

Abstract
Mesenchymal stem cells (MSCs) have been shown to improve outcomes after neonatal hypoxic-ischemic (HI) brain injury possibly by secretion of growth factors stimulating repair processes. We investigated whether MSCs, modified to secrete specific growth factors, can further enhance recovery. Using an in vitro assay, we show that MSC-secreting brain derived neurotrophic factor (BDNF), epidermal growth factor-like 7 (EGFL7), persephin (PSP), or sonic hedgehog (SHH) regulate proliferation and differentiation of neural stem cells. Moreover, mice that received an intranasal application of 100,000 MSC-BDNF showed significantly improved outcomes as demonstrated by improved motor function and decreased lesion volume compared with mice treated with empty vector (EV) MSCs. Treatment with MSC-EGFL7 improved motor function but had no effect on lesion size. Treatment with MSC-PSP or MSC-SHH neither improved outcome nor reduced lesion size in comparison with MSC-EV–treated mice. Moreover, mice treated with MSC-SHH showed even decreased functional outcomes when compared with those treated with MSC-EV. Treatment with MSC-BDNF induced cell proliferation in the ischemic hemisphere lasting at least 18 days after MSC administration, whereas treatment with MSC-EV did not. These data suggest that gene-modified cell therapy might be a useful approach to consider for treatment of neonatal HI brain damage. However, care must be taken when selecting the agent to overexpress.
Introduction
Transplantation of mesenchymal stem cells (MSCs) into both neonatal and adult ischemic brain injury models has been reported to promote endogenous repair processes, to reduce lesion size, and to improve functional outcomes.1,2,3,4,5,6,7 Although it has been shown that MSCs can differentiate into cells of the neuronal or glial lineage, their beneficial effects are not likely to be due to replacement by MSCs of lost cells. Transplanted MSCs rather promote repair of damaged brain tissue via release of trophic factors stimulating endogenous repair processes such as neurogenesis, angiogenesis, and synaptogenesis.3,8
In vitro, there is a low-level basal secretion of neurotrophins and growth factors by MSCs. However, in vitro culture of MSCs with ischemic brain extracts induces the expression of several growth factors and cytokines.1,9,10 In this respect, it is of interest that the type and extent of injury may guide the expression pattern of these MSC-derived growth and differentiation factors after transplantation into the brain.8,9
Perinatal hypoxia-ischemia (HI) often leads to permanent brain damage, causing neurological deficits such as cerebral palsy, mental retardation, and seizures.11 We have previously shown that upon transplantation of MSCs after perinatal HI, graft survival was limited to only ~22% of MSCs surviving until 3 days after transplantation, and 18 days after transplantation, only ~1% of transplanted MSCs were still detectable.8 However, transplanted MSCs were shown to be capable of extensively modulating growth factor production in the brain.
Following the transplantation of MSCs, there is an increased gene expression of factors involved in cell proliferation/differentiation. These specific MSC-induced changes in growth factor environment may have the potential to regulate repair processes in the ischemic brain.
In this article, we investigated whether the overexpression of brain derived neurotrophic factor (BDNF), epidermal growth factor-like 7 (EGFL7), persephin (PSP), or sonic hedgehog (SHH) in MSCs can further reduce HI brain damage. These factors were chosen based on their capacity to act on different repair processes. BDNF is an all-round neurotrophic factor stimulating diverse processes such as neurogenesis, angiogenesis, and synaptic plasticity.12,13 Furthermore, it has been shown that infusion of BDNF can significantly improve outcomes after adult cerebral ischemia.13 EGFL7, also known as vascular endothelial statin (VE statin), Zneu1, or Notch4-like protein, is a secreted antagonist of Notch receptor–mediated signaling that is expressed by endothelial cells, several progenitor cell populations, and a subset of neurons in the adult brain.14,15 Notch signaling is involved in a wide variety of cellular processes in the developing nervous system, including cell proliferation, differentiation, and apoptosis. By inhibiting Notch signaling, EGFL7 has the potential to increase proliferation of progenitor cells and drive neuronal differentiation. PSP is a member of the TGF-β family and mainly known for its neuroprotective properties. By engineering MSCs to express PSP, distressed neurons in the ischemic lesion could potentially be protected. SHH is a molecule that, during development, drives migration and differentiation of neural progenitor cells toward neurons and oligodendrocytes.16,17,18 Neonatal HI causes severe damage, and SHH has a strong potential to stimulate the formation of new oligodrendrocytes, thereby possibly attenuating the deleterious effects of HI brain damage.
Results
Characteristics of gene-transduced MSCs
MSCs were transduced with different adenoviral constructs at multiplicity of infections (MOIs) of 200, 500, 1,000, and 2,000 plaque forming units (pfu) per cell. The level of BDNF secretion by MSCs transfected with BDNF (MSC-BDNF) at an MOI of 500 pfu/cell was 83-fold higher than by uninfected MSCs (Figure 1a). The level of EGFL7 secretion from MSC-EGFL7 infected at an MOI of 500 pfu/cell was 51-fold higher than by uninfected MSCs (Figure 1b). The level of PSP secretion from MSC-PSP infected at an MOI of 500 pfu/cell was 43-fold greater than by uninfected MSCs (Figure 1c). The level of SHH secretion from MSC-SHH infected at an MOI of 500 pfu/cell was 82-fold greater than by uninfected MSCs (Figure 1d).
Figure 1.

Characteristics of modified mesenchymal stem cells (MSCs). Secretion of (a) brain derived neurotrophic factor (BDNF), (b) epidermal growth factor-like 7 (EGFL7), (c) PSPN, and (d) sonic hedgehog (SHH) by MSCs transfected with pAd-HM41-K7-BDNF, pAd-HM41-K7-EGFL7, pAd-HM41-K7-PSPN, or pAd-HM41-K7-SHH at multiplicity of infections (MOIs) of 250, 500, 1,000, or 2,000 pfu/cell. Data represent mean ± SEM. Flow cytometric analysis of surface antigen expression on (e) naïve MSCs, (f) EV-MSC, (g) BDNF-MSC, (h) EGFL7-MSC, (i) PSPN-MSC, and (j) SHH-MSC. EV, empty vector; PSP, persephin.
