

Abstract
Avian pathogenic Escherichia coli (APEC) are responsible for heavy economic losses in poultry industry. Here we investigate DNA methylome of spleen and identify functional DNA methylation changes related to host response to APEC among groups of non-challenged chickens (NC), challenged with mild (MD) and severe pathology (SV). DNA methylation was enriched in the gene bodies and repeats. Promoter and CGIs are hypomethylated. Integration analysis revealed 22, 87, and 9 genes exhibiting inversely changed DNA methylation and gene expression in NC vs. MD, NC vs. SV, and MD vs. SV, respectively. IL8, IL2RB, and IL1RAPL1 were included. Gene network analysis suggested that besides inflammatory response, other networks and pathways such as organismal injury and abnormalities, cell signaling and molecular transport, are probably related to host response to APEC infection. Moreover, methylation changes in cell cycle processes might contribute to the lesion phenotype differences between MD and SV.
Avian colibacillosis is one of the most frequent infectious diseases characterized by multiple organ lesions typically with airsacculitis, pericarditis, perihepatitis and peritonitis1,2. It is responsible for high morbidity, mortality and product contamination in the poultry industry, which induce significant economic losses and pose severe threat to human health2,3. Avian colibacillosis is caused by avian pathogenic Escherichia coli (APEC) infection2. APEC, a gram-negative bacterium, belongs to the extraintestinal pathogenic group of Escherichia coli. Although a large variety of APEC serogroups have been identified so far, the most common and widespread serogroups are O1, O2 and O783. In addition to the control of environmental factors such as ventilation and humidity, antibiotherapy and vaccination are the main methods for the prevention of APEC infection4. However, previous research demonstrated that vaccines against heterologous APEC strains were not fully effective and APEC strains were frequently resistant to a wide range of antibiotics3,5,6. Moreover, there is increasing consumer pressure to substantially decrease the use of antibiotics in food-producing animals. Therefore, the selection of genetically disease resistant poultry population is becoming a topic of great interest in the control of APEC infection. A better understanding of the host response and resistance mechanisms against APEC should be of great value in developing better strategies to further prevent and control APEC infection.
Although a large number of studies in surveying the host-pathogen interactions have been conducted, most of them are focused on the bacterium itself to investigate virulence genes or factors involved in pathogenesis of APEC infection7,8,9,10,11. In recent years, there were several research reported on the host transcriptomes response to APEC and considerable changes in gene expression were found before and after infection12,13,14. However, so far little is done about the epigenetic mechanisms of host response and resistance to APEC infection. DNA methylation is one of the main epigenetic modifications in eukaryotes. In multicellular eukaryotes, methylation occurs primarily at the 5-C position of cytosine within CpG dinucleotides15. Previous studies have shown that cytosine DNA methylation played a central role in many biological processes like gene expression regulation, X chromosome inactivation, genomic imprinting, cancer and disease development16,17,18,19,20,21,22. With the development of technologies, a new valuable approach named Methylated DNA immunoprecipitation-sequencing (MeDIP-seq) has recently been extensively applied to the genome methylation studies in various species23,24,25. Moreover, MeDIP-seq was shown to be highly efficient and reliable for methylome analysis with low DNA concentrations, though the resolution (about 200 bp) was lower than that of whole genome bisulfite sequencing26,27,28,29. In birds, with the use of MeDIP-seq, the DNA methylation landscape was firstly reported in the liver and muscle tissues from the red jungle fowl and avian broiler30. However, their study lacked integrated analysis with gene expression to find the potential functional genes of a certain trait.
The aim of the current study was to investigate the global DNA methylation profiles in chicken spleen and to identify potentially functional DNA methylation changes related to host response and resistance to APEC. Here we firstly characterized the chicken spleen methylomes by MeDIP-seq using Illumina Hiseq 2000. Then, in order to validate the results of MeDIP-seq, eight regions of the genome were selected randomly to perform bisulfite sequencing experiments. Subsequently, in order to identify potentially functionally relevant DNA methylation changes, we integrated the gene expression data and DNA methylation profiles in each contrast of the three groups including birds with non-challenged (NC), challenged with mild (MD) and with severe pathology (SV).
Results
Assemble and blast analysis of MeDIP-seq reads
In this study, spleens of three males were used to produce one pooled DNA sample for each group of NC, MD and SV. A total of 36,734,694 raw reads were generated for each of the three groups, of which more than 73% of the reads could be mapped and about 47% of the reads could be uniquely mapped to the reference chicken genome (Table 1). The uniquely mapping reads of NC, MD, and SV covered 26.6%, 25.6%, and 25.3% of the genome, respectively.
Table 1. Data generated by MeDIP-Seq for each sample.
| Sample | Total number of reads | Total Mapped Reads | Total Unique Mapped Reads | Percentage of mapped reads in total reads | Percentage of unique mapped reads |
|---|---|---|---|---|---|
| NC | 36,734,694 | 27,087,040 | 17,205,739 | 73.74% | 46.84% |
| MD | 36,734,694 | 27,062,238 | 17,460,699 | 73.67% | 47.53% |
| SV | 36,734,694 | 27,375,496 | 17,938,237 | 74.52% | 48.83% |
NC, MD, and SV indicated the group of non-challenged, challenged-mild pathology, and challenged-severe pathology, respectively.
The distribution of MeDIP-seq reads in chicken chromosomes (GGA1-28 and the Z chromosome) for each sample was analyzed and uniquely mapping reads were found in most chromosomal regions except for some gaps. However, in a long region of GGA17 (from 3,180,001 to 11,182,526 bp), no unique-mapped reads but multi-mapped reads could be detected (Supplementary Fig. S1).
