

Abstract
Gene transfer into autologous hematopoietic stem cells by γ-retroviral vectors (gRV) is an effective treatment for adenosine deaminase (ADA)–deficient severe combined immunodeficiency (SCID). However, current gRV have significant potential for insertional mutagenesis as reported in clinical trials for other primary immunodeficiencies. To improve the efficacy and safety of ADA-SCID gene therapy (GT), we generated a self-inactivating lentiviral vector (LV) with a codon-optimized human cADA gene under the control of the short form elongation factor-1α promoter (LV EFS ADA). In ADA−/− mice, LV EFS ADA displayed high-efficiency gene transfer and sufficient ADA expression to rescue ADA−/− mice from their lethal phenotype with good thymic and peripheral T- and B-cell reconstitution. Human ADA-deficient CD34+ cells transduced with 1–5 × 107 TU/ml had 1–3 vector copies/cell and expressed 1–2x of normal endogenous levels of ADA, as assayed in vitro and by transplantation into immune-deficient mice. Importantly, in vitro immortalization assays demonstrated that LV EFS ADA had significantly less transformation potential compared to gRV vectors, and vector integration-site analysis by nrLAM-PCR of transduced human cells grown in immune-deficient mice showed no evidence of clonal skewing. These data demonstrated that the LV EFS ADA vector can effectively transfer the human ADA cDNA and promote immune and metabolic recovery, while reducing the potential for vector-mediated insertional mutagenesis.
Introduction
Adenosine deaminase–deficient severe combined immunodeficiency (ADA-SCID) is a severe primary immunodeficiency characterized by impaired T-, B-, and NK-cell development and accounts for 10–15% of all cases of SCID.1 ADA catalyzes the deamination of deoxyadenosine and adenosine to deoxyinosine and inosine respectively, and the lack of ADA leads to increased intracellular conversion of deoxyadenosine to deoxyadenosine triphosphate (dATP) thus expanding the dATP pool. High levels of dATP affect lymphocyte development, viability, and function causing the immune defects seen in this condition.2 Clinically, patients present with failure to thrive, recurrent and opportunistic infections and death in the first year of life if left untreated.3,4 A murine model recapitulates the human disease with similar metabolic and immunological abnormalities and untreated mice die after 3 weeks from pulmonary insufficiency, which results from the metabolic consequences of the disease.5
Treatment options for ADA SCID are limited and the mainstay of treatment is allogeneic hematopoietic stem cell transplant (HSCT) which offers good survival outcome when well-matched family donors are available. Survival following HSCT from matched unrelated donors (67%), mismatched unrelated donors (29%), or parental donors (43%) are less good.6 Enzyme replacement therapy (ERT) with pegylated bovine ADA (PEG-ADA) results in effective metabolic detoxification, but long-term immune recovery is suboptimal and very poor in some cases.7 Thus, there is a clear need for effective and sustained alternative treatment options.
ADA-SCID has long been held as a model disorder for gene therapy (GT) and was the first genetic disorder for which GT was attempted. Early trials of GT using γ-retroviral vectors (gRVs) targeting correction of peripheral blood (PB) lymphocytes or autologous hematopoietic stem cells (HSCs) or a combination of the two showed limited success, and immune recovery could not be attributed to GT alone, since ERT was continued after the GT procedure.8 Subsequent trials also using gRVs but with the use of nonmyeloablative conditioning and withdrawal of ERT have shown improved outcomes with recovery of immune and metabolic parameters.9,10 In the three studies so far undertaken, 31 of 42 patients (73.8%) have remained off ERT following GT, but immune reconstitution remains suboptimal with T-cell numbers at the lower limit of the normal range and approximately half of the patients remaining on immunoglobulin replacement therapy due to incomplete B-cell reconstitution.11,12,13
More importantly, despite the absence of any adverse events in ADA-SCID patients, the ongoing use of gRVs has raised concerns. In clinical trials of gRV-mediated autologous HSC GT for SCID-X1, X-CGD, and Wiskott–Aldrich syndrome, there has been a high incidence of gRV-mediated insertional mutagenesis.14,15,16,17,18,19 Upon vector integration, the strong enhancer elements that reside in the long terminal repeat (LTR) promoter elements of gRVs can transactivate adjacent genes to initiate the transformation process. In ADA gRV studies, vector insertions near known oncogenes have also been reported, although there have been no clinical clonal outgrowths.20 A number of regulatory agencies have recommended a move away from the continued use of gRVs and the development of safer vector designs. Self-inactivating (SIN) vectors, based on the HIV-1 lentiviral vector (LV), in which the HIV LTR is deleted and transgene expression placed under the control of an internal promoter with minimal or no enhancer activity have received considerable attention.21,22,23 The advantages of a SIN LV include the improved ability of LV to transduce long-term engrafting HSC which may allow improved immune recovery but also the significantly decreased potential for insertional mutagenesis, which has been demonstrated in a number of in vitro24,25 and in vivo studies.26,27
For these reasons, we investigated the use of a SIN LV the for treatment of ADA-SCID. Following in vitro comparative studies using LVs expressing human ADA under the transcriptional control of either the phosphoglycerate kinase promoter, the MND retroviral vector LTR, or the short form of the elongation factor 1α promoter (EFS), the LV EFS ADA vector configuration was selected for further preclinical assessment in relevant models of ADA deficiency.28 We demonstrate that the LV EFS ADA is capable of effective and consistent ADA gene transfer and expression that is equivalent or greater than the currently used gRV, while having a significantly lower capacity for inducing clonal outgrowth. The results obtained from this study provide compelling evidence for the translation of this vector to clinical application.
Results
LV EFS ADA can efficiently transduce murine and human HSC
We have previously cloned the LV EFS ADA vector with a third generation SIN LV design under the transcriptional control of the short version EFS promoter (Figure 1a,b).28 The transduction efficiency and transgene expression of the LV vector was confirmed in multiple cell lines in vitro (data not shown). We then compared transduction efficiency and transgene expression of the LV EFS ADA with the gRVSFada/W vector used in a clinical trial for ADA-SCID GT in primary cells.13 Lineage depleted bone marrow cells (Lin-) isolated from ADA−/− mice (ADA−/− HSC) were isolated and transduced with viral vectors at a multiplicity of infection (MOI) of 20 under optimized protocols. Normalized enzymatic activity (ADA activity/vector copy) showed that LV EFS ADA had similar efficacy of transgene expression in murine ADA−/− HSCs compared with gRVSFada/W (Figure 1c). Similar levels of ADA protein expression were detected in ADA−/− HSC by western blot analysis (Figure 1d) and demonstrated that LV EFS ADA mediates efficient transduction and transgene expression in ADA−/− HSCs.
Figure 1.

Viral constructs and transgene expression in murine lineage depleted bone marrow and human cord blood CD34+ cells. (a,b) Schematic representation of viral vectors. (a) Retroviral vectors (gRV): the γ-retroviral vector, MND-MFG-ADA (gRV MND ADA) contains the MND retroviral LTRs flanking the wild-type human adenosine deaminase cDNA (hADA) with the Moloney murine leukemia virus packaging region (Ψ) and env splice acceptor fragment (env SA). The gRVSFada/W vector contains hADA driven by SFFV LTR. (b) Lentiviral vectors (LV): All lentiviral vectors contain the enhancer-deleted “SIN” LTR (indicated by the X in the U3 region), the primer binding site (Θ), the HIV-1 packaging signal (Ψ), the central polypurine tract (cPPT), the rev-responsive element (RRE). LV MND ADA contains the MND LTR U3 region enhancer/promoter (MND) driving expression of the hADA cDNA. LV EFS ADA contains the human elongation factor-α gene “short” promoter (EFS) driving expression of the codon-optimized human ADA cDNA (co-hADA) and a Woodchuck Hepatitis Virus posttranscriptional regulatory element (wPRE). The LV EFS GFP vector contains the EFS promoter and green fluorescent protein (GFP). (c,d) Murine ADA−/− bone marrow lineage negative (Lin-) progenitors were transduced for 24 hours with lentiviral or retroviral vectors at a multiplicity of infection (MOI) of 20 after 24 or 72 hours preactivation, respectively (Mean ± SD). ADA expression was analyzed 72 hours after transduction by (c) enzymatic activity assay and (d) Western Blot with whole cell lysates. Mean and standard deviation of ADA activities were calculated from experiments performed with cells obtained from three different ADA−/− donors. (e–g) Human cord blood CD34+ cells were transduced with the vectors at the indicated vector concentration, grown for 2 weeks in myeloid differentiation culture, and assayed for (e) vector copy number (VCN) by quantitative polymerase chain reaction (qPCR) and for (f) ADA enzyme activity by colorimetric assay. (g) The ADA enzyme activity present per VC was calculated. Horizontal bars indicate Mean ± SEM.
LV EFS ADA was also compared to another gRV used previously in a clinical trial, gRV MND-ADA,12 and an LV in which the MND LTR U3 enhancer/promoter controls ADA expression (LV MND-ADA). Human cord blood CD34+ cells (CB HSC) were transduced with LV EFS ADA over a range of LV concentrations (106–108 TU/ml; MOI = 10–1,000) and with gRVMND-ADA (generated from a stable PG13 cell clone) at 1.8 × 105 TU/ml. After short-term myeloid culture for 2 weeks, there was a significant dose-dependent trend between LV concentration during transduction and both the resultant vector copy number (VCN) (P = 0.002) and ADA gene expression as measured by the ADA enzyme activity over background activity (P = 0.003) (Figure 1e,f). Transduction of CB HSC with each of the LVs at 1 × 107 TU/ml resulted in 1–3 vector copies per cell, and gRV MND-ADA, applied at a 100-fold lower dose, resulted in only ~0.2–0.8 copies per cell, but when normalized for VCN, activity was similar for LV EFS ADA and gRV MND-ADA (~1–2 ADA U/VC) (Figure 1g). Although, ADA activity/VC was higher with LV MND-ADA (P = 0.03), it is not a preferred choice for clinical HSC GT, as it harbors a strong gRV LTR enhancer/promoter which has increased mutagenic potential.29
Transplantation of LV EFS ADA transduced ADA−/− Lin- cells rescues lethality in ADA−/− mice
ADA−/− mice normally die within 3 weeks following withdrawal of PEG-ADA.5 We transplanted young adult ADA−/− mice with ADA−/− Lin- cells that had been transduced with either LV EFS ADA or SFada/W or with ADA+/+ Lin- cells (WT Lin-group). For all groups, PEG-ADA treatment was carried over for 4 weeks after transplantation to promote engraftment30 before complete withdrawal. The survival rate was 100% in the LV EFS ADA group and the WT group, which was significantly higher than ADA−/− SFada/W group in which the survival rate was 40% (P = 0.02) (Figure 2a).