Flow cytometric analysis of empty vector (EV)-MSC, MSC-BDNF, MSC-EGFL7, MSC-PSP, and MSC-SHH showed that phenotypic expression was essentially identical to primary MSCs. All gene-modified cells and MSCs with EV had the CD29+, CD34+, CD44+, Sca-1+, CD45−, and CD117− cell surface phenotype (Figure 1e).
In vitro effect of gene-transduced MSCs on NSC proliferation and differentiation
To determine the effect of growth factor secretion by gene-transduced MSCs on the process of neurogenesis following neonatal HI in vitro, we used a noncontact coculture model to examine the effects of gene-transduced MSCs on the proliferation and differentiation of neural stem cells (NSCs). MSCs were seeded into a culture insert and placed in a culture of adherent NSCs. The coculture system was then followed for 5 days during which proliferation and differentiation of NSCs was assessed.
At 24 hours after start of culture in the presence of either MSC-EGFL7 or MSC-SHH, more NSCs were Ki67-positive than when cultured in combination with an empty gel or EV-MSC (Table 1 and Figure 2a). MSC-BDNF and MSC-PSP did not stimulate proliferation of NSCs at this time point. Culture with MSC-BDNF significantly increased the percentage of Ki67-positive NSCs at 72 hours after start of coculture when compared with EV-MSC. Culture of NSCs in combination with MSC-EGFL7, MSC-PSP, or MSC-SHH did not increase proliferation of NSCs. At 120 hours after start of coculture, the percentage of Ki67-positive NSCs was still increased when cultured in combination with MSC-BDNF or MSC-EGFL7. Proliferation of NSCs when cultured with MSC-PSP was not altered when compared with EV-MSC at all time points measured.
Table 1. Summarized results of effect of modified MSCs on NSC proliferation and differentiation in vitro.
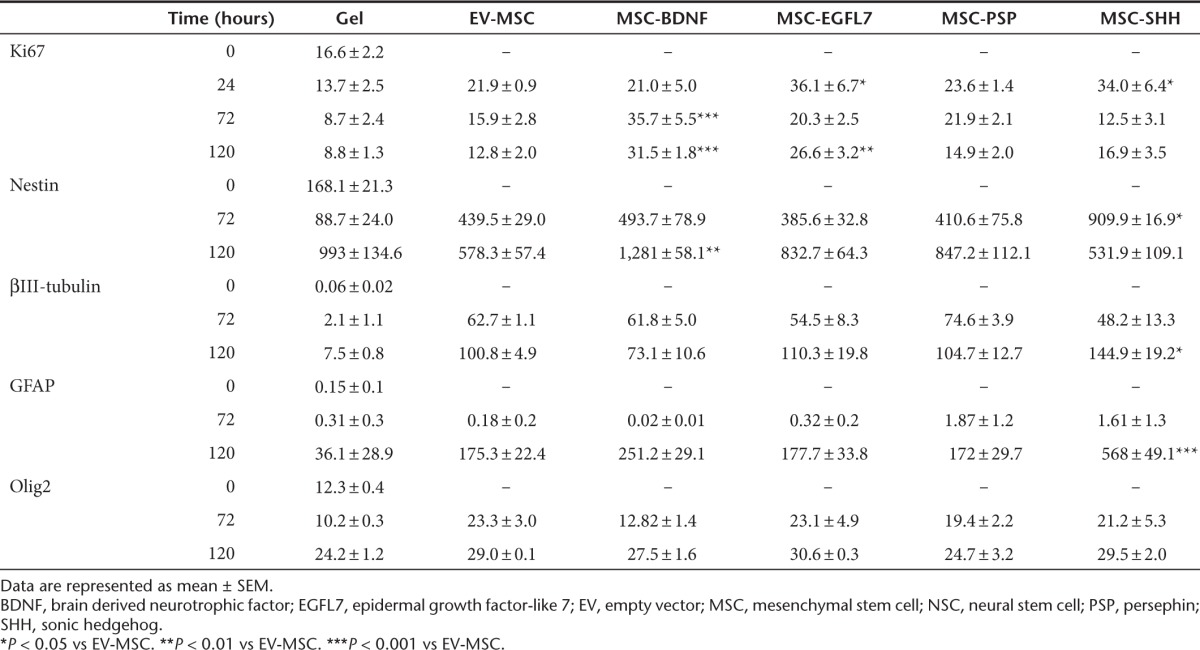
Figure 2.

In vitro effects of modified mesenchymal stem cells (MSCs) on proliferation and differentiation of neural stem cells (NSCs). The effects of growth factor hypersecreting MSCs on NSCs were determined using a noncontact coculture system. (a) Ki67 expression was analyzed to determine the effects of MSCs on proliferation of NSC. The effects of MSCs on differentiation of NSCs were determined by examining the expression of the markers (b) nestin, (c) βIII-tubulin, (d) GFAP, and (e) Olig2 in NSC cultures. (f–j) Representative photomicrographs of Ki67, nestin, βIII-tubulin, GFAP, and Olig2, respectively. Data represent mean ± SEM. n = 3 for all conditions. *P < 0.05 vs EV-MSC. **P < 0.01 vs EV-MSC. ***P < 0.001 vs EV-MSC. Scale bar represent 100 µm. BDNF, brain derived neurotrophic factor; EGFL7, epidermal growth factor-like 7; EV, empty vector; PSP, persephin; SHH, sonic hedgehog.
