

Abstract
Purpose
To elucidate the mechanistic basis for efficacy of intrathecal rituximab. We evaluated complement activation as well as the pharmacokinetics of intraventricular rituximab in patients who participated in two phase 1 multicenter studies.
Experimental Design
We evaluated complement activation as a candidate mediator of rituximab within the CNS. Complement C3 and C5b-9 were quantified by ELISA in serial CSF specimens after intraventricular rituximab administration. We determined rituximab concentration profiles in CSF and serum. A population three- compartment pharmacokinetic model was built to describe the disposition of rituximab following intraventricular administration. The model was derived from results of the first trial and validated with results of the second trial.
Results
Complement C3 and C5b-9 were reproducibly activated in CSF after intraventricular rituximab. Ectopic expression of C3 mRNA and protein within CNS lymphoma lesions was localized to myeloid cells. Constitutive high C3 activation at baseline was associated with adverse prognosis. A PK model was built which contains three distinct compartments to describe the distribution of rituximab within the neuroaxis after intraventricular administration.
Conclusions
We provide the first evidence of C3 activation within the neuroaxis with intraventricular immunotherapy and suggest that complement may contribute to immunotherapeutic responses of rituximab in CNS lymphoma. Penetration of rituximab into neural tissue is supported by this pharmacokinetic model and may contribute to efficacy. These findings have general implications for intraventricular immunotherapy. Our data highlight potential innovations to improve efficacy of intraventricular immunotherapy both via modulation of the innate immune response as well as innovations in drug delivery.
Keywords: Complement, Immunotherapy, Non-Hodgkin’s Lymphoma, Rituximab, Pharmacokinetics
INTRODUCTION
Since the first serotherapy trial using a monoclonal antibody was performed in 1980, immunotherapeutic approaches based upon recombinant antibody technology have come to transform clinical practice in a variety of disciplines.1 In particular, the introduction of the anti-CD20 antibody rituximab has dramatically impacted the management of patients with B-cell malignancies and other disorders related to B-cell pathology.2 The now widespread utilization of recombinant antibodies to treat human disease has prompted significant interest in analyses of their safety, mechanisms of action and resistance, pharmacokinetics and distribution, and combinatorial use with other agents.3, 4
While agents such as trastuzumab and rituximab improve outcomes in systemic HER2-positive breast cancer and B-cell non-Hodgkin’s lymphoma respectively, there is evidence that the blood-brain-barrier significantly attenuates efficacy of immunotherapeutic approaches for brain tumors that involve systemic administration of protein macromolecules.5–9 For this reason, several investigators have evaluated intrathecal administration of antibodies such as rituximab or trastuzumab, to bypass the blood-brain-barrier and induce responses of CNS lymphoma or breast cancer brain metastases.10,11–13,14–27
Our group recently performed the first dose escalation studies of the intrathecal administration of a naked monoclonal antibody (rituximab). An initial study was performed in cynomologus monkeys, followed by two multicenter Phase I trials in patients with recurrent CNS lymphoma.10, 13, 22
In the first clinical study, we evaluated rituximab as monotherapy, administered primarily via the intraventricular route. In the second, we evaluated combination rituximab plus methotrexate as immunochemotherapy. Intraventricular rituximab was well-tolerated at the 10 and 25 mg dose levels, as monotherapy and in combination with methotrexate, and induced responses not only within the cerebrospinal fluid (CSF) but surprisingly, also within the intraocular compartment and in non-bulky lesions (< 2 cm) within brain parenchyma. Activity was detected in patients with disease recurrent or refractory to intravenous rituximab, independent of steroid administration. Similar results using intrathecal rituximab and trastuzumab have been described by other investigators in case reports and small clinical series.10,11–13,14–27
These positive results have been somewhat surprising given the concept of the central nervous system (CNS) as an immunologically privileged site, assumed to be devoid of significant concentrations of complement mediators28 and immune effector cells and because of the presumption that macromolecules administered within the CSF are unable to penetrate brain parenchyma.
Because the CSF is specialized to bathe the brain and spinal cord and is not exposed to the systemic circulation, there is significant representation of CNS-relevant inflammatory cells and protein constituents within its approximate 150 ml volume. We previously described the first large-scale proteomic analysis of CSF to identify CSF proteins that reproducibly distinguish CNS lymphoma from benign conditions.29 While a significant proportion of these peptides are associated with lymphoma invasion and extracellular matrix, most of the upregulated CSF peptides identified involve the innate immune response, including CD14, a marker of monocyte activation, as well as activating and inhibitory components of the complement cascade.29
In addition, Pulido and colleagues recently demonstrated selective penetration of rituximab (molecular weight 145 kD), into full-thickness retina, in contrast to tissue plasminogen activator (70 kD), after intravitreal injection.30 Similar results were obtained with bevacizumab.31 Moreover, the intraventricular administration of neurotrophic proteins has reproducibly been demonstrated to induce neurogenesis in deep brain structures, including thalamus and substantia nigra in mouse models.32–34 Taken together, these findings implicate existence of selective mechanism(s) that facilitate transport of macromolecules from CSF or vitreous into neural tissues.
