

Abstract
The blood–brain barrier (BBB) presents a major challenge to effective treatment of neurological disorders, including lysosomal storage diseases (LSDs), which frequently present with life-shortening and untreatable neurodegeneration. There is considerable interest in methods for intravenous delivery of lysosomal proteins across the BBB but for the most part, levels achievable in the brain of mouse models are modest and increased lifespan remains to be demonstrated. In this study, we have investigated delivery across the BBB using a mouse model of late-infantile neuronal ceroid lipofuscinosis (LINCL), a neurodegenerative LSD caused by loss of tripeptidyl peptidase I (TPP1). We have achieved supraphysiological levels of TPP1 throughout the brain of LINCL mice by intravenous (IV) coadministration of recombinant TPP1 with a 36-residue peptide that contains polylysine and a low-density lipoprotein receptor binding sequence from apolipoprotein E. Importantly, IV administration of TPP1 with the peptide significantly reduces brain lysosomal storage, increases lifespan and improves neurological function. This simple “mix and inject” method is immediately applicable towards evaluation of enzyme replacement therapy to the brain in preclinical models and further exploration of its clinical potential is warranted.
Introduction
Late-infantile neuronal ceroid lipofuscinosis (LINCL) is a fatal, incurable lysosomal storage disease (LSD) of children caused by deficiencies in a lysosomal protease, tripeptidyl peptidase I (TPP1).1 The cellular hallmark of LINCL is an accumulation of a fluorescent storage material in lysosomes of affected individuals, the major component of which is subunit c of mitochondrial ATP synthase (SCMAS).2 Manifestations include seizures, loss of vision and locomotor function, progressive mental decline and premature death, typically at less than 15 years. The incidence of LINCL is ~0.3 per 100,000 live births3,4 and with an average patient lifespan of ~10 years, there are thousands of patients worldwide.
For LSDs in general, enzyme replacement therapy (ERT) is the most successful treatment5,6 and involves administration of a recombinant enzyme into the bloodstream to correct the respective deficiency. The major current limitation of ERT is that the blood–brain barrier (BBB) prevents the passage of therapeutic protein into the CNS. Thus, ERT is currently restricted to treating nonneurological manifestations of LSDs. Given that most LSDs feature neurological deterioration, there is demand for breakthroughs to meet the challenge of the BBB.7
The BBB can be bypassed by administration via the cerebrospinal fluid8,9 and this has shown promise for LINCL.10,11,12 However, cerebrospinal fluid delivery is invasive and may not be suitable for a life-long therapy. There are also concerns13 that widespread distribution throughout the human brain may be difficult given that the cerebrospinal fluid–brain interface is anatomically limited, requiring significant diffusion of a therapeutic for complete coverage.
Intravenous (IV) administration is relatively noninvasive and could potentially achieve widespread distribution throughout the brain given that each cell is in close proximity to the microvasculature. Thus, there are strategies for IV delivery of proteins across the BBB13,14 including permeabilization of the BBB,15 use of molecular Trojan horses in which receptor-binding domains from apolipoproteins are used to construct chimeric enzymes,16,17 and high-dose administration of unmodified or glycan-modified proteins.18,19,20,21 For LSDs, promising progress is being made but to date, only fractional levels, ranging from ~1.5 to 12%, of normal activity, have been achieved in the brain of mouse models.16,17,18,19,20,22,23 Critically, a clear demonstration of therapeutic benefit in terms of extension of lifespan remains to be demonstrated using these approaches. Moreover, methods to promote passage across the BBB require substantial optimization, and may not be universally applicable to all proteins.14,19,21
We have recently described a peptide that can deliver target proteins across the BBB in a “mix and inject” strategy.24 Intriguingly, this peptide works in trans and thus does not require covalent linkage with its target protein. Here, we demonstrate that this approach can achieve supraphysiological levels of TPP1 in the brain after IV administration to an LINCL mouse model. Moreover, we demonstrate that peptide-mediated delivery of TPP1 reduces brain lysosomal storage, ameliorates neurological deficits and significantly extends lifespan of the LINCL mouse.