Figure 2.
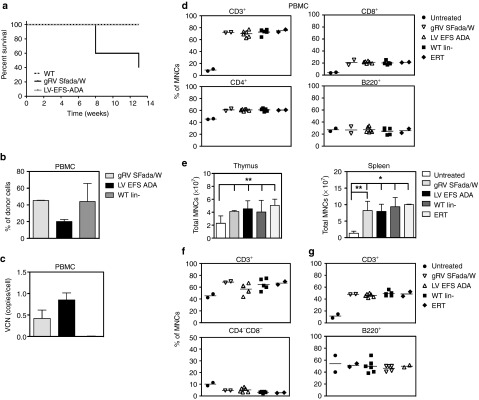
Survival rate, peripheral blood (PB) analysis, and immunophenotype of ADA−/− recipients. (a) Survival: Kaplan–Meier curves of ADA−/− recipients were transplanted with transduced ADA−/− BM Lin- cells (LV EFS ADA, n = 6) and gRVSFada/W (SFada/W, n = 5), respectively, at a multiplicity of infection (MOI) of 20. Control mice were injected with untransduced BM Lin- cells from ADA+/+ donors (WT Lin-, n = 5). All LV EFS ADA and WT mice were alive at 13 weeks compared to gRVSFada/W group, where two mice died at 7 weeks and one died at 12 weeks with the remaining two alive at 13 weeks (P = 0.02). All surviving mice were euthanized for analysis unless otherwise indicated. (b) Percentage of donor cells in total PB mononuclear cells (PBMCs) by quantitative PCR (qPCR). (c) Vector copy number (VCN) in PBMCs of transplanted ADA−/− mice. Percentage of DNA with Y chromosome were evaluated in sex-mismatched transplants indicated in 1a (Mean ± SD). (d) FACS analysis of circulating mature T and B cells in peripheral blood of ADA−/− transplants. Untreated ADA−/− mice (untreated, 18 days old, n = 2) and 4–5 months old PEG-ADA treated ADA−/− mice (ERT, n = 2) were analyzed as controls. Data are displayed as percentage of CD3+, CD4+, CD8+, and B220+ cells in PBMCs. Horizontal bars indicate the average values. (e) Total mononucleated cell counts in thymi and spleens (*P < 0.001; **P < 0.05). Results are given as Mean ± SD. (f) FACS analysis of thymocytes of ADA−/− recipients and control mice. Data are presented as percentage of total CD3+ and CD4−CD8− cells in mononucleated cells. Horizontal bars indicate the average values. (g) FACS analysis of splenocytes in ADA−/− recipients and control mice. Data are presented as percentage of CD3+ and B220+ cells in total mononucleated cells. Horizontal bars indicate the average values.
To evaluate integration of the viral vector and engraftment of donor cells in vivo, we performed quantitative polymerase chain reaction (qPCR) to evaluate VCN and male donor Y chromosome mononuclear cell (MNC) engraftment in PB at 13 weeks after transplantation. Similar levels of donor cells were present in the PB of the gRVSFada/W group (45.3 ± 0.4%) and WT Lin- (44 ± 21.7%) groups, while the level of donor cells was twofold less in the LV EFS ADA group (20.1 ± 2.5%) (Figure 2b). However, in the latter, VCN was 0.85 ± 0.16 copy/cell compared with 0.42 ± 0.2 copy/cell in the gRVSFada/W group (Figure 2c).
Comparison of immune recovery after LV- and gRV-mediated gene delivery in ADA−/− mice
We then analyzed the total numbers of PB mononuclear cells (PBMC) and the percentages of lineage specific populations within the mononuclear cell population (% of MNCs). Compared to all other groups, untreated ADA−/− mice had reduced numbers of total PBMCs and less than 10% of the MNCs were CD3+ (Figure 2d). In the LV EFS ADA or gRVSFada/W groups, more than 69% of the MNCs were CD3+, which included elevated levels of both single positive CD4+ and CD8+ cells which were similar to the levels observed in mice transplanted with WT HSCs (WT Lin- group) or age-matched mice under PEG-ADA treatment (ERT group). Although no significant improvement was detected in the percentage of B220+ cells with any treatment, taking into account the total cell numbers, the absolute number of B220+ cells in all transplants was much improved in comparison to untreated ADA−/− mice. In addition, GR-1+ myeloid cells and NK1.1+ cell numbers were corrected to relatively normal levels (data not shown).
To evaluate the development of the immune system upon ADA restoration, lymphoid organs including thymus and spleen were isolated from untreated and treated ADA−/− mice and lymphoid subpopulations were analyzed by flow cytometry. Overall, untreated ADA−/− mice had the lowest total cell numbers (Figure 2e), as well as the lowest CD3+ and B220+ percentages of total mononuclear cells, in the thymus and spleen (Figure 2f,g). In the thymus, the early CD4−CD8− population was twofold higher in untreated mice, highlighting the block in thymocyte development found in ADA deficiency31 (Figure 2f, lower panel). In transplants with LV- or gRV-transduced HSCs, we found significantly increased MNCs in all spleens (P < 0.05), and to a lesser extent, in thymi (P < 0.05) (Figure 2e). Among thymocytes, the CD3+ proportion was significantly elevated (P < 0.05). More importantly, the average percentage of CD4−CD8− double negative thymocytes was reduced from 12% in untreated ADA−/− mice to only 4.6% in GT groups. This result demonstrates that the block in thymocyte development was overcome with restored ADA expression. In spleens of mice receiving transduced cells, there was an elevated percentage of CD3+ cells compared to untreated controls (P < 0.05) that was similar to WT transplants and mice treated with ERT alone (Figure 2g). B-cell percentages were similar to those in untreated mice but in relation to total cell number, there was a significant increase in absolute number of both CD3+ and B220+ cells in LV and gRV groups with no significant difference between these two groups (Figure 2e,g). These results suggest that the proliferation and differentiation of both T and B cells has been restored in the GT groups at levels that were similar to the levels in the WT transplant and ERT groups.
Systemic detoxification following LV- and gRV-mediated gene delivery
In recipients with LV-transduced cells, we detected a twofold increase in VCN in thymus (0.2 ± 0.15), spleen (1.1 ± 0.34), marrow (0.44 ± 0.32) compared to those with gRV-transduced cells (Figure 3a). Donor cell engraftment was determined in mismatched sex transplants by qPCR for sequences on the Y chromosome. The level of Y chromosome detected in the spleens of mice in the LV EFS ADA group was twofold lower (19.6 ± 2.0%) compared to the gRVSFada/W group. Likewise, the level of donor engraftment or level of Y chromosome in thymi from mice in the LV EFS ADA group were also twofold lower than those with gRVSFada/W, and ten fold lower than engraftment in the WT Lin- group (Figure 3b). Although donor engraftment appears to be lower with transduced (LV or gRV) cells, this may represent dilution of the transplanted donor cells with the host cells that are crosscorrected by the overexpression of ADA from the vector in the primary lymphoid organs. This dilution effect has been described previously and is specific to ADA-SCID because there is crosscorrection of uncorrected ADA-deficient cells with adequate ADA activity provided by ERT, HSCT, or GT.30
Figure 3.

Engraftment of transduced cells, immune reconstitution, and systemic detoxification in ADA−/− recipients. (a) Vector copy number analysis of thymus, spleen, and bone marrow in ADA−/− recipients. (Mean ± SD). (b) Percentage of donor cells in thymus, spleen, and bone marrow by quantitative PCR (qPCR). Percentage of DNA with Y chromosome were evaluated in sex-mismatched transplants indicated in 1a (Mean ± SD). (c) ADA activity and (d) SAHH activity in red blood cells, BM cells, thymocytes, and lung tissue of ADA−/− transplants and control mice were measured by enzymatic activity assay as indicated (Mean ± SD). (e) Histopathologic analysis of lung sections from ADA−/− transplanted with ADA−/− Lin- gRVSFada/W or ADA−/− Lin- LV EFS ADA or ADA+/+ WT Lin- cells compared to lung sections from 18-day-old untreated ADA−/− and ADA−/− mice under ERT (ADA−/− ERT) mice. All sections have been stained with hematoxylin and eosin.
ADA−/− mice have undetectable ADA enzyme activity and decreased SAHH activity in most tissues and organs.5 To confirm the expression of functional ADA by LV EFS ADA and gRVSFada/W in vivo, we analyzed ADA and SAHH activities in multiple systems including nonlymphoid organs such as the lung (Figure 3c,d). In PB and lymphoid organs including spleen and thymus, ADA activities in the LV EFS ADA and gRVSFada/W groups were comparable to activities in WT Lin- transplantation group in (Figure 3c), which is noteworthy given the engraftment of transduced ADA−/− donor cells in the thymus was measured to be ten fold lower compared to ADA+/+ WT Lin- donor cells (Figure 3b).
In a nonimmune organ, such as the lung, ADA enzymatic activity was also equivalent in all GT treated mice in comparison to WT transplants and undetectable in untreated mice (P < 0.05). Similar ADA activities were also found in livers of the transplants (data not shown). Inhibition of S-adenosyl homocysteine hydrolase (SAHH) activity is secondary to the accumulation of dATP in ADA-deficient mice.5 In all transplants, untreated mice showed absent or low levels of SAHH activity in RBC, thymus, spleen, BM, and lung, whereas LV GT treated mice showed increased SAHH activity to levels similar to the WT Lin-–treated mice (P < 0.05) (Figure 3d). These results demonstrate that LV EFS ADA–mediated gene transfer can lead to efficient metabolic correction in the ADA-deficient mouse that is comparable to correction with gRVSFada/W and WT Lin- HSCT.
