the prescribed timeframe. Notably, in the initial phase 2 trial, improvement in the primary clinical endpoint was seen beyond the prescribed assessment time (i.e., 15–18 months versus the prescribed 12 months), offering a surprisingly positive twist to the study's initial disappointment. Additionally, several other motor and quality-of-life endpoints achieved statistical significance at the study's primary, 12 months time point. Moreover, of the ~50% subjects who remained blinded beyond the 12 months timeframe, a statistically significant effect was also seen on the primary endpoint, as well as even more secondary endpoints.37 Also, an exploratory statistical analysis indicated that the differences between AAV/NRTN and sham-surgery were highly unlikely to have occurred by chance (P < 0.007 and P < 0.001 at 12 and 18 months, respectively).5,41
As a perfect example of the look-see paradigm, following additional nonclinical testing,36,42 a follow-up phase 1/2b protocol was designed to improve efficacy by incorporating several important changes, such as increasing dose and volume of the vector to the putamen, additional targeting of the SN directly, and extending the blinded assessment period by several months5; all of these changes were confirmed in animal models to likely improve the biological response to AAV2-NRTN.36,42 Still, the resulting double-blind, controlled phase 2 trial also failed to show statistical significance on the primary endpoint.39 Thus, these results and the AAV/NRTN trial joined the ranks of the preceding phase 2 PD GT trials16,38 in falling far short of what was required to proceed into phase 3 testing.
Again, as we ask the major question of why greater efficacy was not achieved, we return to the issue of delivery. Here, however, the issue perhaps diverges from the previously entertained concern of putaminal coverage, to one of ineffective therapeutic gene transport in the pathological brain tissue. Certainly, one question posed by investigators is whether increasing putaminal coverage of vector and therefore NRTN transgene would be sufficient to improve efficacy. While it is hard to argue against this rationale at face value, a better question is exactly how much transgene expression has been successfully produced previously, and how much is necessary for impact. Recent reexamination of autopsy specimen place estimates of putaminal coverage by NRTN at around 20%,37,43 which due to NRTN-antibody limitations, is likely underestimated.5,44 Perhaps more importantly, and getting to the second concern of brain transport, the more recent phase 2b trial increased the NRTN dose by three- to fourfold and increased the volume per injection by 10-fold in the putamen alone. Additionally, a sizable dose was injected directly into the degenerating cells in the SN in an effort to assure adequate NRTN coverage of cells bodies to help activate repair pathways in these neurons. Together, these changes should have significantly increased NRTN expression and bioactivity events confirmed in animal studies36,42 (though no autopsy tissue is yet available from this study for confirmation); yet the clinical results were still not sufficiently robust. Conversely, in primate MPTP and advanced-age primate models of PD, significant neuronal protection and restoration was achieved with putaminal coverage well below 10% coverage.45,46 Thus, whether the extent of putaminal coverage by vector is the major factor in achieving robust clinical improvement is unclear, for neither that nor the addition of targeting the SN was sufficient to enhance the clinical readout. These data warn that merely improving blanket gene delivery coverage may not hold the key to achieving a desired outcome.
If vector transduction alone is not a major concern, what else might account for the lack of efficacy? One possibility has to do with the degenerative state of PD, and how this may affect therapeutic protein transport through brain circuits. With regard to AAV/NRTN, one possibility is the serious deficiency of axonal transport in PD patients. Such deficiencies have been posited as a major reason why, despite areas of intense NRTN expression in the targeted putamen, little NRTN was seen at the distal SN, where the protein product was intended to be transported to achieve a robust neurotrophic response.37,39,43,44 The topographic relationships between SN neurons and their putaminal projection fibers predicates that even a small area of protein expression in the putamen should produce a proportional area of detectable protein in the SN—a finding confirmed in several nonhuman primate studies with AAV-NRTN.44 However, as very little NRTN was seen in the SN of the PD autopsy tissue, it appeared that neuronal pathology in the aged human PD brain was more severe than that of animal models, preventing NRTN transport from the putamen to the SN. Aside from axonal transport (and related axonopathy) concerns, other pathologies may also diminish the anticipated effect of neurotrophic factor support, even in consideration of direct targeting to the SN cell bodies. For instance, α-synuclein (a protein whose accumulation in dopamine neurons serves as a major pathogenic event in PD) can mediate downregulation of the transcription factor Nurr1 and its downstream target, the GDNF/NRTN RET receptor,47,48,49 directly mitigating GDNF/NRTN signaling, and thus impeding the therapeutic outcome from occurring. These summations strongly argue for animal models that better reflect multiple elements of the clinical condition (e.g., varying degrees of axonopathy, including persistent axonal dysfunction in currently surviving neurons) and/or recruiting less severely advanced patients, which may retain better transport and less-pathological antineurotrophic molecular cascades.