Results
TPP1-ApoE chimeras are not functional
Fusion of lysosomal proteins with receptor-binding domains from apolipoproteins can promote their passage across the BBB.16,17 Using a similar approach, we constructed chimeras with apolipoprotein E (ApoE)-receptor binding sequence inserted between the signal sequence and prodomain of TPP1 or fused to the C-terminus of the preproprotein (Supplementary Figure S1). TPP1 activity and expression were measured in transiently transfected Chinese hamster ovary (CHO) cells (Supplementary Figure S1). The construct with an N-terminal insertion of ApoE was not expressed. The construct with a C-terminal fusion of ApoE was expressed but was inactive and not processed to the mature form. These results suggest that, as engineered in our current set of fusion proteins, the ApoE sequence interferes with TPP1 folding and/or processing.
Effective delivery from the bloodstream to the brain using a trans-acting peptide mediator
A peptide (K16ApoE) comprising 16 lysines followed by 20 amino acids corresponding to a low-density lipoprotein receptor binding sequence from human ApoE (residues 151–170) can facilitate delivery of IV administered IgG, IgM, and E. coli β-galactosidase across the BBB.24 This approach differs from others in that, rather than functioning as part of a fusion protein, K16ApoE acts in trans in mediating delivery of target proteins across the BBB. We investigated whether coinjection of K16ApoE with TPP1 could promote uptake of the latter into the brain of the LINCL mouse.
Using a minimally invasive IV administration route via the tail vein, we conducted dose-response studies in which either low (2 nmol = 0.12 mg; 6 mg/kg) or high (17 nmol = 1 mg; 50 mg/kg) amounts of TPP1 were coinjected with variable amounts of K16ApoE using a total volume of 120 µl. Activity in the brain was dependent upon dose of TPP1 and peptide (Figure 1a,b; circles). Uptake of 2 nmol TPP1 increased with peptide dose, reaching a plateau corresponding to ~20% of normal level. Greater amounts of TPP1 in the brain were achieved using 17 nmol TPP1 and again, uptake increased with peptide up to the highest amount of peptide tested (24 nmol = 0.11 mg; 5 mg/kg). For both doses of TPP1, animals showed signs of toxicity with increasing amounts of K16ApoE. At lower peptide amounts, this typically manifested as lethargy with recovery by the next day, and at higher amounts, death was immediate. Similar toxicity was found with peptide in the absence of TPP1 (Figure 1, legend).
Figure 1.

Dose-response analysis of K16ApoE and tripeptidyl peptidase I (TPP1) mixtures. Six-week-old late-infantile neuronal ceroid lipofuscinosis (LINCL) mice were administered a 120 µl bolus dose of a mixture of the indicated amount of TPP1 and either K16ApoE (circles) or protamine sulfate (filled squares). (a,b) Delivery of TPP1 to the brain. Mice were killed 24 hours after injection and TPP1 activity was assayed in brain. Results were expressed as the percentage of wild-type specific activity after correction for background using untreated LINCL mice. (c,d) Response of animals after administration of indicated dose of TPP1 and K16ApoE was plotted. Note that acute treatment of LINCL mice with 42 nmol K16ApoE in the absence of TPP1 resulted in death (n = 4). Mice administered protamine sulfate did not exhibit any adverse effects.
We also conducted experiments with protamine sulfate, a highly basic polypeptide that has been reported to facilitate passage of proteins across the BBB.25,26 Protamine had no measurable effect in promoting delivery of TPP1 to the brain (Figure 1b, filled squares) and even at the highest amounts tested, had no apparent toxicity.
Therapeutic index is dependent upon peptide administration method
We investigated whether modification of the method of administration could improve delivery of TPP1 to the brain and/or reduce toxic side effects. TPP1 was fixed at 17 nmol for all experiments. A dose of TPP1 with 48 nmol K16ApoE was typically fatal when administered in a 120 µl bolus injection (Figure 1), but was well tolerated as a slow infusion (Figure 2a). Uptake was essentially the same whether the dose was delivered over 3.75 or 15 minutes. Delivery of TPP1 to the brain with slow infusion was proportional to the amount of K16ApoE administered, with a maximum of ~400% of wild-type TPP1 activity in brain with 48 nmol peptide (Figure 2b, circles). This corresponds to delivery of >1% of the administered TPP1 to the brain.
Figure 2.