To ascertain read distribution in different components of the genome, we classified the uniquely mapped reads into four types: the reads uniquely mapped to CpG islands (CGIs), gene bodies, repeats, and others. Here, the criteria for CGIs identification were as follows: length exceeding 200 bp, GC content greater than 50%, and observed-to-expected CpG ratio greater than 0.6. Gene body was defined as the region from transcript starting site to transcript ending site. We found that the uniquely mapped MeDIP-seq reads were mainly present in the gene body regions, whereas less than 7.5% of the reads mapped to the CGIs (Supplementary Fig. S2). About 24% of the uniquely mapped reads in chicken belonged to repeat elements.
Validation of MeDIP-seq data by bisulfite sequencing
In order to confirm quality of the MeDIP-seq results, three regions showing high methylation and one region showing low methylation were selected randomly to perform bisulfite sequencing in the three groups. The results of bisulfite sequencing were consistent with our MeDIP-seq data (Supplementary Fig. S3–S5).
Global DNA methylation profiles in the chicken spleen
To assess overall methylation pattern in the chicken genome, we divided methylated regions into different components including promoters, gene bodies (5′ UTR, 3′ UTR, exon and intron), intergenic regions, CGI, and repeats. Here, the 2 Kb region upstream of the transcription start sites (TSS) were defined as the proximal promoter, while sequences between the 3′ and 5′UTRs of the genes were defined as intergenic regions. In total, there were 55,978 methylated peaks in NC, 54,594 peaks in MD and 63,100 peaks in SV (Table 2). Peak distribution analysis showed that most of the peaks fell into the intergenic regions, 1.14% to 1.70% of the peaks were located at the 5′UTR of genes, 2.01% to 3.13% of the peaks were at the 3′ UTR, 15.53% to 23.61% of the peaks were in the exons, 22.47% to 39.44% of the peaks were in the intron regions, about 11.54% of the peaks were in the CGIs and more than 14.57% of the peaks were located in the repeats in the three groups. In addition, we investigated the relative methylation of each class by calculating the ratio of peaks located in that region to the total area of that region. Consequently, differential methylation levels were observed in different classes. The average methylation of promoters was found to be the lowest among all the classes and repeats exhibited a relative high level of methylation. The average methylation of CGIs was found to be relatively lowly methylated and gene bodies showed a significantly higher (P < 0.05) level of DNA methylation than intergenic regions. Within the gene body, introns showed significantly higher (P < 0.05) methylation than UTRs and exons (Fig. 1A).
Table 2. Peak distribution in different components of the chicken genome.
| Sample | Total peak number | promoter | 5′UTR | Exon | Intron | 3′UTR | Intergenic | CGIs | Repeats |
|---|---|---|---|---|---|---|---|---|---|
| NC | 55978 | 2899 | 952 | 13219 | 22004 | 1752 | 38684 | 6598 | 8158 |
| MD | 54594 | 1856 | 622 | 8479 | 12269 | 1097 | 37370 | 6701 | 8061 |
| SV | 63100 | 3151 | 964 | 14659 | 24887 | 1916 | 43594 | 6668 | 9400 |
NC, MD, and SV indicated the group of non-challenged, challenged-mild pathology, and challenged-severe pathology, respectively.
Figure 1. Methylation distribution in different genomic regions.
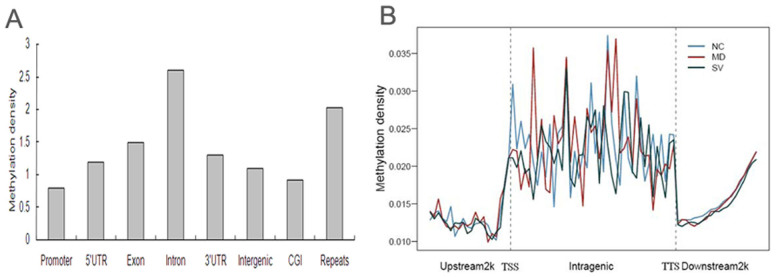
(A). Global patterns of methylation in different genomic regions. Methylation density within promoter, gene body and intergenic regions was calculated by dividing the peak numbers in that region by the total area of that region. (B). Methylation distribution of gene body and flanking regions. Methylation density was calculated by the ratio of methylation peak count vs number of data points. The gene body region referred to the region. In gene body (from TSS to TTS), each gene was split into 40 equal windows and the methylation density was calculated for each window. In upstream and downstream 2 kb regions, the regions were split into 20 non-overlap windows and the methylation density was calculated for each window.
DNA methylation in promoter and gene body
On plotting the methylation density, low methylation levels were found in the proximal promoter regions and there was a sharp increase at the gene body regions, which were stayed at a plateau until the transcription termination site (TTS) (Fig. 1B). In the 2 Kb region downstream of TTS, we observed a clear trend for DNA methylation levels to increase gradually.
DNA methylation in CGIs and repeat elements
CGIs having methylation peaks were termed as methylated islands while the rest were designated unmethylated. Of the 33,915 CGIs reported in the chicken genome, about 19.5% (n = 6,598) were methylated in NC spleen, 19.8% (n = 6,701) in MD spleen, and 19.7% (n = 6,668) in SV spleen (Supplementary Table S1). Among these methylated CGIs, most were located in the intergenic regions. Within the gene body, exons showed higher proportion of methylated CGIs than UTRs and introns. And less than 2% were located in the 5′UTR region. Subsequently, we categorized both methylated and unmethylated CGIs on the basis of their sizes and calculated the CGI numbers in each class. About 29% of methylated CGIs were distributed in the size range of 200–300 bp. On the whole, the number of methylated CGIs decreased with the increase of size (Supplementary Fig. S6). Methylated CGIs were enriched in exons, while unmethylated ones were mainly present in introns. On the other hand, differential methylation was observed in different repeat types of the chicken genome. The predominant type of interspersed repeats was chicken repeat 1 - long interspersed nucleotide element (LINE/CR1), which accounted for more than 48% of the total methylated repeat sequences (Supplementary Table S2). In addition, a relatively high proportion (about 20%) of the methylated repeat sequences was observed in endogenous retrovirus like elements - long terminal repeat (LTR/ERVL).