The effect of modified MSCs on the differentiation potential of NSCs in vitro was determined by analyzing the expression of the markers nestin, βIII-tubulin, GFAP, and Olig2. Culture of EV-MSC with NSCs induced nestin expression when compared with NSCs cultured with empty gel. MSC-SHH significantly increased nestin expression in NSCs at 72 hours in culture compared with EV-MSC (Figure 2b). At 120 hours in culture, MSC-BDNF significantly increased nestin expression when compared with EV-MSC. Culture of NSCs with MSC-EGFL7 or MSC-PSPN did not affect nestin expression in NSCs.
Expression of βIII-tubulin was induced in NSCs after 72 hours in culture with EV-MSC when compared with that of NSCs cultured with an empty gel. At 120 hours in culture, MSC-SHH significantly increased βIII-tubulin expression in NSCs when compared with EV-MSC (Figure 2c). Culture of NSCs with MSC-BDNF, MSC-EGFL7, or MSC-PSP did not increase βIII-tubulin expression when compared with EV-MSC.
GFAP was hardly expressed in NSCs after 72 hours in culture under all culture conditions. At 120 hours in culture of NSCs with MSC-SHH, GFAP expression was significantly increased when compared with EV-MSC (Figure 2d)). MSC-BDNF, MSC-EGFL7, or MSC-PSP did not have any effect on GFAP expression by NSCs. Olig 2 expression in NSCs was not affected by coculture with modified MSCs (Figure 2e).
Effect of gene-transduced MSCs on motor function and lesion volume
Mice underwent HI on postnatal day 9 and were treated with gene-transduced MSCs or vehicle on day 10 after HI. At 10, 21, and 28 days after HI, lateralizing motor deficits were quantified using the cylinder rearing test as the preference to use the unimpaired, ipsilateral forepaw when rearing (Table 2 and Figure 3a). Before MSC treatment on day 10, there was no difference between the different treatment groups. Treatment with EV-MSC, MSC-BDNF, MSC-EGFL7, or MSC-PSP significantly reduced forepaw preference on days 21 and 28 after HI when compared with vehicle treatment. Furthermore, MSC-BDNF– or MSC-EGFL7–treated mice showed less lateralizing deficits than mice treated with EV-MSC. Administration of MSC-SHH resulted in decreased impairment at 21 days after HI when compared with vehicle-treated mice. However, at 28 days after HI, forepaw impairment in these mice was equal to the level of impairment in vehicle-treated mice and significantly worse than mice treated with EV-MSC.
Table 2. Summarized results of in vivo effects of modified MSCs on motor function, lesion volume, and cell proliferation in the hippocampus.
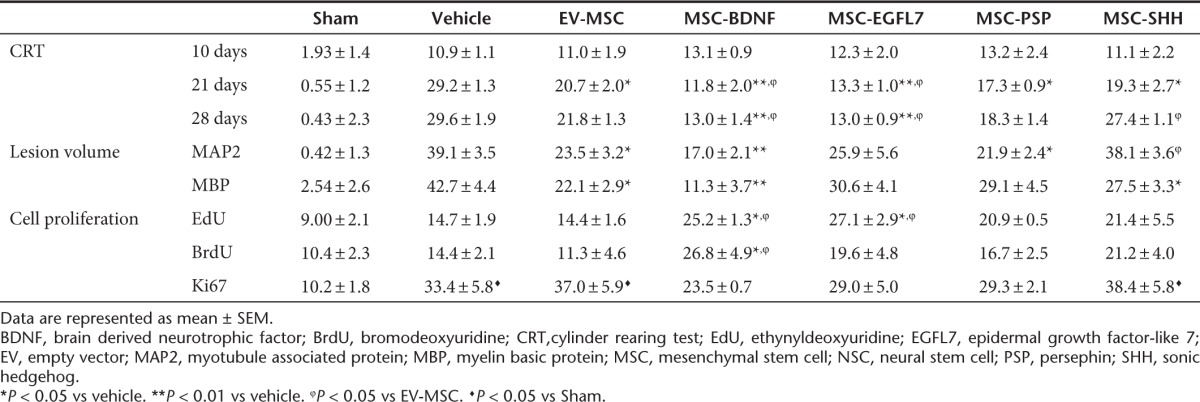
Figure 3.

Effect of treatment with genetically modified mesenchymal stem cells (MSCs) on functional outcomes. Mice received MSCs or vehicle at 10 days after hypoxia-ischemia (HI). (a) Paw preference as a measure of motor disfunction was determined at 10, 21, and 28 days after HI. Quantification of (b) MAP2-positive and (c) MBP-positive area loss expressed as ratio ipsi-/contralateral area. (d,e) Representative photomicrographs of Map2 and MPB respectively. Data represent mean ± SEM. Sham n = 7, vehicle n = 8, EV-MSC n = 9, BDNF-MSC n = 10, EGFL7-MSC n = 9, PSPN-MSC n = 10, and SHH-MSC n = 10. *P < 0.05 vs vehicle. **P < 0.01 vs vehicle. ϕP < 0.05 vs EV-MSC. BDNF, brain derived neurotrophic factor; EGFL7, epidermal growth factor-like 7; EV, empty vector; MAP2, microtubule associated protein; MBP, myelin basic protein; PSP, persephin; SHH, sonic hedgehog; VEH, vehicle.
Effect of treatment with modified MSCs on lesion size was determined at 28 days after HI by analyzing the loss of ipsilateral gray (mouse-anti-microtubule-associated protein (MAP2)) and white (mouse-anti-myelin basic protein (MBP)) matter. In vehicle-treated mice, ipsilateral MAP2 area loss was 39.13 ± 3.5% (Table 2 and Figure 3b,d). Treatment with EV-MSC significantly reduced lesion volume when compared with vehicle treatment (23.48 ± 3.2%; P < 0.05). BDNF- and PSP-secreting MSCs decreased MAP2 area loss when compared with vehicle treatment (17.08 ± 2.07%; P < 0.01 and 21.85 ± 2.38; P < 0.05, respectively) but not when compared with EV-MSC. Treatment with MSC-EGFL7 did not significantly decrease MAP2 area loss when compared with vehicle treatment (25.86 ± 5.6%; P = 0.085). However, ipsilateral MAP2 area loss in MSC-SHH–treated mice was significantly higher than in MSC-EV–treated mice (38.12 ± 3.64%; P < 0.05) and did not differ from vehicle-treated mice.