AAV/GDNF
The questions raised by the AAV/NRTN program and clinical trials are directly pertinent to this nascent clinical program, for despite differential developmental expression profiles,50 supraphysiological levels of GDNF and NRTN produce virtually indistinguishable in vivo efficacy profiles.51,52 Therefore, given the same testing conditions and dosing parameters, expectations that substituting one protein for the other alone will produce a sufficiently different clinical outcome are most likely to disappoint. The investigators pursuing the AAV/GDNF clinical trial could perhaps be adopting a look-see approach by improving upon key aspects of past studies (namely AAV/NRTN). As such, one strategy includes expansion upon putaminal coverage volume via more aggressive CED. This strategy follows the “improved delivery” logic, and is thus prone to the same pitfalls as many past efforts in PD GT. Simply increasing putaminal coverage of GDNF as a sole means to enhance efficacy in the clinic ignores the concerns of compromised axonal transport, for instance. It also overlooks concerns of α-synuclein accumulation on dampening GDNF signaling.47,48,49 Either or both of these factors may account for the higher responsivity in earlier versus later-stage subjects in the second AAV/NRTN phase 2 study39 and are not apparently being accounted for in the nascent AAV2/GDNF protocol. Thus, the AAV/GDNF study in many ways mirrors the past AAV/NRTN studies (but without clear attempts to circumvent transport deficiencies by directly targeting the SN). Thus, without significant further innovation, we worry that this latest clinical endeavor may be relying too heavily on incremental delivery enhancement as the primary means to achieve a substantially superior clinical outcome.
How Far Have We Come?
Now a full decade after the first clinical testing of PD GT began, the disappointments can overshadow the successes. Yet, the collective successes are both important and concrete, and are precisely what the next decade of PD GT research will move forward from: safety, controllability, and quality manufacturing.
Safety of CNS GT
Just a decade ago, when the first PD GT clinical trial was being launched, safety of CNS GT was the major concern to many investigators and nearly all regulatory agencies, investors, and potential Pharma industry partners. However, over the past 10 years, these issues have largely evaporated, in no small part due to the contributions of the programs reviewed in this paper. Despite numerous concerns on nearly everyone's minds, none of the trials noted any troubling safety problems; the most consistent AEs (adverse events) reported for all the studies were related to the stereotactic surgical procedure and those were similar to what has been accepted as tolerable for deep brain stimulation, a stereotactic surgical procedure for PD long-since approved by European regulatory agencies and the FDA. Conversely, all three phase 2 PD studies observed a substantial placebo response (i.e., improvement from baseline in the sham control group), thus indirectly verifying the safety of stereotactic administration of GT products into the brain.16,38,39,53
The CNS GT approach itself can also be deemed as safe—data from the PD GT trials to date show a remarkably clean safety profile, with none of the problems seen when non-GT methods have been used in an attempt to provide chronic, exogenous bioactive protein (e.g., protein aggregation, loss of bioactivity, induction of neutralizing antibodies, etc.) In fact, the safety record for PD GT collectively appears more favorable than several prior approaches intended to deliver chronic protein for human neurodegenerative diseases (e.g., NGF for Alzheimer's; GDNF and fetal tissue transplantation for PD), in that far fewer serious adverse events have been noted in the GT studies compared to the competing delivery approaches.54,55,56,57,58,59 Another concern in the field a decade ago involved the potential impact of preexisting neutralizing antibodies (nAbs) to viral vectors (e.g., AAV, for which humans can have measurable titers). However, with regard to the brain, nAbs so far appear to be inconsequential towards any local inflammatory reaction, transgene expression, or long-term bioactivity.5 Lastly, the need to regulate protein expression in a safe and effective manner, or even shut down expression completely if serious safety issues arise, was a safety issue raised by select researchers. To date, there have been no adverse effects of chronic expression, and regulatory control appears to be more an incremental enhancement than an absolute necessity. However, as doses and transduction overall are increased through improved targeting (e.g., the reinitiated AAV/AADC trial) and/or vector design, the need for a regulatable vector may reemerge as an important consideration. Thus, GT in the context of PD has proven it to be among the safest approaches developed for long-term, targeted delivery of proteins to the brain—a benchmark that has shifted safety assumptions for the GT field as a whole.
Controlled and predictable bioactive protein expression
The PD clinical trials have also generated convincing evidence that GT can meet its most important objective: providing the means to achieve chronically controlled and predictable bioactive protein expression, targeted to specific brain sites in animals and human patients. In animal studies, clear dose–response relationships have been established in both rats and monkeys52,60,61 demonstrating the ability to control amount of protein production and volume of expression by manipulating vector genome dose. Moreover, long-term, targeted expression of biologically active protein has been achieved following a single administration of the vector in rats for at least 4–5 months for GAD,9 and 20 months for NRTN.62 In nonhuman primates, this has been established for at least 56 weeks for GAD,10 1-year for NRTN,61 6–8 years for AADC.25,35 While human expression data are more limited, long-term, bioactive protein expression has nonetheless been confirmed for over 4 years for both AAV-AADC, using a PET imaging surrogate21 and more directly for AAV-NRTN, demonstrating long-term biologically active protein expression via immunohistochemistry in tissue from postmortem brains of previously treated subjects.39
Manufacturing
Advances in process development and manufacturing have made it possible to produce sufficient current good manufacturing practice) quantities of vector in a cost-effective manner to supply multicenter clinical trials as well as product commercialization; thus, cost-of-goods is no longer the impediment to commercialization that it was viewed to be by many just a decade ago. As described in better detail elsewhere,63 one of the great successes in the past decade has been the scalability of vector production, from the original plated HEK293 cell production without the addition of helper virus,64 to the current suspension culture methods,63 baculovirus expression vector systems (e.g.,),65 HSV-based production systems66 and others, with possible titer generation upwards of 1 × 1014 viral genomes per liter of cells. Coupled with the high purity of ion-exchange chromatography, and current good manufacturing practice realization through vector core facilities, vector production can be fast, reliable, highly pure, and affordable.