We also studied other organ pathologies in untreated and treated mice. Nonlymphoid organs including lung, liver, heart, and kidney were harvested and examined histologically. The dominant pathologic improvements were observed in the lungs of treated mice (Figure 3e). Untreated ADA−/− mice show occlusion of the airways and thickening of airway epithelium with accumulation in the airspaces of proteinaceous material and infiltration of alveolar macrophages. There was a striking improvement in all treatment groups, including LV EFS ADA and gRVSFada/W groups, with clearance of interstitial fluid and absence of inflammatory cells with lung histology similar to that seen in ADA−/− WT Lin- mice. There were no predominant findings in the structure or organization of other organs in untreated and treated mice (data not shown).
LV EFS ADA transduction and expression is dose-dependent in human HSC
Human CD34+ cells were isolated from multiple samples of normal ADA-replete human CB (CB HSC) (n = 5) and BM (BM HSC) (n = 4), and BM from infants with ADA-deficient SCID (ADA-SCID HSC) (3-month-old (n = 2) and 9-month-old (n = 1) donors). The prestimulated CD34+ cells were either transduced with LV EFS ADA over a range of vector concentrations (1 × 106 to 1 × 108 TU/ml), corresponding to a range of MOI of 10–1,000, or mock transduced, and analyzed after 14-day culture. There was a significant dose-dependent relationship between the LV EFS ADA concentration during transduction and the resultant VCN in CB HSC (P = 0.012) and ADA-SCID HSC (P = 0.004), but not in BM HSC (Supplementary Figure S1a). Similarly, there were significant dose-dependent correlations between the LV EFS ADA vector concentration during transduction and the resultant ADA expression (ADA activity over background) in CB HSC (P = 0.03) and infant ADA-SCID HSC (P = 0.002), but not in adult BM HSC (Supplementary Figure S1b).
The endogenous ADA enzyme activity measured in mock-transduced ADA-deficient BM cells from the several experiments was 10- to 30-fold lower (Mean ± SEM: 0.05 ± 0.02 U) than in mock-transduced normal CB cells (Mean ± SEM: 0.54 ± 0.07 U) (P = 0.029) or mock-transduced normal BM cells (Mean ± SEM: 1.6 ± 0.82 U) (P < 0.0001) (Supplementary Figure S1c). Across all vector concentrations and MOIs used for transduction by LV EFS ADA, the expression of ADA enzyme activity normalized to single vector copy was 1.69 ± 0.15 U/VC in the normal CB samples; 1.67 ± 0.33 U/VC in the normal BM samples; and 1.2 ± 0.10 U/VC in the ADA-deficient BM samples (Supplementary Figure S1d). The ADA activity in the LV EFS ADA–transduced ADA-deficient SCID BM cells was 24-fold higher than endogenous levels in the mock-transduced samples; ADA activity in LV EFS ADA–transduced normal donor CB and BM HSC was one- to threefold over endogenous levels in mock-transduced ADA-replete donor samples (Table 1).
Table 1. ADA enzyme activity after 2-week in vitro myeloid culture: endogenous in human hematopoietic cells and expressed by EFS-ADA after CD34+ cell transduction.

LV EFS ADA lentiviral transduction and engraftment of human CB HSC and the role of IL-3
To further evaluate LV EFS ADA and to gain insight into the effects of IL-3 on LV transduction of human HSC, we compared transduction and long-term engraftment of the CB CD34+ cells, with and without IL-3 included in the prestimulation and transduction media. CB HSC (n = 2) were thawed, plated (500,000 cell/ml), and prestimulated for 20 hours in medium containing human stem cell factor, human FLT3-L and human TPO (S/F/T), with or without IL-3 (20 ng/ml). The prestimulated cells were transduced with LV EFS ADA (3.0 × 107 TU/ml) or mock transduced. To test the effects of IL-3 exposure on the engraftment of more primitive stem/progenitor cells, LV EFS ADA–transduced or mock-transduced CD34+ cells were xenotransplanted into Nod/SCID/γ C (NSG) primary and secondary mouse recipients.
The VCN measured after 14-day short-term culture was twofold higher with IL-3 (2.5 ± 0.8) compared to without IL-3 (1.2 ± 0.4), but this difference was not significant (Figure 4a). Likewise, there were no significant differences in the total numbers of colony-forming units (CFUs) produced per 1,000 plated CD34+ cells in the LV-transduced group (no IL-3: 87/1,000 = 8.7%, with IL-3: 109/1,000 = 10.9%) compared to the mocktransduced (no IL-3: 109/1,000 = 10.9%, with IL-3: 83/1,000 = 8.3), nor in the different types of colonies formed (Figure 4b,c). Although inclusion or exclusion of IL-3 did not make a significant difference in the percentage of CFU colonies containing LV sequences (39.3% with IL-3 versus 31.1% without IL-3), the mean VCN in DNA from individual CFU was 3.1-fold higher when IL-3 was included (15.1 ± 2.1) than when it was not (4.9 ± 10) (P = 0.001) including, a subset of CFU with an average VCN > 10 only when exposed to IL-3 (Figure 4d,e).
Figure 4.

The role of IL-3 in EFS-ADA lentiviral transduction of human cord blood CD34+ cells and engraftment in NSG mice. Human cord blood CD34+ cells were transduced with LV EFS-ADA (3 × 107 TU/ml) in medium with recombinant human cytokines SCF/ckit ligand, flt-3 ligand, and thrombopoietin (TPO), with or without interleukin-3 (IL-3). (a-d) In vitro: (a) transduced cells cultured for 14 days in vitro under myeloid differentiation conditions and analyzed for VCN (14d VCN) (N.S, not significant). (b,c) Transduced CD34+ cells were grown in colony-forming unit (CFU) assay in methylcellulose and assayed after 2 weeks. Colony (b) enumeration and (c) types formed by CD34+ cells in CFU assay. (d,e) CFU were harvested and DNA analyzed by qPCR for VCN (d) Transduction efficiency was measured by the percentage of colonies positive for vector sequence by PCR for the human ADA cDNA (%PCR (+) CFU). (e) VCN was quantified in DNA extracted from individual CFU by qPCR (*P value = 0.001). (f-h) In Vivo: other portions of the transduced CD34+ cells were transplanted into NSG mice and analyzed after 4 months for engraftment of human cells based on FACS analysis of huCD45 expression and for VCN by qPCR. Engraftment of human cells in (f) bone marrow, thymus (when present) and spleen by FACS of tissue cell suspensions immunostained with anti-human CD45 (%hCD45+). (g) EFS-ADA VCN in bone marrow, thymus (when enough cells were available for analysis; total n = 3),and spleen. (h) Immunophenotypic analysis of human CD45+ cells in NSG bone marrow (CD34+ and CD33+), thymus (CD4−/CD8− double-negative (DN), CD4+/CD8+ double-positive (DP), CD4+ single-positive (SP-4) and CD8+ single-positive (SP-8)) and spleen (CD19+ and CD3+).
Between postnatal day 1 and 3, sublethally irradiated (150cGy) NSG neonates were transplanted with 100,000 CB CD34+ cells (IV), either mock-transduced with IL-3 (n = 5) or without (n = 5), or transduced with LV EFS ADA with IL-3 (n = 13) and without IL-3 (n = 14). Engraftment was not different in tissues isolated from mice transplanted with LV EFS ADA transduced or mock-transduced human CD34+ cells, with or without IL-3 (Figure 4f). VCN was measured in bone marrow, thymus, and spleen and corrected for the level of engraftment (FACs for huCD45) and was not different in tissues isolated from mice transplanted with LV EFS ADA transduced with or without IL-3 (Figure 4g). There were no significant differences in the lineages of the engrafted human cells in the BM (CD34 and CD33), spleen (CD19 and CD3), and thymus (DN, DP, SP-4, SP-8) in any of the groups (Figure 4h). Unfortunately, only 1 out of a total of 35 secondary adult recipient mice from two separate experiments had human cell engraftment, and therefore, we were unable to determine any effects of IL-3 on HSC transduction at the most primitive stem cell level.
LV EFS ADA transduction of human ADA-deficient SCID BM HSC
We further evaluated LV EFS ADA for transduction efficacy, engraftment, and differentiation in human ADA-deficient SCID BM CD34+ cells (ADA-SCID HSC). ADA-SCID HSC were freshly isolated and transduced with LV EFS ADA at 3.3 × 107 TU/ml or mock transduced (n = 3) and analyzed in parallel by in vitro assays and in vivo by transplantation into NSG neonates. CFU assays in methylcellulose were enumerated and characterized by their morphology for lineage type after 12 days. The LV- and mock-transduced cells grew similar numbers and types of colonies: LV EFS ADA 289 colonies/14,000 cells plated (2.1%); mock transduced 50 colonies/2,000 cells plated (2.5%) (Figure 5a,b). Colonies that grew from the LV EFS ADA–transduced cells (n = 2) were 95% positive for the LV vector sequence (Figure 5c). After short-term in vitro myeloid culture, LV EFS ADA–transduced cells had a mean ADA activity of 4.6 ± 1.4 U, which was >92-fold higher than the background ADA activity of mock-transduced ADA-deficient cells (0.05 ± 0.02 U; one-sided P = 0.03) (Figure 5d). The LV EFS ADA–transduced cells had a mean VCN of 2.92 ± 0.75 and expressed 1.55 ± 0.22 ADA U/VC.
Figure 5.