Treatment with EV-MSC reduced MBP area loss in the ispilateral hemisphere when compared with vehicle treatment (22.13 ± 2.9 vs 42.73 ± 4.4%; P < 0.05) (Table 2 and Figure 3c,e). Treatment with MSC-BDNF resulted in a further decrease of MBP area loss when compared with treatment with EV-MSC (11.29 ± 3.7%; P < 0.01). Treatment with either MSC-EGFL7 or MSC-PSP did not result in decreased ipsilateral MBP area loss (30.66 ± 4.1 and 29.10 ± 4.5%; P > 0.05) compared with vehicle treatment. However, ipsilateral MBP area loss in MSC-SHH–treated mice was significantly smaller when compared with vehicle-treated mice (27.45 ± 3.3%; P < 0.05).
Cell proliferation in the ischemic hemisphere after treatment with gene-transduced MSCs
Proliferating cells on days 10, 11, and 12 after HI were labeled using ethynyldeoxyuridine (EdU) (Figure 4a). The number of EdU-positive cells at 28 days after HI as a measure of cell proliferation induced directly after injection of MSCs was increased in the hippocampus of mice treated with MSC-BDNF or MSC-EGFL7 when compared with that of vehicle- or EV-MSC–treated mice (Figure 4b). Treatment with MSC-PSP or MSC-SHH did not result in increased EdU incorporation in the hippocampus when compared with that in vehicle- or EV-MSC–treated mice.
Figure 4.
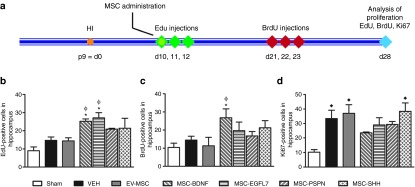
Cell proliferation following treatment with genetically modified mesenchymal stem cells (MSCs). Cell proliferation marker ethynyldeoxyuridine (EdU) was administered for 3 days starting directly after administration of MSCs, and bromodeoxyuridine (BrdU) was administered on days 21–23. Both were analyzed at 28 days after hypoxia-ischemia (HI). (a) Cell proliferation at 28 days after HI was measured using Ki67. (b–d) EdU-, BrdU-, and Ki67-positive cell numbers were analyzed in hippocampus. Data represent mean ± SEM. Sham n = 7, vehicle n = 8, EV-MSC n = 9, BDNF-MSC n = 10, EGFL7-MSC n = 9, PSPN-MSC n = 10, and SHH-MSC n = 10. *P < 0.05 vs vehicle, **P < 0.01 vs vehicle, ϕp<0.05 vs EV-MSC, ♦P < 0.05 vs Sham. BDNF, brain derived neurotrophic factor; d, day; EGFL7, epidermal growth factor-like 7; EV, empty vector; p9, postnatal day 9; PSP, persephin; SHH, sonic hedgehog.
To determine whether there was a lasting effect of growth factor–secreting MSCs on cell proliferation mice were injected with bromodeoxyuridine (BrdU) on days 21, 22, and 23 after HI. Cell proliferation after treatment with BDNF-secreting MSCs was still increased as witnessed by the increased number of BrdU-positive cells in the hippocampus (Figure 4c). Notably, treatment with EGFL7-, PSP-, or SHH-secreting MSCs did not result in a long-lasting induction of cell proliferation in the hippocampus. No difference in EdU- or BrdU-positive cell numbers was observed between sham-operated mice or vehicle- and EV-MSC–treated HI mice.
Cell proliferation at 28 days after HI was analyzed in the hippocampus using Ki67. At 28 days after HI, the number of Ki67-positive cells was significantly higher in mice that underwent HI when compared with that in sham controls (Figure 4d). Treatment with EV-MSC or gene-transduced MSCs did not result in increased cell proliferation in the SVZ at 28 days after HI when compared with vehicle treatment.
Discussion
In the present study, we show that intranasal application of modified MSCs after neonatal HI in mice could be beneficial in reducing long-term functional impairment. For this study, we have chosen four different growth factors to be overexpressed in MSCs, which are all known to play a role in proliferation and/or differentiation of NSCs, BDNF, EGFL7, PSP, and SHH.
Using an in vitro assay, we first determined the effect of modified MSCs on NSC proliferation and differentiation. The NSCs used in the coculture system are derived from the cortices of 14-day-old mouse embryos (E14). Results from our study indicate that NSCs are most responsive toward MSCs modified to express either BDNF or SHH. Notably, BDNF-secreting MSCs stimulate proliferation of NSCs, and MSC-SHH stimulate differentiation of NSCs toward neurons and astrocytes. In line with our current data, it has been shown earlier that application of BDNF to E13-derived NSCs promotes their survival and stimulates proliferation.19,20 Moreover, SHH has been shown to stimulate proliferation of E15 neocortical precursor cells and can promote their differentiation toward all three lineages, i.e., neurons, astrocytes, and oligodendrocytes.21 These data indicate that the MSCs modified to secrete either BDNF or SHH have the same impact as treatment with pure BDNF or SHH.
MSC-EGFL7 stimulate proliferation of E14 NSCs after 5 days in culture but have no effect on differentiation of NSCs. Recent results studying the effect of EGFL7 on adult NSCs have indicated that EGFL7 reduces the proliferative capacity of the NSCs and induces neuronal and oligodendroglial differentiation.22 The increase in NSC proliferation induced by EGFL7 in our study could be explained by the age of the NSCs used, because embryonic NSCs react differently to neurotrophic stimuli than adult NSCs. MSCs hypersecreting PSP do not seem to have any effect on proliferation or differentiation of NSCs in vitro when compared with EV-MSC.