Where Do We Go Next?
In sum, the progress achieved over the past decade with the PD GT programs and their clinical trials has created the irony that GT has worked as it was designed but not as intended. That is, despite successfully providing safely targeted and controlled bioactive protein for very long periods of time in both animal and human brain, the efficacy data generated in the PD clinical trials did not demonstrate sufficiently robust or consistent benefits to patients, compared to that achieved by placebo controls.
Is PD the disease to bank our CNS GT expectations on?
A fundamental question that has emerged following completion of the nine PD trials is whether this disease presents too great a challenge to continue to promote as a prototype platform for launching GT as a transformative approach for broadly treating CNS diseases. PD is recognized to be an extremely complex disease involving many different genetic and environmental etiologic and pathologic variables (Table 1), as well as degeneration of many nondopaminergic and nonmotor systems,3,67 for which the cause-effect relationships are incompletely understood, making the success of any novel interventional approach less certain. Even if we focus narrowly on the continued need for better treatment for the motor impairments, significant practical challenges complicate that endeavor. Some of the most grievous challenges include the heavy reliance on highly subjective and variable clinical assessment tools, the lack of validated biomarkers or clearly effective/predictive surrogate measures (attempts to use PET imaging notwithstanding), high variability in degree and scope of individual-patient symptoms from day to day and even hour to hour, the large but variable placebo response often seen in PD trials, and concerns regarding the predictive validity of currently used animal models. To this last point, the fact that most of the GT PD program produced clear evidence for robust, positive data in the most widely accepted animal models used for PD and yet failed to replicate the magnitude or consistency of these effects in PD patients, necessarily raises a question of whether the predictive validity of these models is sufficient for establishing nonclinical proof of concept (see Table 1).
These difficulties in mind, would the CNS GT community find greater opportunity for growth and success in focusing on alternative diseases? Finding a model disease with which to launch a sea change in CNS GT has been notoriously challenging. Some of the ongoing CNS GT efforts include lysosomal storage diseases, leukodystrophies, amyotrophic lateral sclerosis, Alzheimer's, epilepsies, and cancers. A cursory evaluation of these diseases and their treatment hints at both unique and familiar challenges. A key divergent challenge has been vector targeting issues: some CNS diseases, including lysosomal storage diseases and leukodystrophies warrant nonneuronal cell transduction and/or global CNS delivery, neither of which has been adequately realized with current technology. Some of the convergent challenges include matching the predictive ability of animal models to clinical success. With amyotrophic lateral sclerosis, for instance, the commonly used SOD1 mouse line has benefited from preclinical successes (e.g.,),68 which translated poorly in humans, where patients in the treatment group actually deteriorated more rapidly.69 Disease complexity is not unfamiliar to the Alzheimer's or epilepsy fields, which may benefit most directly from the specific growing pains and lessons of PD GT.
Improving our odds of success
Are we overlooking accessible means of reaching heightened clinical success with PD GT? Two of the major concerns highlighted in the PD GT trials included weak transduction and likely protein product transport issues in the pathological brain tissue. As discussed earlier, in addition to improved delivery technologies such as CED, there are foreseeable advantages of alternating the vector serotype or using engineered vectors to improve transduction directly, rather than resorting simply to higher vector dosing and infusion volumes. The perceived challenge here is in the resources required to obtain regulatory approval for using unique viral entities, whereas this hurdle has long been cleared for AAV2. Recent discussions to streamline the regulatory process for newly developed vectors are formalizing and hopefully may clear a path for improved vector usage in the clinic.70 Regarding the challenges of the pathology itself, if the degenerative state were a major obstacle for therapy, would earlier treatment not be more efficient and potentially successful? A recent exploratory analysis of the AAV2-NRTN phase 2b data suggests so: in the study earlier-stage PD subjects (defined here as those 5 or fewer years postdiagnosis) showed much greater clinical benefit from AAV-NRTN than did those treated at 10 or more years postdiagnosis.39 This elevates the age-old issue in the neurodegeneration field, particularly with regard to neurotrophic factor intervention, regarding whether earlier-stage patients might indeed respond far better and more reliably than those currently enrolled in experimental treatments. Thus, two questions emerge that will command significant research and regulatory attention going forward: how quickly dying neurons reach an irreversibly degenerated state,71 and how early in the disease process experimental treatments will be warranted in patient volunteers.39 Progress in clinical biomarker research (such as that recently described)72 will undoubtedly shape the earlier intervention efforts and may even help establish surrogate endpoints to permit a more biometrically equilibrated scoring system (Table 3).