Identification of differentially methylated regions among the three groups
In our study, a total of 14,396 methylated genes were identified in the three groups, including 9,597 genes observed in all of the three groups. Of these, the methylation gene numbers in the NC, MD, and SV groups were 12,861, 10,292, and 13,418, respectively (Fig. 2A). Here, methylated genes were defined as genes overlapped with peak summits in promoter and gene body regions. Consequently, a total of 4,684 genes showing differential methylation between any two-way comparison of the groups of NC, MD and SV (coverage changes was more than two folds; p value < 0.01) were found, including 1,893 differently methylated genes between NC and MD (NC vs. MD), 2,970 between NC and SV (NC vs. SV), and 2,418 between MD and SV (MD vs. SV) (Fig. 2B). Moreover, 924 differentially methylated genes were found in both NC vs. MD and NC vs. SV, 875 in NC vs. MD and MD vs. SV, as well as 1,291 in NC vs. SV and MD vs. SV. Of these, 493 genes were in all of NC vs. MD, NC vs. SV, and MD vs. SV.
Figure 2. Methylated genes and differentially methylated genes among NC, MD, and SV.
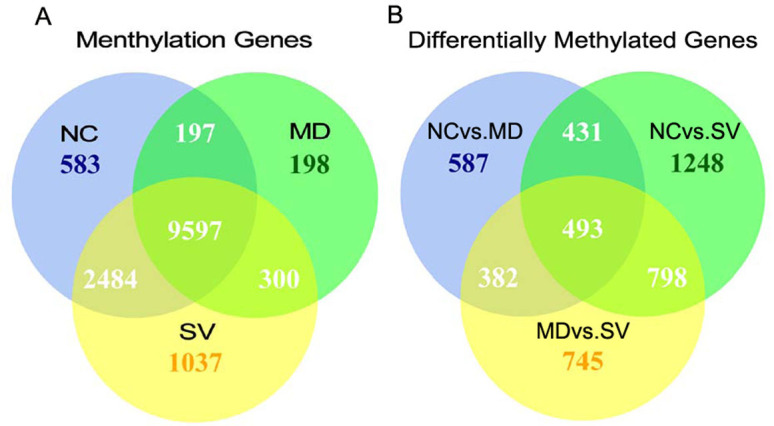
(A). Methylated genes that were unique or shared among three groups of NC, MD, and SV. (B). Differentially methylated genes that were unique or shared among three groups of NC, MD, and SV. Numbers in each section of the figure referred to the numbers of methylated (for A) or differently methylated genes (for B). NC, MD, and SV indicated the group of non-challenged, challenged-mild pathology, and challenged-severe pathology, respectively.
Identification of potentially functional relevant DNA methylation changes
DNA methylation in both promoters and gene bodies was proved to be associated with gene expression31. Here we used the average normalized depth of reads as the measurement of methylation level for each gene. We found that gene expression level was negatively correlated with DNA methylation in the chicken genome of the three groups (Fig. 3).
Figure 3. The correlation between DNA methylation and gene expression.

X-axis is the methylation level measured by the average normalized depth of reads. Here methylations located in the promoter or the genebody region of genes (more than half of the sequences were overlapped with peak summits) were used for integration analysis. Y-axis is the gene expression level measured with RPKM value (Reads Per kb per Million reads). In general, gene expression level was negatively correlated with DNA methylation level.
In order to identify potentially functionally relevant DNA methylation changes, we integrated the gene expression data and DNA methylation profiles in each contrast generated from the three groups of NC, MD, and SV. Differentially methylated genes with differences in the promoter or the gene body regions were selected to investigate concomitant expression changes in each contrast. Using a FDR cutoff of 0.001 and a filter of mean twofold change, 507 genes were found to be differentially expressed in NC vs. MD, 838 in NC vs. SV, and 138 in MD vs. SV. The number of genes exhibiting coordinately changed DNA methylation and gene expression was described in Table 3 for each contrast. In the NC vs. MD contrast, a total of 11 genes were identified to be significantly up-methylated and down-regulated, in which a promoter-methylated gene and 11 body-methylated genes were included, Moreover, there were 11 genes significantly down-methylated and up-regulated, in which a promoter methylated gene named TXNL4B was included (Supplementary Table S3). In the NC vs. SV contrast, a total of 76 genes were significantly up-methylated and down-regulated, of which 4 genes (ACAP3, ADAMTS1, CADPS2, CENPJ) were promoter-methylated and 73 were body-methylated. On the other hand, 11 genes were significantly down-methylated and up-regulated, of which 3 genes were promoter-methylated (IL8, LOC417973, TXNL4B) and 8 were body-methylated (Supplementary Table S3). Of these, five genes, PLEKHA7, ETV1, TXNL4B, SLC2A9, AUTS2, and C10orf11, were found in both of the two comparisons between non-challenged and challenged pathology groups. In the MD vs. SV contrast, 8 genes were statistically significantly up-methylated and down-regulated, while 1 gene was significantly down-methylated and up-regulated (Supplementary Table S3). All these genes were body-methylated. In addition to those genes showing inverse relationships between DNA methylation and gene expression changes, 7, 30, and 7 genes were found to be up-methylated but up-regulated, while 24, 12, and 4 genes were found to be down-methylated and down-regulated in NC vs. MD, NC vs. SV, and MD vs. SV, respectively (Supplementary Table S4).