EFS-ADA transduction of normal and ADA-deficient human cord blood and bone marrow CD34+ cells analyzed in vitro and in vivo. ADA-deficient severe combined immunodeficiency (SCID) bone marrow CD34+ cells from two donors in three separate experiments were isolated and transduced with the EFS-ADA vector at 3 × 107 TU/ml or mock-transduced, cultured in short-term myeloid culture for 2 weeks and then harvested and analyzed. (a–c). In vitro: CFU progenitor assays. (a) Enumeration of lineage committed progenitors (b) the frequency of colonies of different lineages. (c) Transduction efficiency determined by the presence of vector sequence in DNA from isolated colonies. (d) In vitro ADA activity (U) measured in mock-transduced and in EFS-ADA transduced bone marrow CD34+ cells and the VCN and expressed ADA activity (U)/VC measured in the EFS-ADA transduced cultures. (e–i) In Vivo: NSG humanized mice. (e) Engraftment of human (%hCD45+) cells in the bone marrow, thymus, and spleen of NSG mice 4 months after transplantation with mock-transduced or EFS-ADA-transduced human ADA-deficient SCID bone marrow CD34+ cells. (f) Human CD45+ leukocyte populations by immunophenotype in bone marrow, thymus, and spleens from NSG mice (g) ADA enzyme activity (U) (h) EFS-ADA VCN and (I) ADA activity (U)/VC) in cells isolated from the bone marrow (huCD45-selected), thymus (total thymocytes), and spleen (huCD45-selected).
Four months after HSCT of ADA-SCID HSC into NSG mice, engraftment of human cells varied considerably among recipients (2–90%) but was not different with LV EFS ADA compared to mock-transduced cells in bone marrow (mock 31.2%; LV 28.7%), spleen (mock 28.9%; LV 34.7%), or thymus (mock 95.9%; LV 90.0%) (Figure 5e). Notably, transduction by the LV EFS ADA vector did not impair differentiation of the ADA-SCID HSC. In the bone marrow of mice transplanted with mock-transduced cells, 9.5% (±1.2) of the human CD45+ cells expressed the hematopoietic stem/progenitor cell marker CD34 compared to 8.5% (±1.3) in mice transplanted with LV EFS ADA–transduced cells. Similarly, myeloid markers CD14 and CD11b were expressed on 7.3% (±4.5) and 10.4% (±4.4) of the human CD45+ cells from mice transplanted with mock-transduced and LV EFS ADA–transduced cells, respectively (Figure 5f).
Thymocytes isolated from recipients of LV EFS ADA–transduced ADA-SCID HSC had typical proportions of CD4/CD8 double-negative (10.7%), CD4/CD8 double-positive (54.6%), CD4 single-positive (10.1%), and CD8 single-positive cells (24.6%) (Figure 5f). In contrast, thymocytes isolated from recipients of mock-transduced ADA-deficient CD34+ cells had typical proportions of only CD4/CD8 double-negatives (12.4%) and CD4 single-positive cells (11.7%), but had significantly more CD8 single-positive cells, (64.7%; P = 0.020) and significantly less double-positive cells (11.2%; P = 0.028) compared to mock transduced, suggesting abnormal thymopoiesis without ADA gene correction. In mature lymphocyte populations, there was no difference in the percentages of splenic CD3+ human T cells produced from the LV-transduced cells (13.2%) or mock-transduced cells (18.7%), but there was a higher percentage of splenic CD19+ human B cells produced from the LV-transduced cells (68.8%) compared to the mock-transduced cells (51.6%) (P = 0.047).
ADA enzyme activity was analyzed in enriched populations of human CD45+ cells isolated from the bone marrow and spleen, and from total thymocytes of the NSG mice. The mean ADA activity in thymocytes was 0.05 ± 0.01 U from mice transplanted with mock-transduced cells and was 0.52 ± 0.30 U from mice transplanted with LV EFS ADA–transduced cells (Figure 5g). The relatively high ADA activity detected in mock transduced BM and spleen most likely derive from the murine cells, which are replete for ADA expression, contaminating the human CD45-enriched populations. VCN consistently averaged between 3.7 and 5.2 VC per cell in the three organs analyzed, with 1.3–1.8 U/VC ADA enzyme expression (Figure 5h,i).
LV EFS ADA shows significantly decreased transformation potential in comparison to gRV vectors
A major concern regarding the continued clinical use of gRV is the risk of insertional mutagenesis. The in vitro immortalization (IVIM) assay has demonstrated the capability to detect transformation of virally transduced cells under myeloid differentiation conditions.25 In two independent studies, performed in the United Kingdom and in the United States, we adopted this approach and compared the LV EFS ADA to the gRVSFada/W and gRV MND-ADA vectors. A second vector using the promoter/enhancer element of spleen focus-forming virus driving the green fluorescent protein reporter gene (gRVSF91GFP) was also used as a positive control in both studies. In the UK study, another positive control was included using a SIN LV design but with an internal SFFV promoter (LV SF GFP). In both studies, cells were also subject to mock transduction in similar culture conditions to monitor background activity.
In the UK study, the gRV SF91 GFP vector induced positive replating clones in all experiments and the LV SF GFP vector did so in two out of four experiments. Most notably, the clinical gRVSFada/W vector also displayed positive replating activity in all experiments, suggesting that this vector has strong transformation ability. The LV EFS ADA–transduced cells did not produce clones with higher proliferative capacity than mock-transduced cells in any of the four independent experiments (Figure 6a). The replating index (replating frequency/VCN) was calculated and, both gRVSF91GFP and gRVSFada/W vectors had high replating indices. The LV SF GFP vector harboring internal SFFV promoter displayed a relative lower replating index than gRVs. Importantly, the LV EFS ADA vector generated no detectable mutants resulting in a negative replating index. In a modification of previously described IVIM assays,28 cell proliferation was detected using the WST-1 assay method (IVIM-WST1 assay), in which viable cell numbers were determined by the measurement of products generated from cleavage of WST-1 by mitochondrial dehydrogenases thereby allowing a quantitative assessment of the growth of replating clones. Four independent experiments were conducted and the highest reading from mock-transduced clones was set as the threshold, values above which were regarded as positive clones (Supplementary Figure S2).
Figure 6.
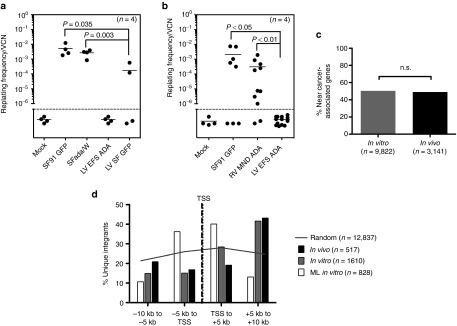
Assessments of genotoxicity of EFS-ADA lentiviral vector. In vitro immortalization (IVIM) Assay. Murine Lin- cells transduced by the indicated vectors were expanded as mass cultures for 2 weeks. An aliquot was taken for qPCR for VCN measurement. On day 15, cells were plated into 96-well plates at a density of 100 cells/well or 1,000 cells/well in 100 µl medium. Two weeks later, the wells showing abundant cell growth were counted as positive, and the frequency of replating cells was calculated based on Poisson statistics using L-Calc Software (Stemcell Technologies, Vancouver, Canada). Horizontal bars indicate mean values. (a) Replating frequency corrected for VCN group by investigators at GOSH, UK. (b) Replating frequency corrected for VCN group by investigators at University of California, Los Angeles, USA (c,d) Vector integration site analysis in human ADA-deficient bone marrow in vitro and in vivo. (c) The percentages of unique integration sites in human cells (isolated from primary NSG mouse recipient bone marrow) near cancer-related genes were determined in vitro (n = 9,822 unique sites) or in vivo (n = 3,141 unique sites). Integration sites in genes or within 300 kb of gene TSS were considered “near” and cancer-related genes were defined as in Higgins et al. 40.(d) The EFS-ADA vector integration sites were mapped relative to transcriptional start sites (TSS) in vitro (n = 1,610 unique sites) and in vivo (n = 517), and compared to a published data set for MLV1 (n = 828). Grey line represents the theoretical random distribution (n = 12,837).
In the US study, four independent experiments (13 assays) were conducted. The gRVSF91.GFP and gRVMND-ADA retroviral vectors produced abundant immortalized clones, with replating frequencies/VCN of 3.36 × 10−3 (or 1 in 306) and 3.68 × 10−4 (or 1 in 2,717), respectively. No colonies were formed by the mock-transduced or the LV EFS ADA–transduced cells across all 13 assays performed. The frequency of replating by LV EFS ADA was significantly lower when compared to gRV SF91.GFP, (P < 0.001) and when compared to gRVMND-ADA (P < 0.001; by two sample nonparametric Wilcoxon rank sum test) (Figure 6b; Supplementary Table S1).
The distribution of unique vector integration sites was determined in LV EFS ADA–transduced human BM and CB CD34+ cells prior to transplant (in vitro) and in cells from the BM of NSG mice 4 months after transplantation (in vivo). LV EFS ADA integration patterns seen in the human cells after the short-term culture and in the cells after xenografting in vivo were essentially identical, with no in vivo clonal skewing toward a higher frequency of vector integrants in cancer-associated genes (Figure 6c), and no dominant clones observed and also no increase of integrants near the 5′ transcriptional start sites of genes (Figure 6d).
Discussion
While past clinical studies using gRV for GT of ADA-deficient SCID have provided clear clinical benefit, the level of immune recovery has, in general been, suboptimal; with T-cell numbers at the lower end of the normal range and less than 50% of patients being able to stop immunoglobulin replacement.12,13 Furthermore, although success has been achieved with no transformation events using gRVs, the potential concern regarding vector-mediated leukemogenesis has remained. Thus, improvement of clinical outcomes and increased safety concerns has necessitated the development of more efficacious and safer vector designs. Ideally, a new vector for future clinical trials for the treatment of ADA-SCID should achieve the following: efficient ADA gene transfer to HSC, high and consistent ADA enzyme activity with low VCN, reduced probability of acute cytotoxicity or genotoxicity, and improved immune reconstitution. In an attempt to fulfill these requirements, we developed and compared the LV EFS ADA vector, in which the codon-optimized hADA cDNA is driven by an internal mammalian EFS promoter in a SIN-configuration background (LV EFS ADA), to the gRV ADA vectors that have been used successfully in ADA GT clinical trials. In our studies, LV EFS ADA led to efficient transduction of HSCs with shortened ex vivo culture time, delivered therapeutic levels of ADA transgene expression via progenitor cells into multiple differentiated lineages, rescued ADA−/− mice from lethality and corrected immune system and metabolic abnormalities. Most importantly, the LV EFS ADA vector showed a significant reduction in transformation potential.