Table 3. Resolving key obstacles to facilitate new Parkinson's disease therapies.

Seeking alternative animal models
In light of the consistent disconnect between preclinical and clinical successes in PD GT, a step back from the clinic might reenvision the problem of delivery to one of predictive power of animal models. The overreliance on acute toxin-based PD models (e.g., the everpopular 6-OHDA and MPTP models) limits our exploration of treatment options and models that encompass more of the pathogenic variables (Table 1) with less focus on limited pathologic or behavioral changes may yield both conventional and biomolecular alternatives. One paradigm gaining momentum is that focusing on the pathological sequelae of α-synuclein overexpression (aggregation of which is a key pathogenic event in PD), which can result in the death of the mid-brain dopamine neurons affected in PD.47,48,73,74,75,76 Perhaps most valuable with regard to GT shortcomings to date is the observation that overexpression of α-synuclein in rats leads to axonal transport deficits and gradual dysfunction of dopamine processes prior to frank cell death, a process akin to that described in PD.73 Moreover, these increased levels of α-synuclein can inhibit the ability of GDNF to protect degenerating dopamine neurons, ostensibly through downregulation of the transcription factor Nurr1 and its downstream target, RET—the GDNF receptor.47,48,75 By inference, such findings provide a putative basis respectively for challenges of retrograde GT transport and neurotrophic factor bioactivity. This model involving overexpression of α-synuclein is still far from fully characterized and is certain to be an incomplete representation of the disease. It is already clear that the protein overexpression required to induce the PD-like pathology far surpasses human pathological levels, while the focus on a singular molecular moiety recapitulates a strategy that has ensnared the Alzheimer's field for years (i.e., β-amyloid transgenics). Nonetheless, progress at the preclinical level, even discoveries well within reach, may provide robust opportunities for substantially increasing the momentum for research and development in GT.
Reaching across the aisle
From regulatory hurdles, to endpoints and even overarching trial design, the conventional pharmaceutical approach to treating PD might seem fully distinct from GT efforts, but yet a solution may come from aligning our strengths—achieving a sum greater than the parts. Where the conventional and GT-based approaches have already been linked to some extent through the prodrug strategy (i.e., AADC—enhancing the therapeutic benefit of oral levodopa treatment) this complementation could seemingly extend beyond the prodrug scenario; as examples, α-synuclein aggregation inhibitors could improve retrograde vector transport, supporting superior nigrostriatal transduction, or Nurr1 agonists could potentiate the effects of neurotrophic factors like NRTN and GDNF. A further envisioning of GT for use in conjunction other nonconventional therapies, such as deep brain stimulation, seems reasonable at a regulatory level (perhaps requiring little additional surgical intervention to achieve both vector delivery and electrode implantation), and might be an outside strategy for achieving the next critical victory in PD GT.
What can we learn from the history of novel therapeutics?
The history surrounding the development of monoclonal antibodies (mAb) into the powerful biopharmaceuticals they have now become provides a comparative perspective regarding the current status of PD GT. With the early fervor over mAbs in the 1980s to early 90s, one company (Janssen Biotech, Horsham, Pennsylvania) spent years of intense effort and hundreds of millions of dollars77 developing its lead mAb product, nebacumab or Centoxin, to treat sepsis. However, following promising data in animal models and even some approvals in Europe for other indications, a pivotal trial failed miserably.78 Centocor's first mAb approval (infliximab; Remicade), initially for Crohn's disease and later for rheumatoid arthritis, was not achieved until 6 years after the Centoxin/sepsis failure, but now enjoys extremely high success both clinically and financially,79 and is just one of nearly 30 different mAb drugs contributing to one of the fastest growing therapeutic areas in biotech.80 This example, and many others involving similarly novel, transformational biomedical technologies (e.g., antisense technology,81 informs us that the types of disappointments seen with GT for PD can absolutely precede major successes, but that such an about-face requires a renewed approach, avoiding the same miscalculations made previously.
Bearing in mind the clear differences between CNS and peripheral targets for GT, the successes in a sister program—namely hemophilia B—should still serve as a valuable model for PD GT researchers. Here, the look-see effect that has proven something of an impediment to PD innovation has been the mainstay component that has allowed for gradual, real progress. To distill a tremendous amount of work for the purposes of example, hemophilia GT saw major preclinical successes, such as AAV-mediated recovery of clotting function in Factor IX (FIX)-deficient dogs,82,83 lead to an unanticipated lack of secreted protein product in humans,84 followed by a shift in preclinical vector targeting from muscle to liver, which, when returned to the clinic led to a short-lived therapeutic benefit,85 again not anticipated from the animal models. With the advent of a higher efficacy vector (i.e., self-complementary AAV),86 codon-optimized genes,87 and testing of more efficacious vectors (i.e., AAV8 for improved liver transduction),87 a recent clinical trial benefited by long-term (albeit low) gene delivery.88 Now using a further optimized point mutant of FIX with sevenfold higher activity, a clinical trial is ongoing.89 Of course, the monogenic nature of hemophilia B, versus the far more complex etiology of PD, has narrowed the prior field's efforts toward improving delivery and persistence, largely avoiding the key challenge of transgene appropriateness. Still, even in the hemophilia GT arena, there clearly continues to be a need for iteration and continuity between bench and clinic, and this serves as proof that the look-see approach can work with perseverance, further innovation and continuing resources.