We previously described a pharmacokinetic (PK) model of rituximab disposition after intraventricular injection in cynomologus monkeys which predicts a three-compartment model with distinct rate constants for rituximab distribution from CSF to serum and from CSF to brain tissue. The goals of this study are to: (1) define the pharmacodynamics of complement activation within the CSF upon intrathecal rituximab administration, and (2) utilize PK data from two recent phase I trials in CNS lymphoma to describe the disposition of rituximab after intraventricular administration in humans. We hypothesize that this information will yield insights into the basis for efficacy of intraventricular rituximab in the treatment of lymphoma within the neuroaxis, as well as be relevant to the development of novel therapeutic strategies for brain diseases that are based upon the intra-CSF administration of therapeutic macromolecules.
METHODS
Study Population
CSF and blood specimens from subjects were obtained after informed consent in accordance with Institutional Review Board–approved protocols at the University of California, San Francisco. Ten subjects were treated in the first phase I trial of intraventricular rituximab (median age 55.5, range 28–82; seven males, three females). Fourteen subjects were treated in the second phase I trial of intraventricular rituximab plus methotrexate (median age 61, range 37–75; seven males, seven females). Traumatic CSF specimens were excluded.
Immunohistochemistry
Frozen sections were immunostained with monoclonal anti-complement C3 (Quidel). Anti-Iba-1 antibody was from Abcam. Immunoreactivity was visualized as described.35 Confocal imaging was performed using a Zeiss LSM510 NLO Meta microscope with multi photon (800nm), 488nm and 543-nm laser excitation.
In situ hybridization
Full-length human complement C3 cDNA in pBluescriptSK(−) was from American Type Culture Collection and verified by resequencing. In situ hybridization was performed using digoxigenin-labeled riboprobes, as described.35
ELISA
C3a ELISA: Quantitative determination of C3a concentration was performed using C3a Enzyme Immuno Assay Kit (Quidel) for the detection of C3-desArg. Albumin ELISA was from Bethyl Laboratories.
Western Blot Analysis
CSF proteins were subjected to SDS-PAGE (10% Bis-Tris) under reducing conditions and transferred to nitrocellulose for immunoblot analysis using an anti-C3 mouse monoclonal antibody (Quidel), peroxidase-conjugated anti-mouse IgG (Jackson Immunoresearch) and ECL (Amersham).
Flow cytometric purification and gene expression analysis of CSF macrophages and B-cells
After collection, CSF was centrifuged at 1500 rpm, and supernatant carefully removed. Cell pellets were resuspended in FACS buffer (PBS, Ca2+/Mg2+-free, with 5% FCS) and incubated with anti-CD11b/Mac-1-APC (BD Biosciences), anti-CD14-AlexaFluor700 (BD Biosciences), and anti-CD19-PE (BD Biosciences) antibodies for 30 minutes, protected from light. Cells were washed twice and resuspended FACS buffer with DAPI. Cells were analyzed and sorted using BD FACS Aria II. Live cells were gated by DAPI exclusion, size and granularity based on forward and side scatter parameters. Cells were sorted directly into Direct Lysis Buffer from NuGEN One-Direct kit and stored at −80°C. Samples were processed using a NuGEN FL-Ovation™ cDNA Biotin Module V2. Quantitative RT-PCR analyses were performed using human complement C3 probe and normalized to GAPDH (ABI).
Pharmacokinetic Sampling
Serial CSF samples for pharmacokinetic analysis were obtained from Ommaya reservoir. During the first week of the trial, matched CSF and venous blood serum samples were obtained immediately prior to treatment and at 1, 2, 4, 8, 24, and 96 hours post-dose. During the following 4 weeks, CSF and blood samples were obtained on Day 1 and Day 4 immediately prior to each dose and again 1 hour post-dose. Blood samples were allowed to clot at room temperature for 45 minutes, then centrifuged at 1300 g. CSF and serum were frozen within one hour of collection and stored at −80°C.
Bioanalysis
CSF and serum concentrations of rituximab were determined using a validated ELISA.36 The lower limit of quantification for rituximab was 0.250 μg/mL for CSF and 0.500 μg/mL for serum.
Pharmacokinetic Data Analysis
Rituximab CSF and serum concentration data were modeled simultaneously using nonlinear mixed effects modeling (NONMEM VII version 7.2.0, ICON). Graphical evaluation of NONMEM outputs was performed using S-PLUS 8.0 for Windows (Insightful). The first-order conditional estimation with interaction (FOCEI) method was used for population PK analyses. PK parameters were derived using POSTHOC step in NONMEM. CSF and serum concentrations below the lower limit of quantitation were assigned as missing.