BDNF is an important neurotrophic factor promoting neurogenesis and angiogenesis. It is neuroprotective, modulates inflammation, and improves synaptic plasticity after ischemic brain injury. Moreover, it has been shown that intracranial infusion of BDNF can reduce the infarct volume after stroke.13,23,24 Recently, we have shown that BDNF-hypersecreting MSCs have modest beneficial effects on outcomes using a neonatal stroke model.25 The present study indicates that MSC-BDNF have beneficial effects also in the neonatal HI mouse model. It is the most potent factor in improving motor function, reducing lesion size, and inducing long-term cell proliferation in the ischemic hemisphere. Besides the neuroprotective and neurogenic properties of BDNF, it also plays a role in refinement of developing neuronal circuits.12,26 The effect of MSC-BDNF might be directed toward refinement of neural circuits, because the neonatal murine brain is still developing during this moment of time. Addition of BDNF to MSCs might lead to more efficient pruning of connections than MSCs alone.
The in vivo data further showed that treatment with MSC-EGFL7 was as effective as treatment with MSC-BDNF in improving motor function. However, this was not combined with reduction in lesion size or increased cell proliferation.
EGFL7 is an antagonist of Notch receptor–mediated signaling. Expression of EGFL7 in NSCs in vitro was shown to decrease Notch signaling, thereby reducing proliferation and self-renewal of NSCs.22 This study also showed that overexpression of EGFL7 in NSCs skewed differentiation of cultured NSCs toward neurons and oligodendrocytes. In our study, we observed that overexpression of EGFL7 by MSCs does not have any effect on differentiation of NSCs but stimulated NSC proliferation after 5 days in culture. In the study of Schmidt et al.,22 NSCs were allowed to differentiate for at least 12 days before analysis, whereas in our study, differentiation analysis was performed after 5 days in culture. The latter could explain the apparent difference in differentiation of NSCs as 5 days might be too short of an interval to determine the expression of neuronal and oligodendroglial markers. In the neonatal HI model, treatment with MSC-EGFL7 induced cell proliferation in the hippocampus. However, these effects were not lasting, because 1 week after application of the MSCs, cell proliferation was not increased in comparison with EV-MSC treatment. Furthermore, reduction of gray matter damage, as measured by ipsilateral MAP2 staining, was similar after treatment with either EV-MSC or MSC. Likewise, white matter injury, as measured by ipsilateral MBP staining, was not reduced after transplantation of MSC-EGFL7. On the other hand, MSC-EGFL7 caused a greater improvement of motor function than EV-MSC. In a previous study, we reported the beneficial effects of MSCs on the corticospinal tract, which is related to the extent of improvement of motor function.27 It is possible that besides the previously described effects of EGFL7 on NSCs, it also affects neuronal signaling/communication in white matter tracts, thereby contributing to improved signaling in motor neurons. However, this is purely speculative, and more research is warranted to define the effects of EGFL7 on mature neurons.
PSP is a member of the TGF-β family and closely related to glial-derived neurotrophic factor. Previously, it has been shown that mice lacking PSP are more sensitive to ischemic brain damage following focal ischemia.28 Damage in these mice could be rescued by a single injection of PSP, and the neuroprotection induced by PSP was 100- to 1,000-fold more effective than glial-derived neurotrophic factor. Furthermore, PSP has been shown to promote survival of midbrain dopamine neurons and motor neurons both in vitro and in vivo.29,30 In our study, adding PSP to MSCs did not result in improved outcomes when compared with EV-MSC. In this study, modified MSCs were transplanted at 10 days after HI injury. Many studies examining neuroprotective agents have indicated that, to be successful, a neuroprotective agent must be administered within a couple of hours after the insult. Because PSP is mainly known for its neuroprotective properties, one could speculate that neuroprotection is not the main mechanism through which MSCs reach their beneficial effects when administered 10 days after onset of injury.
SHH is a powerful factor that stimulates neural progenitor cell proliferation, migration, and differentiation toward neurons and oligodendrocytes.16,17,18 Theoretically, these properties would make SHH an excellent candidate to be overexpressed in MSCs to treat neonatal HI brain damage. However, our results indicate that addition of SHH to MSCs has detrimental effects on outcomes after treatment for neonatal HI. Using the in vitro system, we could detect an early stimulation of proliferation and increased differentiation of NSCs toward neurons and astrocytes when cultured in combination with MSC-SHH. In vivo treatment with MSC-SHH of mice that underwent HI leads to worsened functional outcomes when compared with mice treated with EV-MSC. Furthermore, MSC-SHH–treated mice had significantly more gray matter damage than mice treated with MSC-EV. A recent article by Sirko et al.31 indicated that SHH elicits stem-cell response in reactive astrocytes following ischemic brain injury. High levels of SHH signaling activate reactive gliosis and trigger a proliferative response in the reactive astrocytes. Although these reactive astrocytes could have beneficial effects in that they protect surrounding tissue from injury, they can also limit repair by creating a glial scar.32 It could be speculated that by using MSC-SHH to treat neonatal HI, an astrocyte response has taken place that limits MSC-induced repair of brain injury and, therefore, damage evolves as in vehicle-treated mice.
A nonintegrating adenoviral construct was used to overexpress growth factors in MSCs. Adenovrial vectors are known for their immunogenic properties as they continue to express adenoviral proteins in the infected cell. This raises the potential of an immune response against infected MSCs, which could affect their therapeutic potential. However, when we compare the results of the present study with previously published results with naïve MSCs, the reduction in lesion size and improvement of motor function induced by empty adeno vector–transduced MSCs are equal to the effects of naïve MSCs.1,33 This indicates that an immune response against the adeno-transduced MSCs does not have a major influence on the in vivo effects measured in our present study.