Parting Thoughts
Without proper reflection, the generally disappointing efficacy data generated in the PD GT clinical trials might easily overshadow the significant progress that has been achieved in these same studies. Over the course of translating animal studies into clinic trials, these programs helped transform the entire field of GT. They established that GT could be accurately targeted to the brain in a safe and effective manner, that the viral vector is able to induce neurons to produce controlled and predictable protein for years, and that biological responses can be induced in degenerating neuronal systems in human brains. While it is true that the clinical efficacy achieved to date is not nearly as robust or reliable as required, it is also true that each of the three completed controlled studies has nonetheless produced some evidence for a clinical benefit exceeding that achieved by sham procedure. The fact that the GT approaches to PD worked as designed but did not achieve the magnitude of clinical benefit intended casts caution towards the hurried repetition of modestly-altered procedures, and may warrant a retargeting of the PD GT trajectory, starting back at the laboratory bench. Solid victories, akin to those won over safety and longevity concerns, are the key to continued momentum. Given the therapeutic power that biopharmaceuticals are beginning to show and the value that GT has yielded with regards to its ability to provide safe and effective delivery of proteins, with cautious continuity it is not hard to imagine that GT will be an important contributor and benefactor of more effective treatments for PD and other CNS diseases. In the meantime, questions will and should be raised about the best ways to move forward. For PD specifically, a near-term focus on developing more effective tools for measuring motor performance in people with PD would likely provide many tangible benefits for trial design and execution, as would the development of biomarkers and surrogate endpoints. Moreover, the development of more sophisticated animal models that focus on key pathogenic events rather than symptoms or univariate end-stage pathologies will likely yield improvements in these models' predictive value for GT intervention.
The glass is half full. Attending to these important considerations and implementing the lessons of our past we can reach the success we all envision, and advance the field ever forward.
Acknowledgments
This work was supported by National Institutes of Health Grants 2R01AI072176 and 5R01AI080726 (R.J.S.) and F32NS070356 (M.S.W).
References
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72 suppl. 4:S1–136. doi: 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- Bonnet AM, Jutras MF, Czernecki V, Corvol JC, Vidailhet M. Nonmotor symptoms in Parkinson's disease in 2012: relevant clinical aspects. Parkinsons Dis. 2012;2012:198316. doi: 10.1155/2012/198316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Parkinson's Disease Foundation (2013). Statistics on Parkinson's. http://www.pdf.org/en/parkinson_statistics .
- Bartus RT. Translating the therapeutic potential of neurotrophic factors to clinical ‘proof of concept': a personal saga achieving a career-long quest. Neurobiol Dis. 2012;48:153–178. doi: 10.1016/j.nbd.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Szerlip NJ, Walbridge S, Yang L, Morrison PF, Degen JW, Jarrell ST, et al. Real-time imaging of convection-enhanced delivery of viruses and virus-sized particles. J Neurosurg. 2007;107:560–567. doi: 10.3171/JNS-07/09/0560. [DOI] [PubMed] [Google Scholar]
- Fiandaca MS, Varenika V, Eberling J, McKnight T, Bringas J, Pivirotto P, et al. Real-time MR imaging of adeno-associated viral vector delivery to the primate brain. Neuroimage. 2009;47 suppl. 2:T27–T35. doi: 10.1016/j.neuroimage.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankiewicz KS, Eberling JL, Kohutnicka M, Jagust W, Pivirotto P, Bringas J, et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164:2–14. doi: 10.1006/exnr.2000.7408. [DOI] [PubMed] [Google Scholar]
- Luo J, Kaplitt MG, Fitzsimons HL, Zuzga DS, Liu Y, Oshinsky ML, et al. Subthalamic GAD gene therapy in a Parkinson's disease rat model. Science. 2002;298:425–429. doi: 10.1126/science.1074549. [DOI] [PubMed] [Google Scholar]
- Emborg ME, Carbon M, Holden JE, During MJ, Ma Y, Tang C, et al. Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J Cereb Blood Flow Metab. 2007;27:501–509. doi: 10.1038/sj.jcbfm.9600364. [DOI] [PubMed] [Google Scholar]
- Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M, et al. Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci Transl Med. 2009;1:2ra4. doi: 10.1126/scitranslmed.3000130. [DOI] [PubMed] [Google Scholar]
- Aron L, Klein R. Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci. 2011;34:88–100. doi: 10.1016/j.tins.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Peterson AL, Nutt JG. Treatment of Parkinson's disease with trophic factors. Neurotherapeutics. 2008;5:270–280. doi: 10.1016/j.nurt.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kells AP, Eberling J, Su X, Pivirotto P, Bringas J, Hadaczek P, et al. Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci. 2010;30:9567–9577. doi: 10.1523/JNEUROSCI.0942-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, et al. AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011;10:309–319. doi: 10.1016/S1474-4422(11)70039-4. [DOI] [PubMed] [Google Scholar]
- Coune PG, Schneider BL, Aebischer P. Parkinson's disease: gene therapies. Cold Spring Harb Perspect Med. 2012;2:a009431. doi: 10.1101/cshperspect.a009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Kells AP, Huang EJ, Lee HS, Hadaczek P, Beyer J, et al. Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum Gene Ther. 2009;20:1627–1640. doi: 10.1089/hum.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce G. A phase 1 open-label safety study of intrastriatal infusion of adeno-associated virus encoding human aromatic L-amino acid decarboxylase (AAV-hAADC-2) in subjects with advanced parkinson's disease [AAV-hAADC-2-003]. NIH Recombinant DNA Advisory Committee-Appendix M. 2003.