Tripeptidyl peptidase I (TPP1) uptake in brain with modified administration methods. All experiments were conducted in 6-week-old late-infantile neuronal ceroid lipofuscinosis (LINCL) mice using 17 nmol TPP1, and activity was assayed in brain 24 hours following administration of enzyme. (a) TPP1 and K16ApoE (48 nmol) in 120 µl was administered over the indicated infusion times. Administration over a 1.9-minute interval was fatal. (b) Open circles: TPP1 was coadministered with the indicated amount of peptide in a total volume of 120 µl using a 3.75-minute infusion time. Administration of 96 nmol K16ApoE was fatal. Open squares: Four consecutive 50 µl bolus injections spaced 2 minutes apart were used to deliver 200 µl of a TPP1 mixture with 48 nmol peptide. Open triangles: TPP1 was coadministered with the indicated amount of peptide as a single bolus injection in a total volume of 200 µl. A dose of 56 nmol peptide was fatal.
We investigated an IV administration method where a mixture of 48 nmol K16ApoE and TPP1 was delivered in four consecutive 50 µl bolus doses that were spaced 2 minutes apart. This was well tolerated and gave equivalent results to the slow infusion (Figure 2b, squares). We also examined the effect of administering single 200 µl bolus doses (Figure 2b, triangles). Mice exhibited lethargy when administered TPP1 with 42 nmol K16ApoE while 28 nmol peptide was well tolerated and resulted in delivery of TPP1 to the brain at levels comparable to the slow infusion or divided dose procedure. Currently, 200 µl bolus doses with 28 nmol peptide appears to be the most convenient and well tolerated method for delivering TPP1 to the brain.
Subacute toxicology of K16ApoE
We conducted a subacute study on both LINCL and wild-type mice to determine potential toxicity associated with repeated dosing. Mice were administered daily bolus doses of a K16ApoE (28 nmol) and TPP1 (17 nmol) mixture in a total volume of 200 µl. Neurological function was assessed using a functional observational battery 10 minutes after each dose and after a ~24 hour recovery period. The dosing period was at least five days and was extended until tail-vein administration was no longer viable due to local trauma. There was minimal or no observed toxicity (Supplementary Figure S2). Histopathological analysis also failed to detect any toxicity in brain, kidney, and liver (Supplementary Figure S3a–c).
Insights into mechanism of action
K16ApoE was originally envisaged24 to noncovalently associate with target proteins via the polylysine sequence and mediate passage by binding to the low-density lipoprotein receptor via the 20 amino acid segment of Apo E. K16 was shown to directly interact with target proteins27 and dynamic light scattering analysis confirms that K16ApoE binds TPP1 in vitro (Figure 3a). Delivery of TPP1 to the brain was found to be similar when K16ApoE was premixed with TPP1 or injected either immediately prior to or after TPP1 administration (Supplementary Figure S4). Experiments where K16ApoE was administered at a series of time intervals before administration of TPP1 revealed that the peptide had a functional half-life of ~20 minutes in terms of promoting delivery of enzyme to the brain (Figure 3b). This may suggest that in vivo, K16ApoE exerts its activity by multiple mechanisms. For instance, it is possible that K16ApoE transiently opens the BBB with repair within a relatively short time-frame, as suggested previously,24 and/or associates with proteins in the plasma, promoting transcytosis.
Figure 3.

Potential interactions of K16ApoE and tripeptidyl peptidase I (TPP1). (a) Dynamic light scattering. Particle diameters are measured for TPP1 (80 µl) and K16ApoE alone, or mixed in ratios of 1:1 or 1:5 respectively. (b) TPP1 (8 nmol in 100 µl) was administered to 6-week-old late-infantile neuronal ceroid lipofuscinosis (LINCL) mice at the indicated times after K16ApoE (14 nmol in 100 µl) (open circles). A TPP1 alone control (no peptide) is also shown (filled squares). Animals were killed 24 hours after injection and TPP1 activity was assayed in brain.