RESULTS
Rapid Lymphocytotoxic Effects of Intraventricular Rituximab
During the course of both phase I trials, we observed rapid lymphocytoxic efficacy of intraventricular rituximab in responding patients with cytologically-positive leptomeningeal disease. Marked depletion of B-lymphoma cells within the CSF was demonstrated within hours of intraventricular rituximab therapy, by differential cell counts and cytologic analyses, performed and reported by the clinical laboratory at baseline and at timepoints after intraventricular rituximab. In addition, flow-cytometry of B-cells within CSF specimens at baseline and repeated at one hour reproducibly confirmed depletion of B-cells one hour after rituximab administration. Notably, in several cases in which there was rituximab-associated B-cell lysis, the concentration of monocytes in serial CSF specimens increased in a time-dependent manner, demonstrating the selective anti-B-cell activity of rituximab after intraventricular administration (Figure 1).
Figure 1. Rapid lymphocytotoxic effects within the CSF following intraventricular rituximab.


Figure 1A. Forward versus side scatter FACS plots of CSF cells from a subject with recurrent CNS lymphoma, pre (left) and 1 hour post (right) intraventricular rituximab monotherapy treatment. (3 ml CSF analyzed/condition).
Figure 1B. Anti-CD19 (PE-labeled)-positive populations in the CSF pre (left) and 1 hour post (right) intraventricular rituximab in same patient as in 1A.
Figure 1C. Intra-CSF cell toxicity assessment pre (left) and 1 hour post (right) intraventricular rituximab in a different patient treated with rituximab monotherapy, measured by DAPI positivity, which demonstrated a rapid increase in the number of DAPI+ non-viable cells. The increased number of DAPI+ cells also correlated with a marked decrease in CD19+ B-cells within CSF.
Figure 1D. Degenerated cell quantification (as reported by the UCSF Clinical Laboratory) in CSF of a third subject with recurrent CNS lymphoma, at baseline, 1 hour and 2 hours post-treatment with intraventricular rituximab. Rituximab was associated with a time-dependent increase in CSF monocytosis during this interval.
Figure 1E. Cytologic appearance of malignant cells in CSF at baseline (left), and degenerated cells one hour after intraventricular rituximab (right). (X 400).
Complement Activation in CSF
Given the rapid lymphocytotoxic effects of rituximab observed in the treatment of leptomeningeal lymphoma, we evaluated the possibility of activation of the complement cascade in CSF in CNS lymphoma patients treated with intraventricular rituximab.13 Complement C3 is central to the activation of both the classical and alternative complement pathways, therefore, we measured the concentration of C3a anaphylatoxin as a biomarker of complement activation within the CSF. We used a quantitative ELISA for C3a des-arg to evaluate the temporal relationship between rituximab administration and complement activation in CSF.
Intraventricular rituximab monotherapy was reproducibly associated with complement activation in each of the nine study subjects examined, at each of the three dose levels: 10, 25 and 50 mg rituximab.13 Mean baseline C3a des-arg in CSF in these phase I trial subjects was 28.56 ng/ml (range 5.9–60 ng/ml). Within one-to-two hours of intraventricular rituximab administration, C3a des-arg achieved peak concentrations within CSF (mean 74.6 ng/ml, range 12–181 ng/ml) followed by a decline to baseline or below within 24 hours (Figure 2A). Notably, the rapid onset of complement activation was observed to temporally correlate with anti-lymphoma cytologic effects in responding patients with leptomeningeal disease. Increased C3a generation was confirmed by immunoblot using an anti-C3a-specific monoclonal antibody (Figure 2B). In parallel, generation of the C5b-9 complex within the CSF was also detected after intraventricular rituximab, demonstrating propagation of the complement cascade at the level of C5 and downstream within the CNS lymphoma microenvironment (Figure 2C). Serial measurement of C3a des-arg concentrations over the course of the five week study of twice-weekly intraventricular rituximab confirmed increased C3 activation in CSF at one hour post rituximab administration in CSF with repeat dosing, and demonstrated that C3 protein did not rapidly become depleted (Figure 2E).
Figure 2. Activation of the Complement Pathway in the CSF After Intraventricular Rituximab.

Figure 2A. Intraventricular rituximab administration (10 mg) was reproducibly associated with a time-dependent increase and decline in C3a concentration within the CSF, as demonstrated by ELISA determination of C3a des-arg.
Figure 2B. Immunoblot analysis of C3a concentration in CSF using a monoclonal anti-C3a antibody confirmed C3 cleavage in CSF after rituximab administration.
Figure 2C. Intraventricular rituximab (10 mg) was also reproducibly associated with a time-dependent increase and decline in the concentration of the C5b-9, “membrane attack complex” within the CSF. Figure 2C depicts C5b-9 levels in the same patient as in Figure 2A and with a time course similar to the generation of C3a.