Table 3. Number of genes showing both changed DNA methylation and gene expression in each contrast.
| Contrast | Location | up-regulated | down-regulated | |
|---|---|---|---|---|
| NC vs. MD | promoter | up-methylated | 1 | 1 |
| down-methylated | 1 | 1 | ||
| gene body | up-methylated | 6 | 11 | |
| down-methylated | 10 | 23 | ||
| NC vs. SV | promoter | up-methylated | 8 | 4 |
| down-methylated | 3 | 2 | ||
| gene body | up-methylated | 22 | 73 | |
| down-methylated | 8 | 10 | ||
| MD vs. SV | promoter | up-methylated | 0 | 0 |
| down-methylated | 0 | 0 | ||
| gene body | up-methylated | 7 | 8 | |
| down-methylated | 1 | 4 |
NC vs. MD indicated the comparison between non-challenged and challenged-mild pathology groups; NC vs. SV indicated the comparison between non-challenged and challenged-severe pathology groups; MD vs. SV indicated the comparison between challenged-mild pathology and challenged-severe pathology groups. Location indicated the DNA methylation differences occurred in the promoter or the gene body regions. Up-methylated meant that within the same peak region, there were greater reads in the second group than the first group, while down-methylated meant there were greater reads in the first group than the second group (p value < 0.01). Up-regulated indicated there was higher gene expression level in the second group than the first group, whereas down-regulated meant there was higher gene expression level in the first group than the second group (FDR ≤ 0.001).
Importantly, three well-known cytokine genes, interleukin 8 (IL8), interleukin 2 receptor, beta (IL2RB), and interleukin 1 receptor accessory protein-like 1 (IL1RAPL1), exhibited significant DNA methylation changes and significant inverse gene expression changes between the NC and SV groups (Table 4). The IL8 was methylated at a significantly lower level (p < 0.01) and expressed at a significantly higher level (FDR < 0.001) in SV compared with NC. In contrast, both IL2RB and IL1RAPL1 had a significantly higher level of DNA methylation (p < 0.01) and significantly lower level of expression (FDR < 0.001) in SV than NC.
Table 4. Comparisons of gene expression and DNA methylation between NC and SV for IL8, IL2RB, and IL1RAPL1.
| Gene | Expression | Methylation | ||||||
|---|---|---|---|---|---|---|---|---|
| RPKM (NC) | RPKM (SV) | Change | FDR | Location | Reads (NC) | Reads (SV) | P-value | |
| IL8 | 6.71 | 14.07 | 1.06 | 1.71E-05 | Promoter | 33 | 9 | 1.64E-04 |
| IL2RB | 20.52 | 9.67 | −1.09 | 2.98E-07 | Gene body | 26 | 55 | 2.87E-03 |
| IL1RAPL1 | 1.60 | 0.14 | −3.53 | 5.38E-05 | Gene body | 33 | 74 | 2.73E-04 |
| 23 | 54 | 1.03E-03 | ||||||
NC and SV indicated the group of non-challenged and challenged-severe pathology, respectively. Here change was calculated by log2 ratio (SV/NC). Location referred that the differentially methylation appeared in the promoter or gene body regions of the gene. Reads referred to normalized reads in each incorporated interval of peaks.
Subsequently, we used IPA to investigate which gene networks might be affected by these genes showing significant methylation changes in conjunction with significant inverse gene expression changes. The top gene network identified in the NC vs. MD contrast involved cellular movement, inflammatory disease and inflammatory response (Fig. 4A). Genes of ERK1/2 and IL1B were central of this network. A total of 11 genes showing inverse DNA methylation and gene expression changes were involved in this network, including AOAH, ETV1, ITGA9, LARP6, PLCXD1, PLEKHA7, PTPRZ1, SLC2A9, SLC8A1, SPI1, and TSPAN12. In the NC vs. SV contrast, the top two gene networks identified involved cancer, organismal injury and abnormalities, on the one hand, and cell signaling, molecular transport, on the other (Fig. 4B, 4C). Of those genes showing inverse relationships between DNA methylation and gene expression changes, a total of 18 genes (ADAMTS1, ATP8A2, AUTS2, CD8A, CENPJ, DOCK3, EDAR, FREM1, FSHR, KIAA0226, KIT, MICAL2, NEK3, PDE1C, SLC2A9, ST3GAL1, TACR1, and TPM1) were involved in the first network, while 16 genes (BPI, CACNA1H, CNGA3, ETV1, F3, GRIN2B, IL8, ITPR1, KDM8, LRP5, NFAM1, NFATC1, PRKD1, SLC8A1, TMEFF2, and WASF3) were involved in the second network. Importantly, the cytokine IL8 was one of the central molecules in the second network. On the other hand, one gene network involved in cell cycle was identified in the MD vs. SV contrast and all genes we obtained (CCT5, CENPJ, CAMK2G, DNAAF2, LAP3, MYSM1, PTCHD2, SCUBE1, and VIPR1) were involved in this network (Fig. 4D).
Figure 4. Identification of gene networks in the contrasts of NC vs. MD, NC vs. SV, and MD vs. SV.

(A): A gene network identified in NC vs. MD. (B) and (C): The top two gene networks in NC vs. SV. (D): A gene network identified in MD vs. SV. Gene network was identified through integrative analysis of significant DNA methylation changes in conjunction with significant inverse gene expression changes in each of the three groups. Genes exhibiting down-methylated and up-regulated were shown in red color, while those exhibiting up-methylated and down-regulated were shown in green. The intensity of red and green colors indicated the degree of up- and down-regulated, respectively. Solid lines indicated direct interaction, whereas dashed lines indicated indirect interaction.
Discussion
MeDIP-seq peaks are the methylation enriched regions. Peak scanning and analysis were important to survey the methylome pattern. In the present study, we assessed the chicken spleen methylation pattern through the analysis of the genomic distribution of peaks and found that DNA methylation was enriched in the gene body regions and repeat elements. These findings confirmed the results of previous study conducted by Li et al.30 using the chicken liver and muscle tissues by the MeDIP-Seq method. In mammals, both promoter and CGIs were observed to be hypomethylated25,32, whereas the methylation in gene bodies was found to be higher than that in intergenic regions32,33. All of these results were consistent with the results of the current study. However, in contrast to previous studies in animals25,32,33, we did not observe that exons exhibited higher methylation level than introns in chickens.