Reduction of storage material in brain
LINCL mice exhibit progressive lysosomal storage over time.28 To determine if K16ApoE-mediated delivery of TPP1 could reduce or reverse storage in brain, mice were treated with three weekly doses initiated at age 13 weeks and killed 1 week after the final dose. Western blotting was used to analyze levels of insoluble SCMAS brain storage material in the 16-week-old treated animals and in 13- and 16-week-old untreated LINCL and wild-type controls (Figure 4). The treatment reduced levels below that of the untreated 13-week LINCL controls, although not to the extent of wild-type animals. Immunohistochemical analysis also revealed that treatment diminished SCMAS staining throughout the brain (Supplementary Figure S5). Taken together, this indicates that the treatment not only prevents additional storage, but to some degree, reduces the levels of preexisting storage material.
Figure 4.

Effect of K16ApoE-mediated tripeptidyl peptidase I (TPP1) delivery on SCMAS storage in brain. Starting at 13 weeks of age, late-infantile neuronal ceroid lipofuscinosis (LINCL) mice were administered three weekly doses of K16ApoE (48 nmol) and TPP1 (17 nmol) in a total volume of 120 µl using a 3.75-minute infusion time. Treated mice were killed at 16 weeks of age and analyzed in parallel with untreated 13- and 16-week-old wild-type and LINCL mice. SCMAS storage is significantly different among the three groups of LINCL mice (P < 0.05 for each pairwise comparison using the Newman–Keuls multiple comparison test).
Peptide-mediated delivery to the brain provides therapeutic benefits
Six-week-old LINCL mice were administered five daily doses of TPP1 (17 nmol/dose) in the absence or presence of K16ApoE (12 nmol). Select animals were analyzed 24 hours after the final dose of TPP1 distribution while the remaining animals were monitored for locomotor function and survival.
TPP1 activity in brain was ~800% of wild-type when delivered with K16ApoE, a dramatic increase compared to the ~20% found with TPP1 alone (Figure 5a). In contrast, K16ApoE had no effect on levels of TPP1 delivered to peripheral tissues (Figure 5a). Immunohistochemical analysis revealed that K16ApoE mediated the delivery of TPP1 across the BBB and into neurons throughout the brain (Figure 5b and Supplementary Figure S6).
Figure 5.

Biodistribution of intravenously administered tripeptidyl peptidase I (TPP1). Five daily doses of TPP1 alone (17 nmol) or a mixture of TPP1 (17 nmol) and the K16ApoE peptide (12 nmol) were IV administered to 6-week-old late-infantile neuronal ceroid lipofuscinosis (LINCL) mice. (a) TPP1 activity was assayed in brain and peripheral tissues 24 hours after the final injection. Bars represent mean and range of two animals. (b) Confocal images of TPP1 immunostaining in the cerebral cortex acquired using a 60× objective.
Gait analysis was performed to monitor sensorimotor function. Initially, all treatment groups of LINCL mice resembled wild-type animals. At 105 days, untreated LINCL mice or animals treated with TPP1 alone displayed a decline in sensorimotor function. In contrast, sensorimotor function of the mice treated with the TPP1-K16ApoE mixture appeared normal until a considerably later age, with deficits first observed at 161 days (Figure 6a,b). Consistent with gait analysis, visual inspection indicated that the characteristic hunched appearance and ataxia of LINCL mice was delayed in response to ERT with the TPP1-K16ApoE mixture (Supplementary Videos S1 and S2).
Figure 6.
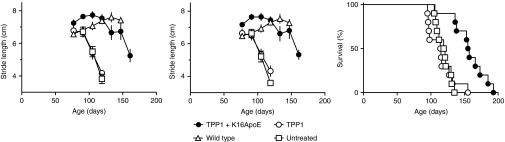
Effect of K16ApoE on ERT in late-infantile neuronal ceroid lipofuscinosis (LINCL) mice. Gait analysis. Graphs represent stride length for (a) fore- and (b) rear-limbs. The tripeptidyl peptidase I (TPP1)+K16ApoE treated group was indistinguishable from wild-type (P > 0.05) but significantly different from the untreated group or group treated with TPP1 alone at 105 and 119 days (P < 0.001 using one-way analysis of variance for each timepoint with the Newman–Keuls multiple comparison test). Error bars indicate standard error of the mean of 3–10 animals per group. (c) Survival analysis.