In summary, this study shows that intranasal application of growth factor hypersecreting MSCs have the potential to contribute to the therapeutic potential of MSCs. Repair processes in the brain such as neurogenesis, oligodendrogenesis, and synaptogenesis are regulated via an intricate balance of intra- and extracellular molecules.34 Meddling in this balance by overexpressing a certain factor could either contribute to repair or impair it. Therefore, care must be taken when selecting the factor to overexpress in the neonatal brain.
Materials and Methods
Adenoviral vectors. Adenoviral vectors (pAd-HM41-K7; Alphagen, Yokohama-shi, Japan) carrying the gene for polylysine-mutated fiberknot were constructed as described previously.35 Mouse BDNF cDNA was cloned using reverse transcriptase–polymerase chain reaction using total RNA isolated from brain as the template. The identity of BDNF cDNA was confirmed by sequencing and comparing it with the GenBank sequence NM_007540. The mouse BDNF primer sequence was forward 5′-TCTAGACACCCCACCATGACCATCCTTTTCCTT-3′, Reverse 5′-TCTTCCCCTTTTAATGGTCAGT-3′. The BDNF cDNA was coupled to an IRES-eGFP sequence derived from pPRIG plasmid by means of polymerase chain reaction to allow labeling of infected cells. The BDNF-IRES-eGFP sequence was inserted into the pShuttle2 vector, which contains a cytomegalovirus promoter, between the XbaI and AflII sites resulting in the pShuttle2-BDNF-IE plasmid. pAd-HM41-K7-BDNF-IE was constructed by ligation of I-CeuI/PI-SceI-digested pShuttle2-BDNF-IE with I-CeuI/PI-SceI-digested pAd-HM41-K7.
Adenoviral vectors carrying mouse EGFL7 (pAd-HM41-K7-EGFL7-IE), PSP (Pspn; pAd-HM41-K7-PSP-IE), or SHH (pAd-HM41-K7-SHH-IE) were constructed as described above. The identity of EGFL7, Pspn, and Shh cDNA was confirmed by sequencing and comparing it with the GenBank sequences NM_008486, NM_008954, and NM_009170, respectively. Primer sequences used are as follows: EGFL7 Forward 5′-CTCTAGACCACCATGCAGACCATGTGGGGC-3′, Reverse 5′-CGGCGCGCCCAGATCTTTTTTGCAGGA-3′; Pspn Forward 5′-GGCGCGCCGCCACCACAGCCACAAGC-3′, Reverse 5′-TCTAGAATGGCTGCAGGAAGACTT-3′; and Shh Forward 5′-CTCTAGACCACCATGCTGCTGCTGCTGGCCAGATGTT-3′, Reverse 5′-CGGCGCGCCGCTGGACTTGACCGCCATTCCCAAGC-3′. An EV control was generated and consisted only of the IRES-eGFP sequence insert into the pShuttle2 plasmid.
Virus particles were generated by transfection of PacI-digested pAd-HM41-K7-BDNF-IE into 293 cells with Lipofectamine 2000 (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Virus particles were purified using Adeno-X virus purification kit (Clontech, Mountain View, CA), and titer was determined by using Adeno-X Rapid Titer Kit (Clontech). Before being used, the titer of the virus particles is determined, and virus stocks are examined for potential contamination with replication-competent viruses.
Mesenchymal stem cells. Mouse C57Bl/6 MSCs (Life Technologies) were purchased and cultured according to the manufacturer's instructions. Cells were positive for CD29, CD34, CD44, and Sca-1 and negative for CD117 (Figure 1e)
For adenovirus-mediated gene transfection, MSCs were seeded at a density of 3 × 106 cells per 25 cm2 flask. MSCs were exposed to the virus particles in 7.5 ml Dulbecco's modified Eagle medium (DMEM; Life Technologies) at 37 °C for 2 hours after which the cultures were washed three times with DMEM (Life Technologies) and recultured with normal medium. After infection, the cells were cultured for an additional 24 hours after which transplantation was performed.
Growth factor ELISAs. To determine the most effective MOI (pfu/cell) to modify MSCs, cell culture supernatants were collected 48 hours after in vitro transduction of MSCs at various MOIs. The following commercial ELISA kits were used to quantify the amount of growth factor produced: BDNF (Promega, Madison, WI), EGFL7 and PSP (USCN Life Science, Wuhan, China), and SHH (R&D systems, Minneapolis, MN).
Noncontact coculture of NSCs with modified MSCs. To determine the effects of growth factor hypersecreting MSCs on NSCs, cells were placed in a noncontact coculture. Mouse cortical CD1 NSCs were purchased and allowed to expand according to the manufacturer's instructions (R&D systems). NSCs were cultured as neurospheres in DMEM:F12 with B27 supplement (Life Technologies) containing 20 ng/ml of bFGF en EGF (Peprotech, Rocky Hill, NJ) until start of coculture with MSCs. One day before start of coculture, NSCs were plated on poly-l-ornithine and laminin (Sigma-Aldrich, Steinheim, Germany) coated 24-well plates. In another plate, 80,000 modified MSCs were embedded into 0.2% HydroMatrix gel in DMEM with 10% fetal calf serum (Sigma-Aldrich), in order for the MSCs to be cultured in their optimal culture medium, and placed into transwell inserts (Millipore, Darmstadt, Germany). Next day, transwell inserts containing MSCs were placed in well containing NSCs. At 0, 72, and 120 hours after start of coculture, NSCs were washed with phosphate-buffered saline and fixed in 3.7% paraformaldehyde. To detect proliferating cells, NSCs were incubated with rabbit-anti-Ki67 (Abcam, Cambridge, UK) and goat-anti-rabbit Alexafluor594 (Life Technologies).