Figure 2D. Serum concentrations of C3a after intraventricular rituximab (same patient as in FIgure 2A). The C3a index (ratio of CSF:serum C3a divided by CSF:serum albumin) was 9 at baseline and increased to 50 at one hour post-rituximab, consistent with intrathecal C3 biosynthesis rather than passage through a disrupted blood-brain barrier. Determination of the C3a index in CSF in three consecutive subjects (treated at 10 mg, 25 mg and 50 rituximab dose levels), each supported the intratumoral production of C3a in tumor and CSF.
Figure 2E. Serial measurement of C3a des-arg concentrations in CSF over the course of the five week study of twice-weekly intraventricular rituximab confirmed reproducible C3 activation at one hour in CSF with each of nine doses (arrows), demonstrating that C3 protein does not rapidly become depleted within the CNS lymphoma microenvironment.
Complement C3 Expression within CNS Lymphoma
Given the stable expression of complement proteins within CSF over serial treatment, we addressed the possibility that complement mediators such as C3 may be synthesized within the CNS lymphoma microenvironment. Immunohistochemical analysis demonstrated strong C3 expression in diagnostic specimens of PCNSL, with particular C3 immunoreactivity associated with tumor vasculature (Figure 3). By contrast, C3 protein immunoreactivity in normal brain was weak or undetectable. We used in situ hybridization with a C3-specific antisense riboprobe to demonstrate intratumoral mRNA expression of C3 by infiltrating cells surrounding tumor vessels. This finding was confirmed in each of five diagnostic specimens of PCNSL analyzed. There was no evidence for C3 gene expression by endothelial cells in these cases. C3 transcripts were minimally detectable in normal brain specimens (not shown). C3 expression within diagnostic specimens of PCNSL was confirmed by quantitative RT-PCR as well (Figure 3). These results demonstrate for the first time the intratumoral production of C3 transcript and protein in the CNS lymphoma microenvironment and implicates complement activation as a potential means by which rituximab may mediate lymphocytoxic responses within CSF and deep brain structures.
Figure 3. Expression of Complement C3 in Primary CNS Lymphoma.

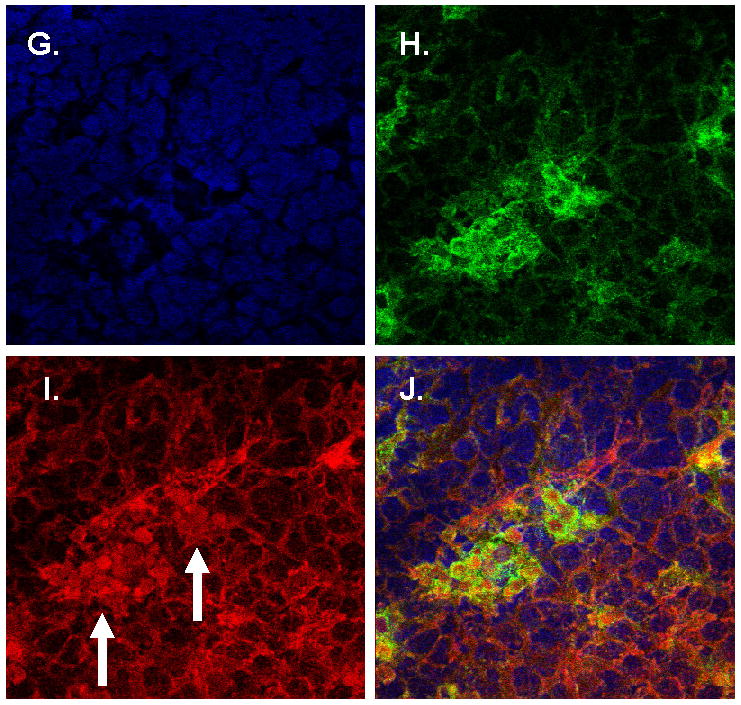

Immunohistochemistry and in situ hybridization studies of complement C3 in PCNSL and normal brain tissues.
Figure 3A. Anti-C3 immunohistochemical analysis of PCNSL specimen reveals strong immunoreactivity associated with tumor vasculature. (x 200).
Figure 3B. Anti-C3 immunohistochemical analysis of normal human brain tissue reveals weak immunoreactivity (x 10).
Figure 3C. In situ hybridization with a C3 anti-sense probe demonstrates C3 mRNA synthesis in PCNSL, with strong expression by cells adjacent to tumor-associated vasculature (x 400).
Figure 3D. In situ hybridization: negative control of PCNSL specimen using sense probe (x 10).
Figure 3E. B-cell and CSF monocyte populations sorted with anti-CD11b and anti-CD14 antibodies.