Treatment with TPP1 alone had no effect on survival of LINCL mice, with median survival 115 days compared to 120 days for untreated controls. Treatment with TPP1 in the presence of K16ApoE significantly increased lifespan (P < 0.001 compared to either TPP1 treated or untreated), with median survival of 156 days (Figure 6c). The increase in survival is comparable to that obtained previously using large-volume intrathecal administration.11
Discussion
Fusions between lysosomal proteins and ApoE have been shown to cross the BBB16,17 but in the case of TPP1, these were either not expressed or nonfunctional, suggesting that this approach may not be applicable to some proteins. Instead, we pursued an alternative method in which TPP1 was IV coinjected with K16ApoE - a peptide that comprises polylysine and an ApoE receptor binding segment that previously had been shown to promote passage of proteins into the brain.24 This “mix and inject” strategy proved to be remarkably effective, achieving ~400% of wild-type levels of TPP1 within the brain by a single IV infusion, which corresponds to ~1% of the administered 17 nmol TPP1. Moreover, administration of the TPP1 and peptide mixture effectively reduced lysosomal storage materials in neurons and also significantly extended the lifespan of the LINCL mouse model. To our knowledge, this is the first study to clearly demonstrate a significant increase in lifespan for a neurological disease when treated with IV ERT after formation of the BBB.
Given the remarkable properties of K16ApoE in promoting delivery of large molecule therapeutics to the mouse brain, we believe that further studies are warranted to determine if the approach could be translated to the clinic. Foremost of these will be to minimize adverse effects, which could be addressed in two complementary approaches. First, it is likely that the delivery method itself is not optimum: over the course of this study, we found that relatively minor modifications to a standard bolus tail-vein injection protocol in mice significantly ameliorated toxicity. It well may be worth exploring the effect of different routes and rates of K16ApoE administration on toxicity in larger animal models that are more amenable to use of indwelling catheters and metered infusion devices. Second, if the desired effect on drug targeting to the brain and toxicity are not mechanistically linked, it should be possible to identify peptide variants that have improved therapeutic indices.
Regardless of the mechanism of action and current levels of toxicity, our results indicate that K16ApoE should have immediate application in studies requiring the delivery of large molecules to the brain of a variety of mouse models. In particular, this will greatly facilitate the evaluation of brain ERT in LSDs with neurological involvement.
Materials and Methods
Animals. Animals were maintained and used following protocols approved by the Robert Wood Johnson Medical School Institutional Animal Care and Use Committee (“Preclinical evaluation of therapy in an animal model for LINCL,” protocol I09-0274-4). LINCL mice were in a C57/BL6 background and genotyped as described previously.28
Materials. Recombinant human TPP1 proenzyme was produced in CHO cells and purified as described.11 For injection, TPP1 was buffer exchanged into phosphate-buffered saline (Gibco) using PD-10 size exclusion columns (GE Healthcare). TPP1 was concentrated using Vivaspin 20 centrifugal concentrators (Sartorius Stedim Biotech, Aubagne, France) to a maximum of 17 mg/ml and stored at −80 °C. TPP1 concentration was estimated from absorbance at 280 nm using an extinction coefficient of 82,195 mol/l−1 cm−1 (1.38 ml mg−1 cm−1). K16ApoE peptide was produced as described24 and stored at −80 °C in phosphate-buffered saline (Gibco) at 22.5 mg/ml wet weight. The net amount of peptide used was calculated based on dry weight after correcting for total nitrogen content. The mixture of TPP1 and K16ApoE was typically prepared by vortexing for five seconds and incubation at room temperature for 1 hour. Protamine Sulfate Injection, USP (10 mg/ml in 0.9 % NaCl) was from APP Pharmaceuticals.
Tail-vein administration. Six-week-old LINCL mice were anesthetized using isoflurane delivered through an inhalation system (VetEquip, Pleasanton, CA). Bolus injection via tail vein was done over ~10 seconds using a 29.5 gauge insulin syringe (Terumo Medical, Somerset, NJ). Slow infusion was conducted using a NE-300 syringe pump (New Era Pump Systems, Wantagh, NY).