NSCs were identified by staining cells with mouse-anti-nestin (BD, San Jose, CA), and differentiation of NSCs toward mature neurons, astrocytes, and oligodendrocytes was analyzed by staining cells with rabbit-anti-βIII-tubulin (Abcam), mouse-anti-GFAP (Cymbus Biotechnology, Southhampton, UK), and rabbit-anti-Olig2 (Millipore), respectively. Primary antibody binding was visualized with goat-anti-mouse Alexafluor488 or goat-anti-rabbit Alexafluor488 (Life Technologies). 4′,6-Diamidino-2-phenylindole (DAPI; Sigma-Aldrich) was used to visualize cell nuclei. For each condition, three randomly selected high magnification fields were selected and photographed. Three independent experiments were analyzed. Ki67- and Olig2-positive cells were calculated as the percentage of total DAPI-positive nuclei per field. Nestin, βIII-tubulin, and GFAP expression were analyzed by calculating the total staining intensity per field divided by the number of DAPI-positive nuclei.
Animals. Experiments were performed according to international guidelines and approved by the local experimental animal committee. At postnatal day 9, C57Bl/6J mice underwent HI by right common carotid artery occlusion under isoflurane anesthesia (3% induction, 1% maintenance in O2:N2O (1:1)) followed by exposure to 10% oxygen in nitrogen for 45 minutes. This procedure induced a lesion involving hippocampus, neocortex, and striatum.36 Sham controls underwent anesthesia and incision only. Pups from at least five different litters were used in each experimental group, and pups of each litter were randomly assigned to all experimental groups taking sex into account in a way that both sexes were equally distributed among experimental groups. Data were obtained from at least two independent experiments. Mortality (~10%) only occurred during or immediately after HI, and there were no sex differences in mortality. All analyses were performed in a blinded set-up.
At 10 days after HI, MSCs or vehicle was delivered intranasally in awake animals. Thirty minutes before MSC or vehicle administration, two doses of 3 µl hyaluronidase (total 100 U, Sigma-Aldrich) in phosphate-buffered saline were applied to each nostril and spontaneously inhaled.37,38 Subsequently, a total of 5 × 105 MSCs in 12 µl phosphate-buffered saline or vehicle were administered as two doses of 3 µl applied to each nostril.
To evaluate cell proliferation/survival, mice received EdU (50 mg/kg intraperitoneally; Life Technologies) on days 10, 11, and 12 after HI and received BrdU (50 mg/kg intraperitoneally; Sigma-Aldrich) on days 21, 22, and 23 after HI. Animals were sacrificed at 28 days after HI and perfused with 4% paraformaldehyde in phosphate-buffered saline.
Functional outcomes. The cylinder rearing test was used to assess forelimb use asymmetry as described.2 Mice were placed in a clear Perspex cylinder (80 mm in diameter and 300 mm in height) for 3 minutes. As they vertically reached up to explore the cylinder wall, the forepaw to touch the wall would be registered. The forepaw to contact the wall during a full rear was recorded as left (impaired), right (nonimpaired), or both. During the 3-minute period, at least 15 weight-bearing contacts should be registered or the mouse would be retested 30 minutes later. Paw preference was calculated as ((nonimpaired–impaired)/(total forelimb usage)) × 100%. Interobserver reliability was 0.87 at 21 or 28 days after HI and 0.76 at 10 days after HI.
Histology and immunohistochemistry. To asses gray and white matter damage, coronal paraffin sections (6 µm) were incubated with MAP2 (Sigma-Aldrich) or mouse-anti-myelin basic protein (Sternberger Monoclonals, Lutherville, MD), and binding was visualized with a Vectastain ABC kit (Vector Laboratories, Burlingame, CA). Brain damage was analyzed at a location equivalent to −1.58 mm from bregma in adult mice by outlining both hemispheres on full-section images using ImageJ software (Rasband WS, ImageJ, NIH, Bethesda, Maryland; http://rsb.info.nih.gov/ij/, 1997–2009). Ipsilateral MAP2 and MBP area loss were calculated as described.2
For cell proliferation analysis, sections were incubated with biotinylated sheep-anti-BrdU and rabbit-anti-Ki67 (both Abcam). Visualization was done with AlexFluor-594–conjugated streptavidin and donkey-anti-rabbit AlexFluor488. EdU incorporation was detected by incubating sections in 100 mmol/l Tris containing 0.5 mmol/l CuSO4, 50 mmol/l ascorbic acid, and 10 µmol/l AlexaFluor-594-azide. BrdU-positive or EdU-positive cells were counted in hippocampus and six cortical high magnification fields in three 200 µm spaced sequential sections per brain.
Statistical analysis. All data are expressed as means ± SEM. Functional outcomes measured with cylinder rearing test was analyzed using two-way ANOVA with Fisher's least significant difference posttests. P < 0.05 was considered statistically significant. Histological measures were analyzed using one-way ANOVA with Bonferroni posttests. P < 0.05 was considered statistically significant.
Acknowledgments
All work was performed in Utrecht, The Netherlands. This study was funded by the European Union (Health-F2-2009-241778, NEUROBID) and Zon-MW Project (No. 116002003).