Figure 3F. Complement C3 mRNA relative expression in sorted CSF B-cell and monocyte populations. C3 transcripts were reproducibly detected in activated, CD14+ CD11b+ monocytes within CSF and in four consecutive diagnostic specimens of primary CNS lymphoma (large B-cell, comprised of at least 80% tumor). There was no C3 expression by C19+ B-cells or by CD11b+/CD14′ myeloid cells within CSF. (* Not detected.)
Figure 3G–J. Confocal microscopy of representative CNS lymphoma specimen: DAPI (blue) identfies all tumor-associated nuclei within the lesion (G); Iba-1 immunoreactivity (green) identifies CNS lymphoma-associated macrophages and microglia (H); Complement C3 immunoreactivity (red) is selectively localized by a monoclonal antibody to the cytoplasm of a subset of CNS lymphoma-associated cells (arrows); (I), merged analysis confirms complement C3 expression predominantly by intratumoral Iba-1+ macrophages and/or microglia (J).
Figure 3K. Spontaneous Complement C3a Activation in the CSF of CNS Lymphoma Patients. C3a des-arg concentration was markedly elevated in the CSF of patients with CNS lymphoma, including newly-diagnosed PCNSL (N=31, (30 DLBCL, 1 T-Cell NHL, median age 62) and relapsed CNS lymphoma (N=26, all DLBCL, median age 48) compared to patients with non-malignant, neuro-inflammatory, or neurodegenerative conditions (N=27, median age 54). (p=0.05) The cohort of patients with non-malignant neurologic conditions included cases of multiple sclerosis (N=6), encephalitides (N=5), transverse myelitis (N=3), two cases each of coccidiomycosis, dementia, normal pressure hydrocephalus, amyotrophic lateral sclerosis, epilepsy and one case each of neurosarcoid, cryptococcal meningitis, and Parkinsonism. Spontaneous C3 activation was also detected in the CSF of patients with brain metastases (N=13, median age 48), including breast cancer (N=6), non-small cell lung cancer (N=2), metastatic carcinoma, unknown primary (N=2), and one case each of metastatic sarcoma, germ cell tumor and small cell carcinoma of the lung.
Figure 3L. Elevated C3a des-arg concentration in CSF is associated with short survival among 17 patients with recurrent CNS lymphoma treated with intraventricular rituximab therapy (10 mg and 25 mg) for recurrent CNS lymphoma. (All ventricular CSF). ECOG >1 was also associated with adverse prognosis in this cohort (P<0.0002). Notably, given that phase I trials are not designed to test efficacy, the significance of C3a as a prognostic variable is limited in this setting. Similar results were obtained in an independent cohort of 35 newly-diagnosed patients with CNS involvement of diffuse large B-cell lymphoma in whom elevated C3a in lumbar CSF at pre-treatment staging, detected in seven patients, also was associated with shorter progression-free survival (P<0.019). (Supplemental data).
While the majority of total complement C3 protein is synthesized by hepatocytes, a variety of evidence has suggested that inflammatory cells such as circulating monocytes may produce C3, particularly in pathologic states such as autoimmune disease.37 To test the hypothesis that monocytes within CNS lymphoma and the leptomeningeal compartment contribute to the elevated C3 and complement activation detected in CNS lymphoma, we used flow-cytometry to isolate, and quantitative RT-PCR to profile, infiltrating leukocytes within the CSF for C3 expression. In three consecutive cases we demonstrated that activated, CD14+ monocytes within the CSF selectively express C3 RNA transcripts, as demonstrated by quantitative RT-PCR. By contrast, C3 transcripts were not detectable in CD14− monocytes or in CD19+ lymphoma isolated from CSF. Given our previous observation that infiltrating macrophages exhibit a perivascular distribution in primary CNS lymphoma,38 these data demonstrate that intratumoral CD14+ monocytes contribute to C3 expression within CNS lymphoma (Figure 3). This conclusion is further supported by dual-color immunofluorescence staining and confocal microscopy (Figure 3G–J) which reproducibly demonstrated immunoreactivity of C3 protein that was localized to the cytoplasm of CNS lymphoma-associated macrophages and microglia, identified by Iba-1 expression.39, 40
Spontaneous Complement Activation in CNS Lymphomas
To further define the relationship between complement activation and the pathogenesis of CNS lymphoma, we compared baseline C3a des-arg concentrations in CSF obtained from 27 HIV-negative patients with non-malignant inflammatory neurologic conditions, 31 patients with a new diagnosis of PCNSL, 26 patients with recurrent CNS lymphoma and 13 patients with brain metastases. Compared to patients with non-malignant neurologic conditions and brain metastases, CNS lymphoma patients exhibited significantly higher spontaneous complement activation within CSF (P=0.05), with the highest concentrations in CSF of patients with recurrent disease. (Figure 3K). We noted a trend between high constitutive complement activation in CSF and shorter progression-free survival: among the 15 CNS lymphoma patients with C3a levels in CSF above 30 ng/ml (greater than two standard deviations above mean C3a in non-malignant neurologic controls), 13 exhibited tumor progression or death within two months of CSF collection. In particular, among 17 relapsed CNS lymphoma patients who were treated with intaventricular rituximab, those with elevated C3a measured in baseline, pre-treatment CSF specimens, experienced shorter overall survival compared to CNS lymphoma patients with lower C3a levels at baseline (P<0.01). (Figure 3L). Similar results were detected in an independent population of 35 newly-diagnosed patients with CNS involvement of diffuse large B-cell lymphoma in whom elevated C3a in CSF at pre-treatment staging, identified in a small subset of patients, also was associated with shorter progression-free survival (P<0.019) (Supplemental Figure 1). Taken together, these data demonstrate that the complement cascade is triggered within the CSF after intraventricular rituximab administration, and may contribute to rituximab-mediated cytotoxicity of CNS lymphoma. In addition, high constitutive levels of complement activation at baseline may be associated with diminished rituximab efficacy. One possible explanation for this paradoxical observation is that C3b, also generated by cleavage of C3, may attenuate the immune response via disruption of NK cell activation and antibody-dependent cellular cytotoxicity.41 The determinants of rituximab response and/or resistance in CNS lymphoma may also relate to its pharmacokinetics and distribution upon intraventricular administration, parameters which have not previously been presented.