Analysis of TPP1 uptake. Animals were deeply anesthetized with sodium pentobarbital/phenytoin (a 1:4 dilution Euthasol; Delmarva Laboratories, Midlothian, VA) and killed by transcardial perfusion with saline. One brain hemisphere was drop-fixed in Bouin's reagent and 10 µm sagittal cryosections were processed for TPP1 immunohistochemistry as described.11 The remaining hemisphere was used to prepare frozen tissue powder using a Bessman pulverizer (Spectrum Laboratories, Rancho Dominguez, CA). For TPP1 enzyme assay, powders were homogenized in 50 volumes (brain, heart, kidney, and lung) or 100 volumes (liver and spleen) of homogenization buffer (0.15 mol/l NaCl /0.1% Triton X-100) using a Polytron homogenizer (Kinematica, Bohemia, NY). Tissue homogenates were centrifuged at 14,000g and supernatants were used for TPP1 activity endpoint assay as described previously.29 Where indicated, samples were preactivated at pH 3.5 to convert proTPP1 to the mature, active enzyme.30 Enzyme activity per milligram of protein was normalized to that of wild-type mice. For calculation of the fraction of dose delivered to the brain, TPP1 activity per wet weight tissue was compared to preactivated purified TPP1 standard. Protein concentrations of tissue extracts were measured using Advanced Protein Assay (Cytoskeleton, Denver, CO).
Construction and analysis of TPP1-ApoE fusion proteins. ApoE with polylinker sequences was inserted between the signal and pro-sequence or at the C-terminus of TPP1. A codon optimized TPP1 sequence in pBlunt31 was amplified using forward (5′-TAC CGG ACT CAG ATC TCG CCA CCA TGG GCC TGC AGG CCT GCC T-3′) and reverse primers (5′-TCT AGA GTC GCG GCC GCT TAC TAT CAG GGG TTC AGT A-3′). TPP1 and fusion derivatives were cloned using Infusion HD cloning kit (Clontech) into expression vector pmCherry N1 (Clontech) in which mCherry was removed by NotI and BglII digestion. Constructs were transiently transfected into CHO-K1 cells using Lipofectamine 2000 (Invitrogen). Cells were harvested 48 hours after the transfection, lysed in 0.15 mol/l NaCl/0.1% Triton X-100 at 4 °C for 1 hour. After centrifugation at 1200g for 20 minutes, supernatants were collected for TPP1 activity assay and immunoblotting. For western blotting, cell lysates were fractionated on reducing 10% Bis-Tris gels (Invitrogen) using MOPS running buffer. Proteins were transferred to PVDF membranes, which were blocked using 5% BSA in phosphate-buffered saline/0.2% Tween 20 and probed with rabbit anti-TPP1 polyclonal antisera Rb72-532 at a 1:1,000 dilution followed by Alexafluor 488 donkey anti-rabbit IgG. Signal was visualized using Typhoon 9400 scanner.
SCMAS immunoblotting. Tissue homogenates (100 µl, ~1.5 mg/ml) were centrifuged at 14000g for 30 minutes. The pellet was resuspended in 100 µl homogenization buffer and centrifuged as before. The pellet was dissolved in reducing lithium dodecyl sulfate polyacrylamide gel electrophoresis buffer and 4- and 8-µg equivalents of the initial homogenates analyzed by immunoblotting as described previously.11
Dynamic light scattering. Particle sizes of TPP1 and K16ApoE were measured in a final volume of 100 µl phosphate-buffered saline in a 3 mm quartz cuvette (Hellma Analytics) using a Zetasizer Nano (Malvern) at room temperature. Particle size was determined from the average of five measurements for each sample. Volume distribution was analyzed by Zetasizer software (Malvern).
Subacute toxicology. Functional observational battery measurements were conducted essentially as described in ref. 33 using a four-point rating scale. For histopathological analysis, mice were killed three days after the final K16ApoE injection as described above except that they were perfused with 4% paraformaldehyde following the saline perfusion. Brains, kidneys and livers were removed and postfixed in the same fixative overnight. The tissues were subsequently dehydrated in graded ethanol solutions, cleared in xylene, and embedded in Paraplast. Coronal and parasagittal 5 μm sections of brain and representative sections of liver and kidney were cut and mounted on polylysine-coated slides. Sections were stained with hematoxylin and eosin.
Survival and behavior analysis. Mice were singly housed from 6 weeks old and gently handled to avoid fatal startle seizures.28 Gait analysis was performed biweekly from 11 weeks and stride length was measured.34 Median lifespan and statistical tests were calculated using Prism 5.03 (Graph-Pad Software, San Diego, CA).