The level of ADA expression is likely to be important in allowing full correction of the disease. In the LV EFS ADA vector, we used a codon-optimized version of the human ADA cDNA to maximize expression and biological activity of transgene, in combination with the internal intron-deleted EFS promoter. Because this shortened version of the human elongation factor 1-α promoter lacks its strong enhancer, expression may be reduced compared to the viral SFFV enhancer/promoter. Nevertheless, LV EFS ADA achieved high ADA expression and activity with low VCN in LV EFS ADA transduced human HSC in vitro and in murine lineage depleted progenitors at levels comparable to those transduced with the gRVs containing viral enhancers. Vectors with this EFS promoter have been shown to have a significantly decreased risk of insertional mutagenesis in both in vitro and in vivo models of transformation.32,33 Clinical trials of GT for SCID-X1 using this promoter are currently ongoing with no evidence of clonal dominance thus far (Thrasher, personal communication).
Since studies in vitro demonstrated that LV EFS ADA had ADA activity/VCN comparable to that from the gRVSFada/W and the gRV MND-ADA vector, we progressed to test the vector in comparative studies in in vivo models of ADA deficiency. The murine ADA−/− model is a highly representative model of the human disease, as untreated mice display severe lymphopenia before dying in the first 3 weeks of life from pulmonary insufficiency, which arises from the metabolic consequences of the disease.5 Correction of the murine disease phenotype by ADA delivery through a variety of GT or ERT approaches has previously been demonstrated and thus this is an appropriate model to test and compare the efficacy of the LV EFS ADA vector.30,34
In ADA−/− murine transplant experiments, ERT was continued for 4 weeks prior to complete withdrawal. Previous studies have demonstrated that ongoing use of PEG-ADA does not inhibit gene-modified cell engraftment and improves thymic reconstitution.30 Although we did not perform comparative studies with cohorts where ERT had been withdrawn prior to GT, the levels of engraftment and immune reconstitution in our GT treated mice cohorts were similar to those achieved in the WT transplant cohort and demonstrate that the short-term use of PEG-ADA post-GT was not detrimental to gene-modified cell engraftment.
After PEG-ADA withdrawal, we only observed deaths in the gRV group, while none were seen in the groups transplanted with LV-transduced or WT cells, indicating that sufficient intracellular ADA production was achieved through gene transfer to correct the pulmonary insufficiency, which is the primary cause of death in deficient animals. Histological analysis of the lungs showed clearance of inflammatory cells and interstitial material and biochemical analysis of lung tissue showed restoration of ADA and SAHH activity. Similar less severe noninfectious pulmonary manifestations have been observed in ADA-deficient patients35, and thus these data are encouraging in demonstrating the effect of GT to correct this nonimmunological manifestation of the disease. All surviving mice also had engraftment of donor gene modified (LV and gRV) cells with comparable VCN in all tissues analyzed and showed evidence of metabolic correction with increased ADA activity and SAHH activity, which indirectly reflects lowered dATP levels in reconstituted mice. The death of mice in the gRV group is likely to be due to the lower engraftment of gene-modified cells in these mice. Animals that died before analysis had lower VCN (<0.1 copy/cell) than the survivors (0.2–0.6 copy/cell). Histopathological examination of deceased mice demonstrated evidence of airway thickening and obstructions in lung.
All cohorts including GT treated, WT treated, and ERT treated ADA−/− mice displayed equivalent levels of immune recovery as reflected by the numbers of total lymphocyte counts and specific subpopulations. Overall, the performance of the LV EFS ADA vector in reconstituting the ADA−/− mouse was comparable to that of gRVSFada/W and gRV MND-ADA and given that these vectors have been used successfully in promoting long-term immune recovery and metabolic correction in human subjects,11,12,13 these findings generate confidence that the LV EFS ADA vector will also be able to promote similar recovery in the clinical setting. However, this hypothesis is difficult to test in animal models and can only be determined in the context of clinical trials.
LV EFS ADA was characterized further by transducing primary human CD34+ cells from normal CB, normal donor BM and, most relevantly, from BM samples from ADA-deficient SCID infants. Efficient vector dose-related transduction of CD34+ cells was achieved, with VCN ranging from 0.1 to 10, using concentrations of LV EFS ADA between 1 × 106 and 1 × 108 TU/ml. Vector concentrations of 1–5 × 107 TU/ml led to 1–3 VCN when analyzed by in vitro assay or in vivo in the NSG mice, which would be an ideal VCN in the clinical setting to achieve the maximal percentage of gene-corrected HSC without reaching excessive burdens of integrated vectors.
ADA expression by the LV EFS ADA was quite consistent across all studies performed, producing 1–3 times of normal ADA activity per single vector copy in the cells from short-term myeloid culture and in human CD45+ leukocytes, including thymic T cells, produced in vivo by engraftment of transduced ADA SCID HSC in NSG mice. Expression by LV EFS ADA was maintained essentially unchanged after in vivo growth. This level of ADA enzyme expression at low VCN indicates that the LV EFS ADA should have therapeutic effect, as a relatively broad range of ADA activity is tolerated and supports immune reconstitution.
In further evaluating LV EFS ADA, we also assessed the role of IL-3 in supporting HSC transduction, as the benefits of including IL-3 in the cytokine cocktail has been of uncertain value.36,37 The addition of IL-3 to the relatively standard combination of cytokines (S/F/T) increased the average VCN in CD34+ cells when analyzed after short-term myeloid culture and in CFUs. However, there were no long-term effects of IL-3 on human HSC engraftment, differentiation or vector transduction using the NSG xenograft model, suggesting that the increased VCN measured in short-term bulk cultures and CFUs was primarily the result of a subset of the progenitors receiving increased copies of the vector, rather than significant transduction of additional cells. Alternatively, high VCN could be toxic in the long term, leading to loss of the highly transduced clones in vivo.
The lack of effects of IL-3 on HSC is consistent with the absence of its receptor (IL3Ra) on primitive human HSC.38 Thus, IL-3 seems to exert its effects on the transduction of committed clonogenic progenitors, which are read-out in short-term assays but has no apparent effect on long-term repopulating cells. In the clinical setting, there may be some advantage to higher transduction of short-term progenitors to provide early expansion of cells expressing ADA during the several months required for long-term HSC to produce lymphocytes de novo.30 Of the many (>40) ADA SCID patients treated previously in clinical trials, some received cells that were transduced in the presence of IL-311,13 and others did not,12 yet the majority realized long-term effective immune recovery without any evidence of lymphoproliferation or other adverse events. The results presented in this manuscript do not make any strong argument for removing IL-3 from the transduction protocol, and therefore to avoid multiple protocol changes, the forthcoming trial with LV EFS ADA will also use IL-3 as part of the transduction cytokine cocktail.
Although there have been no insertional oncogenic events using gRV for GT of ADA-deficient SCID in more than 40 treated subjects, there have been several oncogenic events in other trials using HSC-directed GT to treat immunodeficiencies.14,15,17,19 Furthermore, the safety issue related to the gRVSFada/W has been a concern particularly after common insertion sites have been identified in the MDS1/Evi1 locus in two ADA SCID patients after GT although no malignancies have developed so far (data not shown). To address these concerns, we used a SIN LV designed to reduce the risk of insertional mutagenesis, as this configuration has already been shown to be safer in other studies.24,25,39
To test this specific LV EFS ADA vector, we have used the in vitro platform established by Modlich et al.,25 to assess the in vitro insertional mutagenesis potential of the vectors. The dramatically reduced incidence of transformants in LV EFS ADA transduced cells (no clonal outgrowth observed across multiple assays), in two independent studies, strongly indicates that this LV has significantly reduced genotoxicity compared to the gRVSFada/W or gRV MND-ADA used in prior trials at least in this assay. The improved safety property of LV EFS ADA can be attributed to the SIN design and use of internal mammalian EFS promoter, which reduces the risk of enhancer-mediated neighboring gene transactivation. The integration profile of the LV EFS ADA is also more frequent into active transcribed genes but is not concentrated on promoter-proximal areas.40,41,42 Indeed, the lack of skewing of LV EFS ADA integration sites during in vivo growth of transduced human CD34+ cells in NGS mice is consistent with other reports of undetectable genotoxicity from similar LVs in in vitro assays. Clinical trials using SIN LVS are already underway for a number of monogenic diseases of the bone marrow including immunodeficiencies and metabolic diseases, and the data thus far show a polyclonal pattern of vector integration with no evidence of clonal dominance.26,43,44
Taken together, these studies demonstrate that LV EFS ADA has the potential fulfill the requirements deemed essential to provide increased safety and clinical benefit for ADA SCID patients. Based on these data presented here, two clinical trials using autologous BM or PBSCs HSC transduced with LV EFS ADA, have recently been initiated, one in the United Kingdom (University College London, London) following approval by GT Advisory Committee (GTAC) and Medicines and Healthcare Products Regulatory Agency (MHRA) (EudraCT No: 2010-024253-36) and one in the United States. (University of California, Los Angeles, Los Angeles and the National Institutes of Health, Bethesda) following approval by the US Food and Drug Administration (FDA; IND #BB15440; ClinicalTrials.gov NCT#01852071). These studies will allow us to determine the efficacy and safety of LV EFS ADA in the clinical setting and to see if LV EFS ADA can provide more effective HSC transduction and engraftment than that observed in gRV studies, and result in more rapid and robust immune reconstitution without genotoxicity.