Intraventricular Rituximab Pharmacokinetics
Eighty-five (85) rituximab serum samples and 135 CSF samples were collected from 10 patients in the first trial for PK analyses.13 Three patients (Patient No. 7, 8 and 9), were excluded from the overall PK analyses due to the detection of high levels of existing rituximab concentrations at pre-treatment. These data were visually inspected for outliers in which we identified and omitted six outlier data points from the final dataset. The final dataset contained data from 7 patients (3 from the 10 mg dose group and 4 from the 25 mg dose group), with 58 serum PK samples and 118 CSF PK samples.
Rituximab concentration in the CSF displayed a bi-phasic profile after intraventricular administration. A steady accumulation was observed for serum rituximab after multiple administrations. Representative rituximab concentration-time profiles in CSF and serum are presented in Figure 4.
Figure 4. Compartmental PK Model of Intraventricular Rituximab.
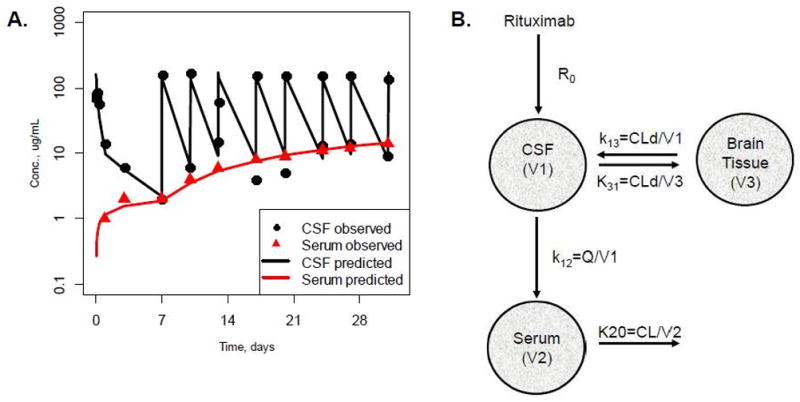
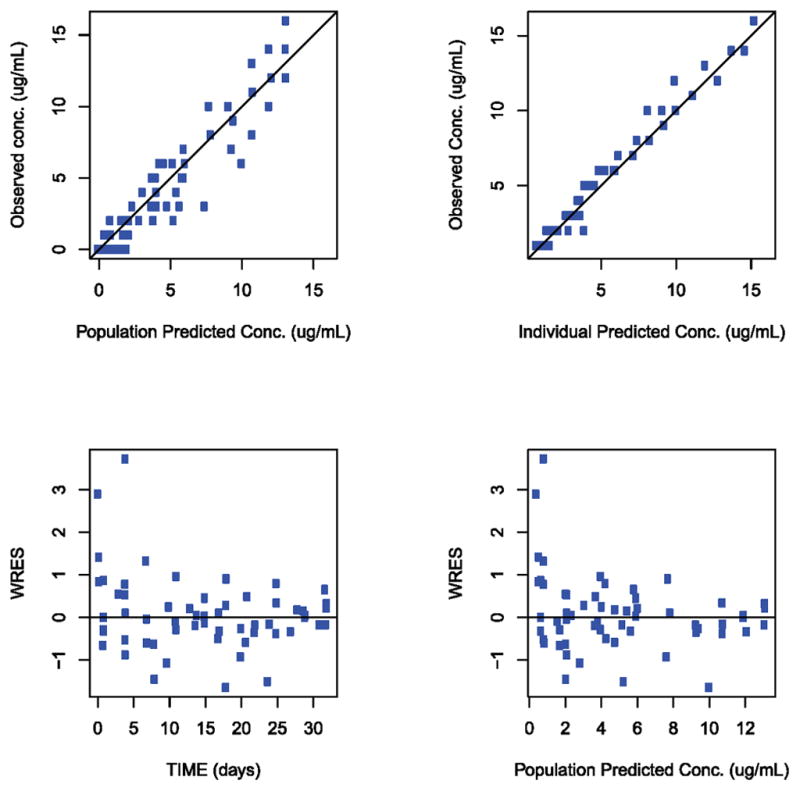
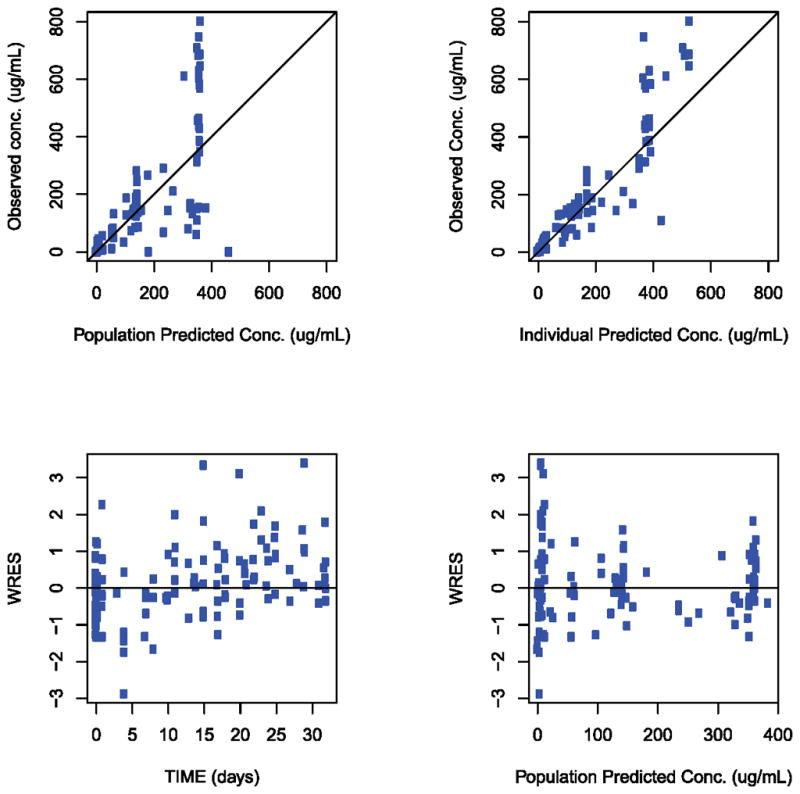
Figure 4A. Representative individual concentration-time profiles of rituximab in CSF and serum and compartmental PK model of intraventricular rituximab.
Black circles were observed rituximab concentration in CSF; Red triangles were observed rituximab concentrations in serum. Black line was model predicted rituximab concentration-time profile in CSF; Red line was model predicted rituximab concentration-time profile in serum.
Figure 4B. Rituximab compartmental PK model after intraventricular injection in patients with CNS lymphoma. R0 = drug intraventricular injection; k13 = rate constant from CSF to brain tissue compartment; k31 = rate constant from brain tissue compartment to CSF; k12 = distribution rate constant from CSF to serum; k10 = elimination rate constant; CL = elimination clearance; Q = distribution clearance from CSF to serum; CLd=distribution clearance from CSF to brain tissue; V1 = CSF volume; V2 = serum volume; V3 = brain tissue volume.