- Avigen Genzyme Acquires Significant Portion of Avigen's Gene Therapy Technology. 21 December 2005 Press Release 2005.
- Mittermeyer G, Christine CW, Rosenbluth KH, Baker SL, Starr P, Larson P, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson's disease. Hum Gene Ther. 2012;23:377–381. doi: 10.1089/hum.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009;73:1662–1669. doi: 10.1212/WNL.0b013e3181c29356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankiewicz KS, Forsayeth J, Eberling JL, Sanchez-Pernaute R, Pivirotto P, Bringas J, et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther. 2006;14:564–570. doi: 10.1016/j.ymthe.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Forsayeth JR, Eberling JL, Sanftner LM, Zhen Z, Pivirotto P, Bringas J, et al. A dose-ranging study of AAV-hAADC therapy in Parkinsonian monkeys. Mol Ther. 2006;14:571–577. doi: 10.1016/j.ymthe.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadaczek P, Eberling JL, Pivirotto P, Bringas J, Forsayeth J, Bankiewicz KS. Eight years of clinical improvement in MPTP-lesioned primates after gene therapy with AAV2-hAADC. Mol Ther. 2010;18:1458–1461. doi: 10.1038/mt.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund T, Kordower JH. Gene therapy for Parkinson's disease. Mov Disord. 2010;25 suppl. 1:S161–S173. doi: 10.1002/mds.22785. [DOI] [PubMed] [Google Scholar]
- Herpich N.2013Novel partnership between academia and industry drives forward gene therapy approach to treat Parkinson's. FoxFeed Blog: http://www.michaeljfox.org/foundation/news-detail.php?novel-partnership-between-academia-and-industry-drives-forward-gene-therapy-approach-to-treat-parkinson .
- Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson's disease. Mol Ther. 2010;18:1731–1735. doi: 10.1038/mt.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer R, Douglas MR. Gene therapy for Parkinson's disease. Parkinsons Dis. 2012;2012:757305. doi: 10.1155/2012/757305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwu WL, Muramatsu S, Tseng SH, Tzen KY, Lee NC, Chien YH, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med. 2012;4:134ra61. doi: 10.1126/scitranslmed.3003640. [DOI] [PubMed] [Google Scholar]
- Markakis EA, Vives KP, Bober J, Leichtle S, Leranth C, Beecham J, et al. Comparative transduction efficiency of AAV vector serotypes 1-6 in the substantia nigra and striatum of the primate brain. Mol Ther. 2010;18:588–593. doi: 10.1038/mt.2009.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg MS, Samulski RJ, McCown TJ. Adeno-associated virus (AAV) gene therapy for neurological disease. Neuropharmacology. 2013;69:82–88. doi: 10.1016/j.neuropharm.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TC, Dickinson PJ, Roberts BN, Yendluri S, Gonzalez-Edick M, Lecouteur RA, et al. Enhanced gene transfer efficiency in the murine striatum and an orthotopic glioblastoma tumor model, using AAV-7- and AAV-8-pseudotyped vectors. Hum Gene Ther. 2006;17:807–820. doi: 10.1089/hum.2006.17.807. [DOI] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol Ther. 2008;16:89–96. doi: 10.1038/sj.mt.6300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankiewicz KS, Daadi M, Pivirotto P, Bringas J, Sanftner L, Cunningham J, et al. Focal striatal dopamine may potentiate dyskinesias in parkinsonian monkeys. Exp Neurol. 2006;197:363–372. doi: 10.1016/j.expneurol.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Brown L, Wilson A, Kruegel B, Siffert J, Johnson EM, Jr, et al. Properly scaled and targeted AAV2-NRTN (neurturin) to the substantia nigra is safe, effective and causes no weight loss: support for nigral targeting in Parkinson's disease. Neurobiol Dis. 2011;44:38–52. doi: 10.1016/j.nbd.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Bartus R, Baumann TL, Siffert J, Herzog CD, Alterman R, Boulis N, et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson's patients. Neurology. 2013;80:1698–1702. doi: 10.1212/WNL.0b013e3182904faa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks WJ, Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, et al. Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2010;9:1164–1172. doi: 10.1016/S1474-4422(10)70254-4. [DOI] [PubMed] [Google Scholar]
- Bartus R.2013CERE-120 (AAV-neurturin) for the Treatment of Parkinson's Disease: Experience from 4 Clinical Trials and Human Autopsy Data.American Society of Gene and Cell Therapy 16th Annual Meeting: Salt Palace Convention Center in Salt Lake City, Utah, USA.