Materials and Methods
Experimental animals All animals were handled in laminar flow hoods and housed in microinsulator cages in pathogen-free colonies. Animal procedures and housing were in accordance with Home Office animal welfare legislation at University College London (UCL) in the United Kingdom, and in accordance with the Animal Research Committee and Division of Laboratory Animal Medicine and the National Institutes of Health guidelines at University of California, Los Angeles (UCLA) in the United States.
ADA mice. The ADA colony, maintained at UCL (FVB; 129-Adatm1Mw Tg(PLADA)4118Rkmb/J) was purchased from the Jackson Laboratory (Bar Harbor, ME) and was described previously.5 ADA+/+ and ADA−/− mice were generated by intercrossing ADA+/− females with ADA+/− males. Progeny were genotyped by polymerase chain reaction (PCR) assay (http://jaxmice.jax.org). ADA−/− mice were maintained by weekly intraperitoneal injection (i.p.) of PEG-ADA (kind gift from Sigma-Tau Pharmaceuticals) at a dose of 1,000 units/kg until transplanted and then remained on ERT for 1 month after transplant.30
NSG mice: The NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ strain (NSG) was purchased from the Jackson Laboratory (Bar Harbor, MA) in UCLA and were bred as mutant pairs.45
C57BL/6 mice. For IVIM assays at UCL and UCLA, 7-week-old C57BL/6 inbred mice were purchased from Harlan Laboratories (UK) and from Jackson Laboratory.
Viral vector construction The LV EFS ADA vector was constructed as described before28 at UCL. Briefly, a codon-optimized human ADA cDNA sequence linked with an EFS fragment was inserted into ClaI/SalI sites in the pCCLsincpptW1.6hWasp-WPRE backbone. The LV MND-ADA was constructed by inserted a blunted Hind III human ADA cDNA fragment into the SmaI site of pCCLc-MNDU3-X2 backbone, which contains the retroviral MND LTR U3 region driving expression of human ADAcDNA.
The SFada/W vector was described in Gaspar et al.,46 with a wild-type human ADA cDNA controlled by the gRV SFFV LTR. The gRV MND-ADA was constructed as described in Candotti et al.,12 and contains the human ADA cDNA under the control of the MND LTR. The LV EFS GFP vector was cloned with a HincII/BamHI fragment containing EFS promoter into the P'HR-cppt-SEW (LV SF GFP) vector.47 The gRV SF91 GFP vector was a kind gift from Professor Christopher Baum.48
Viral vector production and titer determination
LV. The LVs were packaged in HEK293 T cells by triple transfection of the packaging plasmids pMD.G2 (VSV-G envelope) and pCMVΔ8.91 (gag-pol plasmid) with the corresponding viral construct, using polyethylenimine (Sigma-Aldrich), with sodium butyrate stimulation for the first 24 hours (Sigma Aldrich). Virus supernatant was collected 48–72 hours after transfection, and viral particles were concentrated by ultracentrifugation or tangential flow.49 Evaluation of LV EFS ADA in the NSG mice was performed with vector produced in two batches produced under Good Manufacturing Practice at the Indiana University Vector Production Facility.50 The LV vector DNA titer was determined on murine SC-1 fibroblasts and human HT29 colon carcinoma cells which were harvested at 72 hours after transduction and DNA was extracted with DNeasy Blood and Tissue kit (Qiagen, UK) following the manufacturer's instructions. qPCRs were performed with primers and probe to detect the HIV psi region specific for the packaging region of LVs (sense primer 5′-acctgaaagcgaaagggaaac-3′, antisense primer 5′-cgcacccatctctctccttct-3′, and probe FAM-agctctctcgacgcaggactcggc-TAMRA).
gRV. The gRVSFada/W and gRV SF91 GFP vectors were packaged in HEK293T cells by triple transfection of the packaging plasmids pEco (murine ecotropic envelope; Clontech, Europe) and M13 (MuLV gag/pol expression plasmid, kind gift from Professor Christopher Baum) with corresponding construct using Calcium Phosphate Transfection Kit (CAPHOS, Sigma) under manufacturer's instructions. Supernatants were collected 48–72 hours after transfection and filtered through a 0.45 μm filter. The vector titer was determined on murine SC-1 fibroblasts by spinoculation with serial dilutions of supernatant for 40 minutes at 1,000xg, 4 °C in the presence of 8 μg/ml polybrene. Viral transduced cells were harvested after 72 hours and DNA extracted with DNeasy Blood and Tissue kit (Qiagen) following the manufacturer's instructions. qPCR were performed with primers and probe to detect a common region in wPRE fragment in the gRV GFP vectors28 or viral integrations, Titin for murine cells or b-actin for human cells as DNA-loading control. The gRV SF91 GFP vector was also packaged from a stable clone of the GP+E86 ecotropic packaging cell line as a positive control for the in vitro insertional mutagenesis (IVIM) assay at UCLA. Titer was determined on HT29 cells and DNA was extracted with DNeasy Blood and Tissue kit (Qiagen) following the manufacturer's instructions. qPCR were performed with primers and probe to detect GFP (sense primer is 5′-ctgctgcccgacaacca-3′, antisense primer is 5′-gaactccagcaggaccatgtg-3′, and probe 5′-FAM - ccctgagcaaagaccccaacgaga-Tamra-3′). The gRV-MND-ADA vector supernatant was produced from a stable clone of the PG13 GALV-packaging line as described.12
All experiments were performed with thawed vector stocks of known titers (LV: 0.6–10 × 109 transducing units (TU)/ml; gRVSFada/W and gRV SF91 GFP: 1–10 × 106 TU/ml); gRV MND-ADA: 1.8 × 105 TU/ml).
Enrichment, transduction, and transplantation of murine ADA−/− BM Lin- HSCs
Enrichment and transduction of ADA−/− HSC. The BM cells were harvested by flushing tibias, femora, and pelvis of age-matched male donor ADA+/+ or ADA−/− mice. BM lineage negative (Lin-) cells were enriched with the BDIMag Mouse Hematopoietic Progenitor Cell Enrichment Set (BD Biosciences, San Jose, CA) and preactivated in Stemspan serum-free expansion medium (SFEM) (StemCell Technologies, UK) in the presence of 100 ng/ml of murine stem cell factor, human Flt3 ligand (Flt3-L), murine thrombopoietin (mTPO), and 20 ng/ml of murine interleukin-3 (IL-3). After 24-hour preactivation, the LV EFS ADA or LV EFS GFP vectors were directly added to cells at a MOI of 20 and incubated for 16–24 hours. For SFada/W gRV vector, the cells were preactivated for 72 hours and then underwent a two-round transduction protocol with a 6-hour gap. In each round, viral particles corresponding to a MOI of 20 were spinoculated for 40 minutes at 1,000 g, 4 °C onto a retronectin-coated plate. The cells were added into virus-coated plates after removal of supernatant. After 24 hours after transduction, all cells were injected via the tail vein into 4–12 weeks old sublethally irradiated (5 Gy, split dose) female ADA−/− recipients at a dose of 5 × 105 cells/mouse. In ADA−/− WT group, isolated ADA+/+ BM Lin- cells were injected instead. All transplants were maintained on ERT with weekly i.p. injection of PEG-ADA at 1,000 units/kg for 4 weeks posttransplantation. A group of age-matched ADA−/− mice under continuous PEG-ADA injection were used as a positive control. The negative control group of untreated ADA−/− mice were euthanized at day 18-20 after birth.
Isolation, transduction, and transplantation of human HSC
Isolation. Human CD34+ cells (HSC) were isolated from anonymous waste normal human cord blood and bone marrow, which has been deemed exempt from IRB review as not constituting human subjects research, and from ADA-deficient SCID bone marrow, under approved UCLA IRB #10-001399 with informed consent provided by parents of the subjects. Normal human adult bone marrow samples (100 ml/donor) were also purchased from AllCells, LLC (Emeryville, CA). Human CD34+ cell isolation was performed as described.51 Briefly, human cord blood or human bone marrow was diluted 1:2 with Dulbecco's phosphate-buffered saline and distributed into 50 ml conical tubes containing 15 ml of Ficoll-Paque PLUS (GE HealthCare Life Sciences, Piscataway, NJ) and centrifuged (no brake) at 400xg for 30 minutes at room temperature. The mononuclear cells (the buffy coat) were harvested and CD34+ cells were isolated by immunomagnetic separation with the Miltenyi MACS CD34+ Cell Isolation Kit (Miltenyi Biotech, Auburn, CA). Cells were counted and either transduced as freshly isolated CD34+ cells or cryopreserved (freezing medium: 90% serum and 10% DMSO) and then transduced after thawing.
Transduction. Human CD34+ cells (100,000 or 500,000 cell/ml), were plated on Retronectin coated six-well plates (20 µg/ml; Takara/Clontech, Mountain View, CA) and prestimulated for 24 hours in X-Vivo 15 serum-free medium (Biowhittaker/Lonza, Walkersville, MD) supplemented with L-glutamine (2 mmol/l), human TPO (100 ng/ml), human stem cell factor (300 ng/ml), human Flt3 ligand (Flt3-L; 300 ng/ml), and with or without IL-3 (20 ng/ml) (all cytokines from BioLegend, San Diego, CA). The cells were transduced with the EFS-ADA LV at a concentration of 3.0 × 107 TU/ml (except where indicated otherwise) for 18–20 hours at 37 °C with 5% CO2. gRV transductions were done following 2 days of prestimulation, as above, by adding unconcentrated gRV-MND-ADA vector supernatant to cells daily × 3 days.12
Transplantation. Irradiated (150 cGy) neonatal NSG mice were transplanted with 50,000 to 100,000 transduced (LV EFS ADA) or nontransduced (mock) human CD34+ cells by intravenous injection into the superficial temporal (facial) vein between postnatal day 1 and 3. The mice were euthanized 4 months after transplant, and the thymus, spleen, and bone marrow were harvested and analyzed for the presence of human cells (engraftment) and vector (VCN and expression). Bone marrow cells were isolated from each primary recipient, red blood cells were lysed, and 1 × 10e7 nucleated cells were serially transplanted into a conditioned secondary recipient (250cGy).