The repressive effects of promoter-associated methylation on gene expression have been well demonstrated34. Earlier reports revealed that the methylation pattern of promoter regions showed a V shaped curve indicative of low methylation levels at the TSS in human, mice, as well as rat, whereas genes in the puffer fish exhibited a prominent dip in methylation just upstream of the TSS25,32,33,35. In many plants such as Arabidopsis and rice, CG methylation exhibited a characteristic peak in the body of protein-coding genes33. In the present study, DNA methylation was lowest at about 400 bp upstream of the TSS, then increased sharply and stayed at a plateau in the gene body regions, which were in accordance with the earlier investigation for chicken30. Previous studies demonstrated that DNA methylation in the gene body regions might alter transcription elongation efficiency and regulate cell context-specific alternative promoters in gene bodies36,37,38. The relatively high methylation level of the gene body in the chicken suggested that the methylation of the gene body regions played an important role in regulating gene expression.
In contrast to the 40–50% density observed in mammalian genomes, the density of interspersed repeats in chicken genome is less than 9%39,40,41. The predominant interspersed repeat element is the CR1 long interspersed nucleotide element (LINE) and it accounts for over 80% of all interspersed repeats in the chicken genome39. Based on our methylome analysis in chicken spleens, we identified LINE/CR1 to be the predominant target of DNA methylation, accounting for more than 48% of the total methylated repeats and these data were consistent with the recent study of the chicken DNA methylome30. High-throughput DNA sequencing revealed similar numbers of CGIs in humans and mice: 25,495 and 23,021 per haploid genome, respectively42. In this study, a total number of 33,915 CGIs were identified in the chicken genome. Previous reports demonstrated that most of the CGIs were unmethylated and our results for chicken confirmed these earlier conclusions43,44. The methylation of the 5′UTR region usually led to the suppression of gene expression. Our data showed that less than 2% of methylated CGIs were located in this region in the chicken genome. Although slightly lower, these results were consistent with the observations reported by previous study30. It has been shown that the methylation of promoter CGIs would induce transcriptional silencing of their downstream genes by changing chromatin structures and blocking transcription initiation19,45. Recently, many studies have uncovered a large class of CGIs named orphan CGIs, which were remote from annotated promoters and could be categorized as intragenic or intergenic CGIs, and confirmed that these CGIs had the characteristics of functional promoters38,42,46. Moreover, further analysis suggested that the majority of methylated CGIs were located in intragenic and intergenic regions38,47. In the present study, we also found that methylated CGIs in chicken were mainly present in the intergenic regions following by exons. On the other hand, a recent study conducted in rat showed that a large proportion of methylated CGI were located in the size range of 200–300 bp and the number of CGIs decreased with increase in the size of the islands, which were in accordance with the results of the current study.
The whole chicken genome sequenced in 2004 is 1,063 Mb in total length and contains 20,000–23,000 predicted genes39. In our study, a total of 14,396 methylated genes were identified and about two-thirds of them were found in all of the three groups. These findings suggested that DNA methylation is a common feature of the chicken genome. Moreover, our results demonstrated that DNA methylation level was negatively correlated with gene expression level. The spleen is an important immunological organ in chicken and it has been successfully utilized for studies about host immune response to disease13,14,48. Therefore, here we used spleen tissues to detect DNA methylation differences after challenged with APEC. Based on the comparison analysis among the NC, MD and SV groups, we identified 4,684 genes showing significant differential DNA methylation in spleens. These genes were useful for the following detection of potential genes affecting host response and resistance to APEC. Sandford et al.13 studied the spleen transcriptome response to APEC infection in chicken and found very little expression difference between mildly infected and non-infected groups on either day 1 or day 5 post infection. Nie et al.14 also found fewer significantly differentially expressed unigenes in the chicken spleen transcriptome of NC vs. MD than that of NC vs. SV. In the current study, methylome comparison analysis among different treatment groups indicated that the birds with severe pathology would produce larger DNA methylation changes.
By integrating DNA methylation and mRNA expression data, a number of genes exhibiting coordinately changed DNA methylation and gene expression were identified in this study. Of these, 22, 87, and 9 genes showed significantly inversely correlation between changes of DNA methylation and changes of expression in the NC vs. MD, NC vs. SV, and MD vs. SV contrasts, respectively. These DNA methylation changes might be functional relevant changes and those genes might be useful for future epigenetic studies about host response and resistant to APEC infection. Some of those genes, such as FABP3 and IL8, were investigated in previous studies of chicken response to virus infection49,50. However, for most of them, so far little was known about DNA methylation-based deregulation in chickens challenged by pathogen.
Our study revealed that genes with inversely changed DNA methylation and gene expression in different contrasts enriched in different biological pathways. These results indicated that the possible mechanism of host response to pathogen challenge might be distinct in a particular pathology state. The network related to inflammatory and cellular movement appeared in the contrast compared between birds with mild infection and control, and 11 genes were implicated in this network. Inflammation is the first response to infection and it is an essential component of the organism's defense mechanism against pathogens and tissue damage51. The purpose of inflammatory response is to control or eradicate the infection and heal the damage. The network we found in NC vs. MD suggested that DNA methylation changes might play important roles in modulating the host immune/inflammatory response to APEC via the regulation of gene expression. On the other hand, in the contrast compared between birds with severe lesions and control, top gene network associated with organismal injury and abnormalities was observed and 18 genes were implicated in it, suggesting that protective responses to organismal injury were activated after severe infection and methylation changes in those genes might have functional consequences in organismal injury/survival. In addition, network involving cell signaling and molecular transport in the NC vs. SV contrast highlighted the importance of proper signaling cascades to fight severe infection. In the current study, a network involving cell cycle was found in the comparison between the MD and SV chickens, which showed different response to APEC infection, and 9 of the genes we identified were contained in this network. These findings indicated that the DNA methylation changes in the host cell cycle processes might contribute to the lesion phenotype differences.