- Marks WJ, Jr, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol. 2008;7:400–408. doi: 10.1016/S1474-4422(08)70065-6. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Baumann TL, Brown L, Kruegel BR, Ostrove JM, Herzog CD. Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: developing and demonstrating “clinical proof-of-concept” for AAV-neurturin (CERE-120) in Parkinson's disease. Neurobiol Aging. 2013;34:35–61. doi: 10.1016/j.neurobiolaging.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Brown L, Kruegel BR, Wilson A, Tansey MG, Gage FH, et al. Enhanced neurotrophic distribution, cell signaling and neuroprotection following substantia nigral versus striatal delivery of AAV2-NRTN (CERE-120). Neurobiol Dis. 2013;58:38–48. doi: 10.1016/j.nbd.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Brown L, Chu Y, Baumann T, Kordower JH, Bartus RT. Society for Neuroscience; New Orleans, LA; 2012. Robust, stable, targeted, long-term neurturin expression and enhanced tyosine hyroxylase labeling in Parkinson's disease brain 4 years following delivery of CERE-120 (AAV2-neurturin) to the human putamen. [Google Scholar]
- Bartus RT, Herzog CD, Chu Y, Wilson A, Brown L, Siffert J, et al. Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson's disease and nonhuman primate brains. Mov Disord. 2011;26:27–36. doi: 10.1002/mds.23442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Herzog CD, Dass B, Bakay RA, Stansell J, 3rd, Gasmi M, et al. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006;60:706–715. doi: 10.1002/ana.21032. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Dass B, Holden JE, Stansell J, 3rd, Gasmi M, Tuszynski MH, et al. Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov Disord. 2007;22:1124–1132. doi: 10.1002/mds.21503. [DOI] [PubMed] [Google Scholar]
- Decressac M, Ulusoy A, Mattsson B, Georgievska B, Romero-Ramos M, Kirik D, et al. GDNF fails to exert neuroprotection in a rat a-synuclein model of Parkinson's disease. Brain. 2011;134 Pt 8:2302–2311. doi: 10.1093/brain/awr149. [DOI] [PubMed] [Google Scholar]
- Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A. a-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med. 2012;4:163ra156. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Déglon N, Pralong W, Aebischer P. Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson's disease. Neurobiol Dis. 2004;17:283–289. doi: 10.1016/j.nbd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Akerud P, Alberch J, Eketjäll S, Wagner J, Arenas E. Differential effects of glial cell line-derived neurotrophic factor and neurturin on developing and adult substantia nigra dopaminergic neurons. J Neurochem. 1999;73:70–78. doi: 10.1046/j.1471-4159.1999.0730070.x. [DOI] [PubMed] [Google Scholar]
- Rosenblad C, Kirik D, Devaux B, Moffat B, Phillips HS, Björklund A. Protection and regeneration of nigral dopaminergic neurons by neurturin or GDNF in a partial lesion model of Parkinson's disease after administration into the striatum or the lateral ventricle. Eur J Neurosci. 1999;11:1554–1566. doi: 10.1046/j.1460-9568.1999.00566.x. [DOI] [PubMed] [Google Scholar]
- Gasmi M, Brandon EP, Herzog CD, Wilson A, Bishop KM, Hofer EK, et al. AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson's disease. Neurobiol Dis. 2007;27:67–76. doi: 10.1016/j.nbd.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Ceregene I.2013Ceregene reports data from Parkinson's disease phase 2b study. http://www.ceregene.com/press_041913.asp .