Figure 4C and 4D. Goodness-of-fit plots for the pharmacokinetic model. (4C) Serum. (4D) CSF. IPRED individual predicted conc. (unit in μg/mL). PRED population predicted conc. (unit in μg/mL), TIME study time (unit in days), WRES population-weighted residuals. Solid lines indicate line of unity.
Pharmacokinetic Model and Parameters
A compartmental pharmacokinetic model was built to describe the rituximab concentration time profiles in both CSF and serum after intraventricular administration in 7 patients. The model consists of three compartments: CSF, serum and brain tissue compartments (Figure 4). Clearance of drug from the CSF occurs by distribution clearance, Q, with the serum compartment, and another distribution clearance, CLd, with the brain tissue compartment. This model assumes that rituximab elimination occurs only from the serum compartment, which is through FcRn recycling and catabolism for monoclonal antibodies, the same characteristics elimination mechanism as with intravenous injection. The concentrations of rituximab in each of the three compartments are described by the following set of first-order differential equations:
Where t is the sample time, A1, A2, and A3 are the amount of drug in the CSF, serum and brain tissue respectively. K12, K13, and K31 represent the distribution rate constants from CSF to serum (12), from CSF to brain tissue (13), and from brain tissue to CSF (31), respectively. Potential efflux of the drug from the serum compartment to CSF was ignored in this model, justified based on the experimental observation that the concentration of monoclonal antibody in the CSF after intravenous dosing is greater than 1,000 times lower than the serum concentration.42 Therefore, the potential contribution to the CSF concentration from serum compartment efflux to CSF is expected to be insignificant. A representative individual fit of the model to both CSF and serum concentration is depicted in Figure 4 as well as the goodness-of-fit plots for the full data set and the population-based compartmental PK model used (Figure 4C and D). Overall, the three-compartmental PK model with the brain tissue compartment effectively describes both serum and CSF PK data.