SUPPLEMENTARY MATERIAL Figure S1. TPP1 expression constructs. Figure S2. Subacute toxicology behavioral analysis. Figure S3. Subacute toxicology histological analysis of brain. Figure S4. Order of injection of K16ApoE and TPP1. Figure S5. Immunostaining for SCMAS in the cerebellum. Figure S6. Immunostaining for TPP1 in the brain. Video S1. Representative 164-day-old LINCL mouse treated with TPP1 and K16ApoE as described in Figure 5. Video S2. Representative untreated 126-day-old LINCL mouse.
Acknowledgments
We thank Vikas Nanda and Arvanish Parmar for helpful advice and Mukarram El-Banna for excellent technical assistance. This work was supported in part by a Johnson and Johnson Focused Giving Award and NIH R01 NS 37918 to P.L. and a Batten Disease Support and Research Association fellowship to Y.M. Additional support was provided by NIH P30ES005022. Work at the Mayo Clinic was supported by Bernie and Edith Waterman and the Ting Tsung and Wei Fong Chao Family Foundation. The authors declare no conflict of interest.
Supplementary Material
Representative 164-day-old LINCL mouse treated with TPP1 and K16ApoE as described in Figure 5.
Representative untreated 126-day-old LINCL mouse.
References
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, et al. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, et al. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease) Am J Med Genet. 1992;42:561–567. doi: 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- Poupetová H, Ledvinová J, Berná L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis. 2010;33:387–396. doi: 10.1007/s10545-010-9093-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santorelli FM, Garavaglia B, Cardona F, Nardocci N, Bernardina BD, Sartori S, et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J Rare Dis. 2013;8:19. doi: 10.1186/1750-1172-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335. doi: 10.1146/annurev-genom-090711-163739. [DOI] [PubMed] [Google Scholar]
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- Sly WS, Vogler C. The final frontier – crossing the blood-brain barrier. EMBO Mol Med. 2013;5:655–657. doi: 10.1002/emmm.201302668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson PI. Novel treatments and future perspectives: outcomes of intrathecal drug delivery. Int J Clin Pharmacol Ther. 2009;47(suppl. 1):S124–S127. [PubMed] [Google Scholar]
- Hemsley KM, Hopwood JJ. Delivery of recombinant proteins via the cerebrospinal fluid as a therapy option for neurodegenerative lysosomal storage diseases. Int J Clin Pharmacol Ther. 2009;47(suppl. 1):S118–S123. doi: 10.5414/cpp47118. [DOI] [PubMed] [Google Scholar]
- Vuillemenot BR, Katz ML, Coates JR, Kennedy D, Tiger P, Kanazono S, et al. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104:325–337. doi: 10.1016/j.ymgme.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Xu S, Wang L, El-Banna M, Sohar I, Sleat DE, Lobel P. Large-volume intrathecal enzyme delivery increases survival of a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2011;19:1842–1848. doi: 10.1038/mt.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Cooper JD, Sleat DE, Cheng SH, Dodge JC, Passini MA, et al. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. doi: 10.1038/mt.2008.9. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Boado RJ. Reengineering biopharmaceuticals for targeted delivery across the blood-brain barrier. Meth Enzymol. 2012;503:269–292. doi: 10.1016/B978-0-12-396962-0.00011-2. [DOI] [PubMed] [Google Scholar]
- Rozaklis T, Beard H, Hassiotis S, Garcia AR, Tonini M, Luck A, et al. Impact of high-dose, chemically modified sulfamidase on pathology in a murine model of MPS IIIA. Exp Neurol. 2011;230:123–130. doi: 10.1016/j.expneurol.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Barranger JA, Brady RO, Pagel M, Furbish FS, Quirk JM, et al. Delivery of hexosaminidase A to the cerebrum after osmotic modification of the blood–brain barrier. Proc Natl Acad Sci USA. 1981;78:5838–5841. doi: 10.1073/pnas.78.9.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino NC, D'Orsi L, Sambri I, Nusco E, Monaco C, Spampanato C, et al. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. 2013;5:675–690. doi: 10.1002/emmm.201202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, El-Amouri SS, Dai M, Kuan CY, Hui DY, Brady RO, et al. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc Natl Acad Sci USA. 2013;110:2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb JH, Vogler C, Levy B, Galvin N, Tan Y, Sly WS. Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA. 2008;105:2616–2621. doi: 10.1073/pnas.0712147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh HT, Grubb JH, Vogler C, Sly WS. Biochemical evidence for superior correction of neuronal storage by chemically modified enzyme in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA. 2012;109:17022–17027. doi: 10.1073/pnas.1214779109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler C, Levy B, Grubb JH, Galvin N, Tan Y, Kakkis E, et al. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc Natl Acad Sci USA. 2005;102:14777–14782. doi: 10.1073/pnas.0506892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Sohar I, Wang L, Sleat DE, Lobel P. Systemic administration of tripeptidyl peptidase I in a mouse model of late infantile neuronal ceroid lipofuscinosis: effect of glycan modification. PLoS ONE. 2012;7:e40509. doi: 10.1371/journal.pone.0040509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roces DP, Lüllmann-Rauch R, Peng J, Balducci C, Andersson C, Tollersrud O, et al. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004;13:1979–1988. doi: 10.1093/hmg/ddh220. [DOI] [PubMed] [Google Scholar]
- Polito VA, Abbondante S, Polishchuk RS, Nusco E, Salvia R, Cosma MP. Correction of CNS defects in the MPSII mouse model via systemic enzyme replacement therapy. Hum Mol Genet. 2010;19:4871–4885. doi: 10.1093/hmg/ddq420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar G, Curran GL, Mahlum E, Decklever T, Wengenack TM, Blahnik A, et al. A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and IgM to the brain. PLoS ONE. 2011;6:e28881. doi: 10.1371/journal.pone.0028881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausbaugh LJ. Intracarotid infusions of protamine sulfate disrupt the blood-brain barrier of rabbits. Brain Res. 1987;409:221–226. doi: 10.1016/0006-8993(87)90705-0. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Buciak JL, Kang YS, Boado RJ. Protamine-mediated transport of albumin into brain and other organs of the rat. Binding and endocytosis of protamine-albumin complex by microvascular endothelium. J Clin Invest. 1993;92:2224–2229. doi: 10.1172/JCI116825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlum E, Mandal D, Halder C, Maran A, Yaszemski MJ, Jenkins RB, et al. Engineering a noncarrier to a highly efficient carrier peptide for noncovalently delivering biologically active proteins into human cells. Anal Biochem. 2007;365:215–221. doi: 10.1016/j.ab.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohar I, Lin L, Lobel P. Enzyme-based diagnosis of classical late infantile neuronal ceroid lipofuscinosis: comparison of tripeptidyl peptidase I and pepstatin-insensitive protease assays. Clin Chem. 2000;46:1005–1008. [PubMed] [Google Scholar]
- Guhaniyogi J, Sohar I, Das K, Stock AM, Lobel P. Crystal structure and autoactivation pathway of the precursor form of human tripeptidyl-peptidase 1, the enzyme deficient in late infantile ceroid lipofuscinosis. J Biol Chem. 2009;284:3985–3997. doi: 10.1074/jbc.M806943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, et al. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther. 2007;15:1782–1788. doi: 10.1038/sj.mt.6300249. [DOI] [PubMed] [Google Scholar]
- Lin L, Lobel P. Production and characterization of recombinant human CLN2 protein for enzyme-replacement therapy in late infantile neuronal ceroid lipofuscinosis. Biochem J. 2001;357 Pt 1:49–55. doi: 10.1042/0264-6021:3570049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusich JA, Grant KR, Blough BE, Wiley JL. Effects of synthetic cathinones contained in “bath salts” on motor behavior and a functional observational battery in mice. Neurotoxicology. 2012;33:1305–1313. doi: 10.1016/j.neuro.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Labattu B, Tison F. A simple method to measure stride length as an index of nigrostriatal dysfunction in mice. J Neurosci Methods. 2002;113:123–130. doi: 10.1016/s0165-0270(01)00485-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative 164-day-old LINCL mouse treated with TPP1 and K16ApoE as described in Figure 5.
Representative untreated 126-day-old LINCL mouse.