- Eriksdotter Jönhagen M, Nordberg A, Amberla K, Bäckman L, Ebendal T, Meyerson B, et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 1998;9:246–257. doi: 10.1159/000017069. [DOI] [PubMed] [Google Scholar]
- Nauta HJ, Wehman JC, Koliatsos VE, Terrell MA, Chung K. Intraventricular infusion of nerve growth factor as the cause of sympathetic fiber sprouting in sensory ganglia. J Neurosurg. 1999;91:447–453. doi: 10.3171/jns.1999.91.3.0447. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Burchiel KJ, Comella CL, Jankovic J, Lang AE, Laws ER, Jr, et al. ICV GDNF Study Group. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69–73. doi: 10.1212/wnl.60.1.69. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann Neurol. 2003;54:403–414. doi: 10.1002/ana.10720. [DOI] [PubMed] [Google Scholar]
- Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59:459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- Hovland DN, Jr, Boyd RB, Butt MT, Engelhardt JA, Moxness MS, Ma MH, et al. Six-month continuous intraputamenal infusion toxicity study of recombinant methionyl human glial cell line-derived neurotrophic factor (r-metHuGDNF in rhesus monkeys. Toxicol Pathol. 2007;35:1013–1029. doi: 10.1177/01926230701481899. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Dass B, Gasmi M, Bakay R, Stansell JE, Tuszynski M, et al. Transgene expression, bioactivity, and safety of CERE-120 (AAV2-neurturin) following delivery to the monkey striatum. Mol Ther. 2008;16:1737–1744. doi: 10.1038/mt.2008.170. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Brown L, Gammon D, Kruegel B, Lin R, Wilson A, et al. Expression, bioactivity, and safety 1 year after adeno-associated viral vector type 2-mediated delivery of neurturin to the monkey nigrostriatal system support cere-120 for Parkinson's disease. Neurosurgery. 2009;64:602–12; discussion 612. doi: 10.1227/01.NEU.0000340682.06068.01. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Bishop K, Brown L, Wilson A, Kordower JH, Bartus RT. Gene transfer provides a practical means for safe, long-term, targeted delivery of biologically-active neurotrophic factor proteins for neurodegenerative diseases. Drug Deliv and Transl Res. 2011;1:361–382. doi: 10.1007/s13346-011-0037-z. [DOI] [PubMed] [Google Scholar]
- Grieger JC, Samulski RJ. Adeno-associated virus vectorology, manufacturing, and clinical applications. Meth Enzymol. 2012;507:229–254. doi: 10.1016/B978-0-12-386509-0.00012-0. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidi G, Lamb K, Zolotukhin S. An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc Natl Acad Sci USA. 2009;106:5059–5064. doi: 10.1073/pnas.0810614106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther. 2009;20:796–806. doi: 10.1089/hum.2009.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Obeso JA. Challenges in Parkinson's disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004;3:309–316. doi: 10.1016/S1474-4422(04)00740-9. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Western ALS Study Group Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- O'Reilly M, Kohn DB, Bartlett J, Benson J, Brooks PJ, Byrne BJ, et al. Gene therapy for rare diseases: summary of a national institutes of health workshop, september 13, 2012. Hum Gene Ther. 2013;24:355–362. doi: 10.1089/hum.2013.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136 Pt 8:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Irwin DJ, Chen-Plotkin AS, Siderowf A, Caspell C, Coffey CS, et al. Association of cerebrospinal fluid beta-amyloid 1-42, T-tau, P-tau181, and alpha-Synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 2013. [DOI] [PMC free article] [PubMed]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci USA. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centocor Inc H. Centocor Inc History. vol. 14. St. James Press: International Directory of Company Histories 1996.
- McCloskey RV, Straube RC, Sanders C, Smith SM, Smith CR. Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann Intern Med. 1994;121:1–5. doi: 10.7326/0003-4819-121-1-199407010-00001. [DOI] [PubMed] [Google Scholar]
- Marks L. The birth pangs of monoclonal antibody therapeutics: the failure and legacy of Centoxin. MAbs. 2012;4:403–412. doi: 10.4161/mabs.19909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z.2009. Therapeutic Monoclonal Antibodies: From bench to clinic: Hoboken, New Jersey.
- Jones D. The long march of antisense. Nat Rev Drug Discov. 2011;10:401–402. doi: 10.1038/nrd3474. [DOI] [PubMed] [Google Scholar]
- Mount JD, Herzog RW, Tillson DM, Goodman SA, Robinson N, McCleland ML, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99:2670–2676. doi: 10.1182/blood.v99.8.2670. [DOI] [PubMed] [Google Scholar]
- Wang L, Nichols TC, Read MS, Bellinger DA, Verma IM. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. 2000;1:154–158. doi: 10.1006/mthe.2000.0031. [DOI] [PubMed] [Google Scholar]
- Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- Wu Z, Sun J, Zhang T, Yin C, Yin F, Van Dyke T, et al. Optimization of self-complementary AAV vectors for liver-directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280–289. doi: 10.1038/sj.mt.6300355. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan PE, Walsh C, Powell J, Gomperts E, Samulski RJ, Mcphee S. A phase 1/2 open-label trial of an adeno-associated virus (AAV) serotype 8 factor IX gene therapy (AskBio009) in adults with hemophilia B. Haemophilia. 2013;19:468–469. [Google Scholar]
- Lonser RR. A phase 1 open-label dose escalation safety study of convection-enhanced delivery (CED) of adeno-associated virus encoding glial cell line-derived neurotrophic factor (AAV2-GDNF) in subjects with advanced Parkinson's disease. Recombinant DNA Advisory Committee Protocol. 2009. pp. 0901–962.
- Palfi S, Gurruchaga JM, Ralph GS, Lepetit H, Lavisse S, Buttery PC.2014Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy of Parkinson's disease: a dose escalation, open-label, phase 1/2 trial. The Lancet , http://dx.doi.org/10.1016/S0140-6736(13)61939-X . [DOI] [PubMed]