Among the genes showing significant methylation changes in conjunction with significant inverse gene expression changes, three pro-inflammatory cytokine genes, IL8, IL2RB, and IL1RAPL1, were of particular interest. Cytokines were small proteins released by immune system cells and other cells, and they were an important part of the intercellular communication system responsible for immune response52. Generally, cytokines fulfilled the biological processes by binding their specific receptors. Chemokines were a special type of cytokines that induced the migration of cells to sites of infection or injury52. The interleukin molecules (ILs) were an important class of cytokines. Some ILs, such as IL8, also belonged to chemokines52. Recently, IL8 was reported to play an important role in immune response to Salmonella in chicken53. In addition, it was demonstrated to be one of important genes involved in chicken MD-resistance or -susceptibility50. In the present study, IL8 was a central gene of the network involving cell signaling and molecular transport in NC vs. SV. Birds with severe pathology had significantly lower levels of DNA methylation and significantly higher levels of expression of IL8 than birds with non-challenged. Moreover, the altered DNA methylation was occurred in the promoter region of the IL8 gene. These indicated that the DNA methylation change of IL8 might play a critical role in modulating the host response and resistance to APEC. IL1RAPL1, similar to the interleukin 1 accessory proteins, was a member of the interleukin 1 receptor family. Earlier study has demonstrated an involvement of IL1RAPL1 in the immune proinflammatory response to pathogen infection in pigs54. On the other hand, IL2RB was reported to play a key role in T-cell mediated immune response and its genetic polymorphisms were associated with various diseases55,56. In our study, both IL1RAPL1 and IL2RB were significantly up-methylated and down-regulated in SV compared to NC, which supports the critical function of DNA methylation changes of these two genes in the immune response to APEC in chicken.
In conclusion, we have generated the splenic DNA methylome for the chicken and the methylated regions obtained from MeDIP-seq could be validated by bis-seq. The genome-wide DNA methylation profiles were compared among the NC, MD, and SV groups. By integrating DNA methylation and mRNA expression data, a number of potentially functional relevant DNA methylation changes were identified, of which some played important roles in the regulation of the expression of genes involved in the host inflammatory response and organismal injury/survival after APEC infection. Methylation changes of genes involving cell signaling and molecular transport were also important in fighting severe infection. Moreover, DNA methylation changes of genes in the cell cycle processes might contribute to the lesion phenotype differences between MD and SV chickens.
Methods
Ethics statement
All experiments of this study were approved by the Iowa State University Institutional Animal Care and Use Committee (# 11-07-6460-G). And the experiment was performed according to regulations and guidelines established by this committee. All efforts were made to minimize suffering.
Animals and sample collection
A total of 360 non-vaccinated commercial male broilers were reared in cages with ad libitum access to water and food. All chickens were subjected to a 22:2 hour light:dark cycle for the first 15 days and then changed to a 16:8 hour cycle. At 4 weeks of age, birds were divided into two groups. One group (n = 240) was challenged with 0.1 ml (containing 108 colony forming units) APEC O1. The others (n = 120) were assigned to the control group, in which birds were non-challenged (NC) but treated with 0.1 ml phosphate buffered saline (PBS). At one day post challenge, birds were sacrificed and necropsied to determine their lesion scores as described by previous study13. Birds with scores ranging from 0 to 2 were designated as mild pathology, while those with scores ranging from 4 to 7 were regarded as severe pathology. The average lesion scores for NC, MD, and SV were 0.00 ± 0.00, 0.50 ± 0.58, and 5.25 ± 1.26, respectively. In each group, the whole spleens from 3 individuals were collected and placed in RNAlater immediately, and then stored at −80°C until DNA extraction.
DNA preparation and MeDIP-seq
Genomic DNA was extracted using Qiagen DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer's recommendation and then treated with 10 μg/ml RNase enzyme (Qiagen, Valencia, CA, USA) to degrade RNA. The quality and purity of DNA were determined by agarose gel electrophoresis and spectrophotometer. DNA quantity was measured with Quant-iT dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA). DNA from three individuals within each group was pooled in equal amounts to produce a mixed sample per group. Subsequently, 5 μg mixed DNA sample was sonicated to generate random fragments ranging from 100 to 500 bp. Then end-repair, phosphorylating and A-tailing were performed with Paired-End DNA Sample Prep kit (Illumina, San Diego, CA, USA) according to the manufacturer's protocol. After ligation with Illumina adaptors, the fragments were used for subsequent MeDIP enrichment with Magnetic Methylated DNA Immunoprecipitation kit (Diagenod, Liège, Belgium) using 1 μg 5-methylcytosine antibody. DNA from qualifying MeDIP experiment was amplified to produce libraries with insert sizes between 200 and 300 bp. Products were quantified using an Agilent 2100 Analyzer (Agilent Technologies, Santa Clara, CA, USA) and the Quant-iT dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA). Sequencing was carried out on the Illumina Hiseq 2000 (Illumina, San Diego, CA, USA) following the standard protocol to generate paired-end 50-bp reads in a commercial company (BGI, Shenzhen, Guangdong, China).
Bisulfite sequencing
One microgram of pooled DNA from each group was bisulfite-treated using the EpiTect Bisulfite kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Four pairs of primers were designed with Methyl Primer Express Software v1.0. P1 (F: TTGTTTTTGTTTTGGATGGTTA, R: ATTAACCACCCCCACCTAC), P2 (F: AGGGTTGTAGATTTGTATTTGGA, R: ACTTAATACCTTTCTCCCCATCT) and P3 (F: GTTGATTGTAGTTTTTTGAAGG, R: CACATCCTCAATAATAATATCCC) were used for the validation of regions with high methylation, whereas P4 (F: ATGTGGTATTGAGGGATATAGGT, R: TAAATAATTTCCCCCCCTATC) was used for the validation of regions with relatively low methylation (Supplementary Table S5). Semi-nested PCR was performed in 50-μl reaction volumes containing 50 ng template DNA, 1 μM of each primer and 2.5 U of LA Taq HS (TaKaRa, Osaka, Japan). The first round and the second round of amplification conditions were the same as follows: 94°C for 3 min; 35 cycles of 94°C for 30 s, 56°C for 30 s and 72°C for 30 s; and 72°C for 5 min. PCR products were gel-purified with a Gel Extraction Kit (Tiangen, Beijing, China) and then cloned into the pMD18-T vector (Takara, Osaka, Japan). Ten clones were sequenced and then analyzed using ClustalW.