References
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Repeated mesenchymal stem cell treatment after neonatal hypoxia-ischemia has distinct effects on formation and maturation of new neurons and oligodendrocytes leading to restoration of damage, corticospinal motor tract activity, and sensorimotor function. J Neurosci. 2010;30:9603–9611. doi: 10.1523/JNEUROSCI.1835-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav Immun. 2010;24:387–393. doi: 10.1016/j.bbi.2009.10.017. [DOI] [PubMed] [Google Scholar]
- van Velthoven CT, Kavelaars A, Heijnen CJ. Mesenchymal stem cells as a treatment for neonatal ischemic brain damage. Pediatr Res. 2012;71 4 Pt 2:474–481. doi: 10.1038/pr.2011.64. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Matsukawa N, Yu G, Xu L, Mays RW, Kovach J, et al. Behavioral and histological characterization of intrahippocampal grafts of human bone marrow-derived multipotent progenitor cells in neonatal rats with hypoxic-ischemic injury. Cell Transplant. 2006;15:231–238. doi: 10.3727/000000006783982034. [DOI] [PubMed] [Google Scholar]
- Lee JA, Kim BI, Jo CH, Choi CW, Kim EK, Kim HS, et al. Mesenchymal stem-cell transplantation for hypoxic-ischemic brain injury in neonatal rat model. Pediatr Res. 2010;67:42–46. doi: 10.1203/PDR.0b013e3181bf594b. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Wang L, Lu M, Zhang X, Chopp M. Therapeutic benefit of intracerebral transplantation of bone marrow stromal cells after cerebral ischemia in rats. J Neurol Sci. 2001;189:49–57. doi: 10.1016/s0022-510x(01)00557-3. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Mesenchymal stem cell transplantation changes the gene expression profile of the neonatal ischemic brain. Brain Behav Immun. 2011;25:1342–1348. doi: 10.1016/j.bbi.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Qu R, Li Y, Gao Q, Shen L, Zhang J, Liu Z, et al. Neurotrophic and growth factor gene expression profiling of mouse bone marrow stromal cells induced by ischemic brain extracts. Neuropathology. 2007;27:355–363. doi: 10.1111/j.1440-1789.2007.00792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li Y, Wang L, Katakowski M, Zhang L, Chen J, et al. Ischemic rat brain extracts induce human marrow stromal cell growth factor production. Neuropathology. 2002;22:275–279. doi: 10.1046/j.1440-1789.2002.00450.x. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Steigleder T, Cooper-Kuhn CM, Schwab S, Sommer C, Schneider A, et al. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. 2007;38:2165–2172. doi: 10.1161/STROKEAHA.106.477331. [DOI] [PubMed] [Google Scholar]
- Dikic I, Schmidt MH. Notch: implications of endogenous inhibitors for therapy. Bioessays. 2010;32:481–487. doi: 10.1002/bies.200900140. [DOI] [PubMed] [Google Scholar]
- Nichol D, Stuhlmann H. EGFL7: a unique angiogenic signaling factor in vascular development and disease. Blood. 2012;119:1345–1352. doi: 10.1182/blood-2011-10-322446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- Sims JR, Lee SW, Topalkara K, Qiu J, Xu J, Zhou Z, et al. Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke. 2009;40:3618–3626. doi: 10.1161/STROKEAHA.109.561951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Islam O, Loo TX, Heese K. Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr Neurovasc Res. 2009;6:42–53. doi: 10.2174/156720209787466028. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131:337–345. doi: 10.1242/dev.00930. [DOI] [PubMed] [Google Scholar]
- Schmidt MH, Bicker F, Nikolic I, Meister J, Babuke T, Picuric S, et al. Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol. 2009;11:873–880. doi: 10.1038/ncb1896. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Wei N, Lu T, Zhu J, Xu G, Liu X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience. 2011;172:398–405. doi: 10.1016/j.neuroscience.2010.10.054. [DOI] [PubMed] [Google Scholar]
- Ploughman M, Windle V, MacLellan CL, White N, Doré JJ, Corbett D. Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke. 2009;40:1490–1495. doi: 10.1161/STROKEAHA.108.531806. [DOI] [PubMed] [Google Scholar]
- van Velthoven CT, Sheldon RA, Kavelaars A, Derugin N, Vexler ZS, Willemen HL, et al. Mesenchymal stem cell transplantation attenuates brain injury after neonatal stroke. Stroke. 2013;44:1426–1432. doi: 10.1161/STROKEAHA.111.000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- van Velthoven CT, van de Looij Y, Kavelaars A, Zijlstra J, van Bel F, Huppi PS, et al. Mesenchymal stem cells restore cortical rewiring after neonatal ischemia in mice. Ann Neurol. 2012;71:785–796. doi: 10.1002/ana.23543. [DOI] [PubMed] [Google Scholar]
- Tomac AC, Agulnick AD, Haughey N, Chang CF, Zhang Y, Bäckman C, et al. Effects of cerebral ischemia in mice deficient in Persephin. Proc Natl Acad Sci USA. 2002;99:9521–9526. doi: 10.1073/pnas.152535899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilak MM, Shifrin DA, Corse AM, Bilak SR, Kuncl RW. Neuroprotective utility and neurotrophic action of neurturin in postnatal motor neurons: comparison with GDNF and persephin. Mol Cell Neurosci. 1999;13:326–336. doi: 10.1006/mcne.1999.0756. [DOI] [PubMed] [Google Scholar]
- Milbrandt J, de Sauvage FJ, Fahrner TJ, Baloh RH, Leitner ML, Tansey MG, et al. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron. 1998;20:245–253. doi: 10.1016/s0896-6273(00)80453-5. [DOI] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected]. Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donega V, van Velthoven CT, Nijboer CH, van Bel F, Kas MJ, Kavelaars A, et al. Intranasal mesenchymal stem cell treatment for neonatal brain damage: long-term cognitive and sensorimotor improvement. PLoS ONE. 2013;8:e51253. doi: 10.1371/journal.pone.0051253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi H, Sasaki T, Kawabata K, Sakurai F, Hayakawa T. Fiber-modified adenovirus vectors mediate efficient gene transfer into undifferentiated and adipogenic-differentiated human mesenchymal stem cells. Biochem Biophys Res Commun. 2005;332:1101–1106. doi: 10.1016/j.bbrc.2005.05.055. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, Ohl F, Arndt SS, Kavelaars A, van Bel F, Heijnen CJ. Mild neonatal hypoxia-ischemia induces long-term motor- and cognitive impairments in mice. Brain Behav Immun. 2010;24:850–856. doi: 10.1016/j.bbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Nasal administration of stem cells: a promising novel route to treat neonatal ischemic brain damage. Pediatr Res. 2010;68:419–422. doi: 10.1203/PDR.0b013e3181f1c289. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, Klopfer T, et al. Intranasal delivery of cells to the brain. Eur J Cell Biol. 2009;88:315–324. doi: 10.1016/j.ejcb.2009.02.001. [DOI] [PubMed] [Google Scholar]