Analysis in vitro of transduced human HSC
Myeloid culture. Immediately following the transduction period, the LV-transduced and mock-transduced cultures were maintained in Iscove's modified Dulbecco's medium supplemented with 20% fetal calf serum (Omega Scientific, Tarzana, CA), 0.5% human serum albumin (AlbuRx 25; CSL Behring LLC, Kankakee, IL), L-glutamine (2 mmol/l), penicillin/streptomycin (100 U/ml), human IL-3 (5 ng/ml), IL-6 (10 ng/ml), and stem cell factor (25 ng/ml) (all cytokines from BioLegend). On day 7, one half of the medium was exchanged for fresh medium with freshly diluted cytokines. On day 14 of posttransduction culture, 1 × 106 cells were harvested for DNA extraction and 0.5 × 106 cells were harvested for ADA enzyme activity assay. DNA was purified using the DNAeasy kit (Qiagen, Valencia, CA) and ADA enzyme activity was determined with the ADA enzyme assay by Diazyme (San Diego, CA).
Colony assays. Samples of the transduced CD34+ cells were also plated for progenitor assays (CFU) (two to three dilutions in duplicate) in semisolid methylcellulose medium supplemented with cytokines (Stem Cell Technologies, Vancouver, BC, Canada). Between days 11 and 14, colonies were counted and characterized by progenitor type. Single colonies were aspirated from the methylcellulose and placed into a microcentrifuge tube containing 1 ml of Dulbecco's phosphate-buffered saline for 1 hour at 37 °C. The tubes were centrifuged for 10 minutes at 400xg and cell pellets stored at −20 °C for later DNA extraction and VCN analysis. Colony DNA was purified with a single phenol/chloroform extraction, precipitated in the presence of glycogen (20 mg/ml, Roche Diagnostics, Mannheim, Germany; Invitrogen, Carlsbad, CA) and resuspended in 25 µl of Tris-EDTA (pH 7.4). To determine CFU VCN, 5 µl of the extracted DNA were analyzed by Multiplex qPCR using primers/probe for the human ADA cDNA and the human SDC4 gene (to normalize for DNA concentration) and compared to the EFS-ADA copy number standard described above.
Flow cytometry analysis for immunophenotype and engraftment
ADA mice. The percentage of T cells (CD3+, CD4+, and CD8+), B cells (B220+), myeloid cells (GR-1+), and natural killer (NK1.1+) cells were analyzed in the PB, thymus, spleen, or bone marrow of ADA mice. For flow cytometry, 2 × 105 cells from red cell lysed PB, lymphoid organs, or bone marrow were preincubated for 15 minutes at room temperature with murine serum followed by staining for 30 minutes at 4 °C with anti-mouse antibodies all from BD Pharmingen including: PE-CD3, PE Cy7-CD4, APC-CD8a, APC-B220, APC-GR-1, and APC-NK1.1. After washing, cells were analyzed using CyAn ADP Analyzer (Beckman Coulter) and Summit software.
NSG mice. In transplanted NSG mice, the level of engraftment and the immunophenotype of human cells were determined by flow cytometry (FACS) immunostaining with anti-human antibodies from BD Biosciences and flow cytometry on a BD LSRII instrument with DIVA (BD Biosciences) Software. Percent engraftment was determined on bone marrow cell suspensions (flushed from femur and tibia bones) immunostained with anti-human CD45 (PerCp or APC). The percentage of engrafted human cell lineages was determined on tissue cell suspensions immunostained as follows: thymus-anti-human CD4-PE, anti-human CD8-APC; spleen-anti-human CD3-PE, anti-human CD19-APC; and bone marrow anti-human CD11b-APC, anti-human gran-1-PE.
Quantification by qPCR for VCN and donor cell engraftment All amplification reactions were performed in the 7,500 Fast Real-Time PCR System (Applied Biosystems/Life Technologies (LT) UK and USA) under default conditions and analyzed using Manufacturer's software.
ADA mice. Genomic DNA was extracted from murine tissues and PB by DNeasy Blood & Tissue Kit (Qiagen). VCN in total cells from different organs was detected by qPCR using primers amplifying sequences in wPRE or Titin.28 Known copies of wPRE from LV-transduced MEL cells serially diluted into irrelevant genomic DNA was used to set up a standard curve. The frequency of male donor cells was determined by qPCR for the Y chromosome using primers described previously.34 These data were calculated using a standard curve of serially diluted male cells into female cells from ADA mice.
NSG mice. Genomic DNA was extracted from murine spleen and bone marrow with DNeasy Blood & Tissue Kit (Qiagen). From smaller thymic tissue samples (0.5–1 × 105 cells), DNA was extracted with phenol chloroform extraction as described previously.12 The human ADA gene in both gRV (not codon optimized) and LV vectors (codon optimized) was amplified using primers and probe that span exon 6 and 7 of the human ADA gene (sense primer 5′-ggtccatcctgtgctgcat-3′, anti-sense primer 5′-cggtctgctgctggtacttctt-3′, and probe 5′-FAM-ccagcccaactggtcccccaag-tamra-3′). VCN was normalized by qPCR of the human syndecan 4 gene (SDC4) (sense primer 5′-cagggtctgggagccaagt-3′, anti-sense primer 5′-gcacagtgctggacattgaca-3′, and probe 5-HEX-cccaccgaacccaagaaactagaggagaat-Iowa Black FQ. DNA extracted from a cellular clone containing four copies of integrated LV EFS ADA vector was serially diluted into equally concentrated DNA from nontransduced cells to make the standard curve used to quantify the VCN per cell.
ADA and SAHH activity assay
ADA mice. ADA activity assay was performed with cell lysates from transplanted ADA−/− and controls prepared in 200–500 μl of H2O per sample. 12.5 μl of the lysate was incubated with the reaction mix containing 50 μl of phosphate-buffered saline (Invitrogen/Life technologies), 37.5 μl of 10 mmol/l adenosine (Sigma-Aldrich) for 0 or 20 minutes in 37 °C water bath. Then, the reaction was stopped by 12.5 μl of 40% trichloroacetic acid (Sigma-Aldrich). The precipitations were spun down, and trichloroacetic acid in the supernatant was extracted by H2O-saturated diethyl ether.
ADA mice. For SAHH activity assay, 100 μl of master mix (50 μl of 62.5 mmol/l KH2PO4, 5 μl of 20 mmol/l DTT, 10 μl of 10 mmol/l EDTA, 20 μl of 37.5 mmol/l homocysteine, and 15 μl of H2O) was added into each tube with 10 μl of 150 μmol/l deoxycoformycin (Pentostatin; TOCRIS Bioscience, UK) and 25 μl of lysate. The tubes were preincubated in a 37 °C water bath for 5 minutes. To start the reaction, 10 μl of 6.5 mmol/l adenosine was added into the mixture and incubated for 0 or 60 minutes at 37 °C until stopped by adding in 25 μl of 40% trichloroacetic acid. The precipitations were spun down and trichloroacetic acid in the supernatant was extracted by H2O-saturated diethyl ether. The level of substrates in ADA or SAHH activity assays was measured on anion-pair HPLC Waters 2795 system with PDA detection (Waters, Milford, MA). The final ADA activity was normalized with protein concentration or hemoglobin concentration.
NSG mice. ADA enzyme activity was measured in human cells isolated from total tissue cell suspensions of spleen and bone marrow using the anti-human CD45 Miltenyi MACs Cell Separation System (Miltenyi Biotech, Auburn, CA). Mice transplanted with mock-transduced cells were also used to determine a baseline of ADA activity in the engrafted human cells, as the CD34+ cells from ADA replete cord blood or bone marrow will have background ADA expression. A colorimetric ADA enzyme assay kit (Diazyme Laboratories, Poway, CA) was used to determine the amount of ADA enzyme activity in the primary human CD34+ cells from in vitro culture and from human cells isolated from NSG mice. Cells (0.5 × 106 cells) were centrifuged at 400 × g for 5 minutes and a dry pellet was frozen at −80 °C for batch assays. The kit uses a calibrator that is serially diluted to make a standard curve for quantification. The catalytic conversion of adenosine by ADA enzyme is ultimately read-out by the conversion of hypoxanthine to uric acid and hydrogen peroxide that reacts with N-Ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline and 4-aminoantipyrine in the presence of peroxidase to generate quinone dye, which is detected spectrophotometrically at 550 nm.
Western blot. The cell lysate was prepared with 1 × 106 cells in RIPA lysis buffer by standard method and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins were transferred to nitrocellulose membrane (Sigma-Aldrich). Anti-ADA antibody was a kind gift from Dr. M. Hershfield (Duke University, Durham, NC). Anti-GAPDH monoclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, Germany).
Histological analysis. Tissues and organs of ADA mice including lung, liver, heart, and kidney were harvested and examined histologically. Tissues and organs were rinsed in phosphate-buffered saline and then fixed in 10% formalin for more than 24 hours at 4 °C. Then, tissues were dehydrated, cleared, and embedded in paraffin following routine procedures. Sections of 4 µm in size were cut and stained with hematoxylin and eosin and mounted using standard protocols for histopathological analysis under an optical microscope (Olympus BX50).
IVIM-WST1 assay. In vitro immortalization assay was adopted from Modlich et al.25 Briefly, BM Lin- cells of C57BL6 mice were isolated with the BDIMag Mouse Hematopoietic Progenitor Cell Enrichment Set (BD Biosciences, 558451) and preactivated in Stemspan serum-free expansion medium (StemCell Technologies) containing 50 ng/ml murine stem cell factor, 100 ng/ml hFlt-3 ligand, 100 ng/ml hIL-11, and 10 ng/ml mIL-3 (PeproTech, UK) at a density of 5 × 105 cells/ml. 1 × 105 cells were transduced on day 4 and 5 at an MOI of 20 for each viral vector. LVs were directly added to cells. gRVs were preloaded on retronectin-coated plate (TaKaRa, Japan) by spinoculation for 40 minutes at 4 °C and then incubated with cells for 16–24 hours.