RNA-Seq
The process of RNA-Seq was described in our previous study14. Briefly, total RNA of spleens used for the DNA methylation analysis was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Then, three spleens in each group were pooled in equal amounts to generate one mixed sample and these three mixed samples were subsequently used for cDNA library construction with the TruseqTM RNA sample prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. After the steps of size selection, PCR amplification, product validation and quantification, libraries were subjected to Illumina deep sequencing on the Genome Analyzer IIx (Illumina, San Diego, CA, USA) in Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China).
MeDIP-Seq data analysis
Reads containing adapters, unknown or low quality bases were filtered out from raw data generated by Illumina sequencing. Then the retained reads were mapped to the chicken reference genome downloaded from Ensembl genome browser (ftp://ftp.ensembl.org/pub/release-63/fasta/gallus_gallus/dna/) using the software SOAPaligner v 2.21 (http://soap.genomics.org.cn/)57. Mismatches no more than 2 bp were allowed in the alignment and uniquely mapped reads were retained for further analysis. Gene information was also obtained from the public FTP site of Ensembl (ftp://ftp.ensembl.org/pub/release-63/gtf/gallus_gallus/). Repeat annotations were downloaded from the UCSC database (http://hgdownload.cse.ucsc.edu/goldenPath/rn4/bigZips/chromOut.tar.gz) and reads distribution on repetitive elements was analyzed by the software RepeatMasker (http://www.repeatmasker.org/). The CGIs were scanned by CpGPlot (https://gcg.gwdg.de/emboss/cpgplot.html) in this study. The peak detection was carried out with the software MACS V 1.4.2 (http://liulab.dfci.harvard.edu/MACS/)58. Statistical analyses of methylation level differences in different classes was performed with least square method using SAS 8.0 software (SAS Institute Inc., Cary, NC, USA). All genes with peaks were employed for the subsequent identification of differentially methylated genes that exhibited more than 2-fold changes in the three samples while compared to each other and p value < 0.01. Then GO enrichment analysis was conducted for those genes exhibiting altered DNA methylation using the DAVID Functional Annotation Tool with a 0.05 cutoff for Benjamini adjusted p-value59.
Integration of gene expression analysis
For the raw reads obtained from RNA-Seq, the sequencing adaptors, reads with unknown nucleotides larger than 5%, and low quality bases (more than half of the bases' qualities were less than 10) were trimmed. Then all the clean data was mapped to the chicken genome with no more than 5 bp mismatches using SOAPaligner v 2.21 (http://soap.genomics.org.cn/)57. Sequences uniquely mapped to the chicken genome were used for subsequent analysis. Gene expression level was calculated with RPKM method (Reads Per kb per Million reads)60 and genes with more than twofold changes and false discovery rate (FDR) ≤ 0.001 were regarded as significant differentially expressed. Differentially methylated genes located in the promoter or the genebody region genes (more than half of the sequences were overlapped with peak summits) were used to identify potentially functional genes affecting host response and resistance to APEC by integrating with mRNA expression data. Additionally, genes of which expression levels changed in accordance with DNA methylation changes were used for gene network and biological processes enrichment analysis with Ingenuity Pathways Analysis (IPA) (Ingenuity Systems; http://www.ingenuity.com).
Author Contributions
Conceived and designed the experiments: X.Q.Z., Q.N. Performed the experiments: H.X., X.N.Z., Y.H., Z.L. Analyzed the data: H.X. Y.H., Q.N. Contributed materials: Q.N. Wrote the paper: H.X.
Supplementary Material
Supplementary Table S3
Supplementary Table S4
Supplementary Info File #1
Acknowledgments
We thank Susan J. Lamont (Department of Animal Science, Iowa State University, USA) for providing the APEC-challenged samples. This research was supported by grants from the National High Technology Research and Development Program (863) of China (2011AA100301; 2010AA10A102), the National Broiler Industry Technology System (nycytx-42-G1-04) and the National Natural Scientific Foundation of China (31000544). Animal studies were supported by National Research Initiative Competitive Grant no.2008-35604-18805 from the USDA National Institute of Food and Agriculture Microbial Genome Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Gross W. B. Coliobacillosis. In Diseases of poultry. Iowa State University Press (Iowa, 1991). [Google Scholar]
- Barnes H. J., Nolan L. K. & Vaillancourt J. F. Colibacillosis. In Diseases of poultry. Blackwell Publishing (Iowa, 2008). [Google Scholar]
- Lutful Kabir S. M. Avian colibacillosis and salmonellosis: a closer look at epidemiology, pathogenesis, diagnosis, control and public health concerns. Int. J. Environ. Res. Public Health 7, 89–114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dho-Moulin M. & Fairbrother J. M. Avian pathogenic Escherichia coli (APEC). Vet. Res. 30, 299–316 (1999). [PubMed] [Google Scholar]
- Cloud S. S., Rosenberger J. K., Fries P. A., Wilson R. A. & Odor E. M. In vitro and in vivo characterization of avian Escherichia coli. I. Serotypes, metabolic activity, and antibiotic sensitivity. Avian Dis. 29, 1084–1093 (1985). [PubMed] [Google Scholar]
- Zhao S. et al. Antimicrobial susceptibility and molecular characterization of avian pathogenic Escherichia coli isolates. Vet. Microbiol. 107, 215–224 (2005). [DOI] [PubMed] [Google Scholar]
- Dai J. et al. Suppression subtractive hybridization identifies an autotransporter adhesin gene of E. coli IMT5155 specifically associated with avian pathogenic Escherichia coli (APEC). BMC Microbiol. 10, 236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pace F. et al. The type VI secretion system plays a role in type 1 fimbria expression and pathogenesis of an avian pathogenic Escherichia coli strain. Infect. Immun. 78, 4990–4998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. et al. Transcriptome analysis of avian pathogenic Escherichia coli O1 in chicken serum reveals adaptive responses to systemic infection. Infect. Immun. 79, 1951–1960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. Novel roles for autotransporter adhesin AatA of avian pathogenic Escherichia coli: colonization during infection and cell aggregation. FEMS Immunol. Med. Microbiol. 63, 328–338 (2011). [DOI] [PubMed] [Google Scholar]