After two-round transductions, cells were expanded as bulk populations for 2 weeks in Iscove's modified Dulbecco's medium containing the same cytokine cocktail as above with 10% fetal calf serum. DNA samples were taken at day 9 for vector copy analyses by qPCR. Two weeks later, cells were plated into 96-well plates at a density of 100 cells per well and incubated at 37 °C for another 14 days. Subsequently, half of cells from each well were incubated with 10 μl of WST-1 (Roche, Europe) for 4 hours at 37 °C. The absorbance was measured at 450 nm in a FLUOstar Optima luminometer (BMG Labtech, Ortenberg, Germany). The highest absorbance from mock-transduced clones was set as the baseline above which all clones were counted as positive ones. The frequency of replating cells was calculated using L-Calc software (Stem Cell Technologies) and normalized with VCN. Selected clones were expanded for further characterization.
Statistical analysis. Descriptive statistics of continuous outcome variables, such as the means and standard error by experimental groups, are presented in figures and data tables. For continuous outcome measurements, group differences were assessed by unpaired t-test (for two experimental groups) or one-way/two-way analysis of variance with interaction (for more than two groups)52 followed by pairwise comparisons. Linear mixed models53 were used for dose-dependent analysis. Concentration and MOI were modeled as the fixed effects, while experiments or donors were modeled as random effects to account for the random variations in the data. Wilcoxon rank sum test was used for IVIM assay analysis. For all statistical investigations, tests for significance were two-tailed unless otherwise specified. A P value less than the 0.05 significance level was considered to be statistically significant (*). All statistical analyses were carried out using SAS version 9.3.54
SUPPLEMENTARY MATERIAL Figure S1. Transduction and expression by EFS-ADA in human CD34+ cells. Figure S2. The LV EFS ADA vector shows significantly decreased transformation potential than the SFada/W vector. Table S1. In vitro immortalization assay cell replating frequency.
Acknowledgments
In the United Kingdom, studies were funded by grants from the Medical Research Council (L.Z and C.M-E.) and the NIHR Biomedical Research Centre at Great Ormond Street Hospital (C.M-E. and A.J.T). The authors acknowledge the support of GOSHCC (H.B.G), Wellcome Trust (A.J.T and M.P.B.) and the European Commission's 7th Framework Program Contract 261387 (CELL-PID). In the United States, studies were supported by research grants from the National Institutes of Health (2P01 HL073104, 1R01 A1074043, and U01 AI100801), and by the University of California, Los Angeles Jonsson Comprehensive Cancer Center and the Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research. Vector production in the Indiana University Vector Production Facility was supported by the NHLBI Gene Therapy Resource Program (HHSN2682012000051). The Flow Cytometry Core and High-Throughput Sequencing Core of the Broad Stem Cell Research Center were essential to performing these studies. Ute Modlich and Chris Baum, Hannover Medical School, Germany generously provided reagents, protocols, and advice for establishing the in vitro immortalization assay. Finally, we would like to thank the parents of the ADA SCID subjects who gave consent for provision of bone marrow aspirates for these studies.
Supplementary Material
Transduction and expression by EFS-ADA in human CD34+ cells.
The LV EFS ADA vector shows significantly decreased transformation potential than the SFada/W vector.
In vitro immortalization assay cell replating frequency.
References
- Hershfield MS. Adenosine deaminase deficiency: clinical expression, molecular basis, and therapy. Semin Hematol. 1998;35:291–298. [PubMed] [Google Scholar]
- Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J Clin Invest. 2001;108:131–141. doi: 10.1172/JCI10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque W, Gaspar HB. Bilateral sensorineural deafness in adenosine deaminase-deficient severe combined immunodeficiency. J Pediatr. 2004;144:278–280. doi: 10.1016/j.jpeds.2003.10.055. [DOI] [PubMed] [Google Scholar]
- Ratech H, Greco MA, Gallo G, Rimoin DL, Kamino H, Hirschhorn R. Pathologic findings in adenosine deaminase-deficient severe combined immunodeficiency. I. Kidney, adrenal, and chondro-osseous tissue alterations. Am J Pathol. 1985;120:157–169. [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- Booth C, Hershfield M, Notarangelo L, Buckley R, Hoenig M, Mahlaoui N, et al. Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006) Clin Immunol. 2007;123:139–147. doi: 10.1016/j.clim.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114:3524–3532. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Vai S, Mortellaro A, Casorati G, Ficara F, Andolfi G, et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8:423–425. doi: 10.1038/nm0502-423. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–3646. doi: 10.1182/blood-2012-02-400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cassani B, Andolfi G, Mirolo M, Biasco L, Recchia A, et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest. 2007;117:2233–2240. doi: 10.1172/JCI31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knöss S, Schambach A, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Von Kalle C, Bougnères P, Fischer A, Cavazzana-Calvo M, et al. Gene therapy of x-linked adrenoleukodystrophy using hematopoietic stem cells and a lentiviral vector. Bull Acad Natl Med. 2010;194:255–64; discussion 264. [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Equihua CA, Zhang L, Knight S, Saadeh H, Scholz S, Carmo M, et al. The beta-globin locus control region in combination with the EF1alpha short promoter allows enhanced lentiviral vector-mediated erythroid gene expression with conserved multilineage activity. Mol Ther. 2012;20:1400–1409. doi: 10.1038/mt.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan-Curay J, Cohen-Haguenauer O, O'Reilly M, Ross SR, Fan H, Rosenberg N, et al. Challenges in vector and trial design using retroviral vectors for long-term gene correction in hematopoietic stem cell gene therapy. Mol Ther. 2012;20:1084–1094. doi: 10.1038/mt.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonaro DA, Jin X, Wang X, Yu XJ, Rozengurt N, Kaufman ML, et al. Gene therapy/bone marrow transplantation in ADA-deficient mice: roles of enzyme-replacement therapy and cytoreduction. Blood. 2012;120:3677–3687. doi: 10.1182/blood-2012-02-408591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LF, Van de Wiele CJ, Laurent AB, Hooker SW, Vaughn JG, Jiang H, et al. Metabolites from apoptotic thymocytes inhibit thymopoiesis in adenosine deaminase-deficient fetal thymic organ cultures. J Clin Invest. 2000;106:1149–1157. doi: 10.1172/JCI9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Frost AR, Blundell MP, Bales O, Antoniou MN, Thrasher AJ. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, et al. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]
- Mortellaro A, Hernandez RJ, Guerrini MM, Carlucci F, Tabucchi A, Ponzoni M, et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108:2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
- Booth C, Algar VE, Xu-Bayford J, Fairbanks L, Owens C, Gaspar HB. Non-infectious lung disease in patients with adenosine deaminase deficient severe combined immunodeficiency. J Clin Immunol. 2012;32:449–453. doi: 10.1007/s10875-012-9658-3. [DOI] [PubMed] [Google Scholar]
- Ficara F, Superchi DB, Hernández RJ, Mocchetti C, Carballido-Perrig N, Andolfi G, et al. IL-3 or IL-7 increases ex vivo gene transfer efficiency in ADA-SCID BM CD34+ cells while maintaining in vivo lymphoid potential. Mol Ther. 2004;10:1096–1108. doi: 10.1016/j.ymthe.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Uchida N, Hsieh MM, Hayakawa J, Madison C, Washington KN, Tisdale JF. Optimal conditions for lentiviral transduction of engrafting human CD34+ cells. Gene Ther. 2011;18:1078–1086. doi: 10.1038/gt.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn LA, Hao QL, Sasidharan R, Parekh C, Ge S, Zhu Y, et al. Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat Immunol. 2012;13:963–971. doi: 10.1038/ni.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Beard BC, Dickerson D, Beebe K, Gooch C, Fletcher J, Okbinoglu T, et al. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol Ther. 2007;15:1356–1365. doi: 10.1038/sj.mt.6300159. [DOI] [PubMed] [Google Scholar]
- Beard BC, Keyser KA, Trobridge GD, Peterson LJ, Miller DG, Jacobs M, et al. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum Gene Ther. 2007;18:423–434. doi: 10.1089/hum.2007.011. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cossu G, de Felipe P, Galli MC, Narayanan G, Renner M, et al. The committee for advanced therapies' of the European Medicines Agency reflection paper on management of clinical risks deriving from insertional mutagenesis. Hum Gene Ther Clin Dev. 2013;24:47–54. doi: 10.1089/humc.2013.119. [DOI] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, Bjorkegren E, Parsley K, Gilmour KC, King D, Sinclair J, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505–513. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, et al. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther. 2002;13:803–813. doi: 10.1089/10430340252898984. [DOI] [PubMed] [Google Scholar]
- Schambach A, Wodrich H, Hildinger M, Bohne J, Kräusslich HG, Baum C. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol Ther. 2000;2:435–445. doi: 10.1006/mthe.2000.0191. [DOI] [PubMed] [Google Scholar]
- Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, Hollis RP. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J Virol Methods. 2011;177:1–9. doi: 10.1016/j.jviromet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leath A, Cornetta K. Developing novel lentiviral vectors into clinical products. Meth Enzymol. 2012;507:89–108. doi: 10.1016/B978-0-12-386509-0.00005-3. [DOI] [PubMed] [Google Scholar]
- Nightingale SJ, Hollis RP, Pepper KA, Petersen D, Yu XJ, Yang C, et al. Transient gene expression by nonintegrating lentiviral vectors. Mol Ther. 2006;13:1121–1132. doi: 10.1016/j.ymthe.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Tukey J. Variances of variance components: II. Unbalanced single classifications. The Annals of Mathematical Statistics. 1957;28:43–56. [Google Scholar]
- West BT, Welch KB, Galecki AT. Linear Mixed Models: A Practical Guide to Using Statistical Software. Chapman & Hall/CRC; New York; 2007. [Google Scholar]
- SAS Institute; Cary, NC; SAS Institute (2012). SAS/STAT user's guide, Version 9.3. 9.3 ed. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transduction and expression by EFS-ADA in human CD34+ cells.
The LV EFS ADA vector shows significantly decreased transformation potential than the SFada/W vector.
In vitro immortalization assay cell replating frequency.