- Holden K. M., Browning G. F., Noormohammadi A. H., Markham P. F. & Marenda M. S. TonB is essential for virulence in avian pathogenic Escherichia coli. Comp. Immunol. Microbiol. Infect. Dis. 35, 129–138 (2012). [DOI] [PubMed] [Google Scholar]
- Lavric M. et al. Gene expression modulation in chicken macrophages exposed to Mycoplasma synoviae or Escherichia coli. Vet. Microbiol. 126, 111–121 (2008). [DOI] [PubMed] [Google Scholar]
- Sandford E. E. et al. Spleen transcriptome response to infection with avian pathogenic Escherichia coli in broiler chickens. BMC Genomics 12, 469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Q., Sandford E. E., Zhang X., Nolan L. K. & Lamont S. J. Deep Sequencing-Based Transcriptome Analysis of Chicken Spleen in Response to Avian Pathogenic Escherichia coli (APEC) Infection. PLoS One 7, e41645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D. & Henikoff S. Genome-wide analysis of DNA methylation patterns. Development 134, 3959–3965 (2007). [DOI] [PubMed] [Google Scholar]
- Sasaki H., Allen N. D. & Surani M. A. DNA methylation and genomic imprinting in mammals. EXS 64, 469–486 (1993). [DOI] [PubMed] [Google Scholar]
- Courtier B., Heard E. & Avner P. Xce haplotypes show modified methylation in a region of the active X chromosome lying 3′ to Xist. Proc Natl Acad Sci U S A 92, 3531–3535 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried Z., Eden S., Mendelsohn M., Feng X., Tsuberi B. Z. & Cedar H. DNA methylation represses transcription in vivo. Nat. Genet. 22, 203–206 (1999). [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002). [DOI] [PubMed] [Google Scholar]
- Robertson K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005). [DOI] [PubMed] [Google Scholar]
- Conerly M. & Grady W. M. Insights into the role of DNA methylation in disease through the use of mouse models. Dis. Model Mech. 3, 290–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M. & Esteller M. DNA methylation and cancer. Adv. Genet. 70, 27–56 (2010). [DOI] [PubMed] [Google Scholar]
- Zhang X. et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201 (2006). [DOI] [PubMed] [Google Scholar]
- Ruike Y., Imanaka Y., Sato F., Shimizu K. & Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 11, 137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sati S. et al. High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region. PLoS One 7, e31621 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird P. W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 11, 191–203 (2010). [DOI] [PubMed] [Google Scholar]
- Li N. et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 52, 203–212 (2010). [DOI] [PubMed] [Google Scholar]
- Feber A. et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res. 21, 515–524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo O. et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 7, 617–636 (2012). [DOI] [PubMed] [Google Scholar]
- Li Q. et al. Genome-wide mapping of DNA methylation in chicken. PLoS One 6, e19428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics 13, 300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L. et al. Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 107, 8689–8694 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R. J. & Bird A. P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97 (2006). [DOI] [PubMed] [Google Scholar]
- Zemach A., McDaniel I. E., Silva P. & Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919 (2010). [DOI] [PubMed] [Google Scholar]
- Lorincz M. C., Dickerson D. R., Schmitt M. & Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11, 1068–1075 (2004). [DOI] [PubMed] [Google Scholar]
- Zilberman D., Gehring M., Tran R. K., Ballinger T. & Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39, 61–69 (2007). [DOI] [PubMed] [Google Scholar]
- Maunakea A. K. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004). [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- Illingworth R. S. et al. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6, e1001134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichman-Almashanu L. Z. et al. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 12, 543–554 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F. et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 38, 1378–1385 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards E. J. & Elgin S. C. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108, 489–500 (2002). [DOI] [PubMed] [Google Scholar]
- Deaton A. M. & Bird A. CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth R. et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 6, e22 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano R., Konnai S., Onuma M. & Ohashi K. Microarray analysis of host immune responses to Marek's disease virus infection in vaccinated chickens. J. Vet. Med. Sci. 71, 603–610 (2009). [DOI] [PubMed] [Google Scholar]
- Luo J., Yu Y., Chang S., Tian F., Zhang H. & Song J. DNA Methylation Fluctuation Induced by Virus Infection Differs between MD-resistant and -susceptible Chickens. Front. Genet. 3, 20(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. Temporal transcriptome changes induced by MDV in Marek's disease-resistant and -susceptible inbred chickens. BMC Genomics 12, 501(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Ji Y., Kersten S. & Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutr. 32, 261–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke J. W. & Borish L. Cytokines and chemokines. J. Allergy Clin Immunol. 117, S441–445 (2006). [DOI] [PubMed] [Google Scholar]
- Ciraci C., Tuggle C. K., Wannemuehler M. J., Nettleton D. & Lamont S. J. Unique genome-wide transcriptome profiles of chicken macrophages exposed to Salmonella-derived endotoxin. BMC Genomics 11, 545 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo R. C. et al. Gene expression profile suggests that pigs (Sus scrofa) are susceptible to Anaplasma phagocytophilum but control infection. Parasit. Vectors 5, 181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál Z. et al. A novel galectin-1 and interleukin 2 receptor β haplotype is associated with autoimmune myasthenia gravis. J. Neuroimmunol. 229, 107–111 (2010). [DOI] [PubMed] [Google Scholar]
- Spitz M. R. et al. Multistage analysis of variants in the inflammation pathway and lung cancer risk in smokers. Cancer Epidemiol. Biomarkers Prev. 21, 1213–1221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Li Y., Kristiansen K. & Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B. T. & Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L. & Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S3
Supplementary Table S4
Supplementary Info File #1