Rituximab CSF and serum data after intraventricular dosing were fitted simultaneously to estimate critical PK parameters. (Table 1). The estimated volume of distribution of rituximab in CSF (V1) and in serum (V2) were 62.2 (22.3% SEE) and 9250 mL (23.4% SEE), respectively. Distribution rate constants from CSF to serum and brain tissue in the model were 3.57 /day (24.2% SEE) and 2.61 /day (34.3% SEE) respectively, and the elimination rate constant from serum was 0.0329 /day (57.4 %SE). The calculated distribution clearance between CSF and serum (Q) and between CSF and brain tissue (CLd) were 208 and 189 mL/day respectively, and the calculated volume of distribution in brain tissue (V3) was 320 mL. The elimination clearance from the serum compartment, CL was 332 mL/day, associated with median terminal elimination half-life from serum was 21.1 days, which was consistent with rituximab elimination half-life previously described after intravenous administration.43
Table 1.
Population pharmacokinetic parameters for rituximab after intraventricular administration in patients with CNS lymphoma.
| Parameter | Population Estimated (% SEE) | Inter-Patient Variability (%SEE) |
|---|---|---|
| Volume of distribution in CSF (V1), mL | 62.2 (22.3) | 50.6 (39.9) |
| Volume of distribution in Serum (V2), mL | 9250 (23.4) | 33.6 (39.9) |
| Distribution rate constant from CSF to Serum (K12), /day | 3.57 (24.2) | 61,6 (26.8) |
| Elimination rate constant from Serum (K20), /day | 0.0329 (57.4) | 56.7 (37.0) |
| Distribution rate constant from CSF to brain tissue (K13), /day | 2.61 (34.3) | |
| Distribution rate constant from brain tissue to Serum (K31), /day | 0.638 (69.4) | |
| Residual error for CSF data (proportional), % | 62.3 (14.7) | |
| Residual error for serum data (Proportional), % | 17.4 (39.1) | |
| Elimination Half-life from Serum, day | 21.1 | |
SEE=standard error of estimate
Pharmacokinetic Evidence for Rituximab Penetration in Brain
The observed biphasic disposition profiles in CSF provide supportive evidence for rituximab penetration in brain tissue. In order to better understand if the biphasic curve is a consequence of rituximab penetration within brain tissue after intraventricular administration, the final compartmental pharmacokinetic model was used to perform a theoretical sensitivity analysis on the V3 parameter. When V3 is equal to 0 mL, meaning to assume that there is no brain tissue compartment, a much faster decline (i.e., mono exponential decline) of rituximab concentration is observed in CSF, indicating that there does not exist a second biological compartment besides the CSF for drug disposition. As the V3 increases, a second elimination phase (bi-phasic) in the CSF appears (Supplemental Figure 2). The biphasic disposition profiles in CSF observed using this model also provide supportive evidence for rituximab penetration in brain tissue.
Validation of the PK Model in a Second Phase I Study
The utility of this three-compartment pharmacokinetic model is highlighted by the fact that it predicted an approximation of the pharmacokinetics of rituximab disposition following intraventricular administration in an independent cohort of seven patients treated at the 25 mg dose level in the second multicenter phase I trial of intraventricular rituximab plus methotrexate (Figure 5).22 This result supports the general applicability of this model structure to the design of clinical interventions that use intraventricular monoclonal antibodies to treat disease within the CNS.
Figure 5.

Figure 5A and 5B. Rituximab concentration-time profiles in (5A) CSF and (5B) serum from 7 patients in 25 mg dose group from second phase I trial of intraventricular rituximab. Overall disposition of rituximab in the second study was successfully predicted based upon the three compartment model derived from analysis of rituximab pharmacokinetics in the first phase I trial of intraventricular rituximab. Open circles represent the original observations; shaded area represents the 95% prediction internal according to the model; the black lines represent the 97.5, 50, and 2.5th percentiles of the simulated data.
DISCUSSION
Here we provide the first demonstration of the activation of the complement cascade within the brain microenvironment after intra-CSF administration of rituximab, a monoclonal antibody containing IgG1 constant domains. These observations provide a potential mechanistic explanation for some of the immunotherapeutic responses of CNS lymphomas with intrathecal rituximab that have been described by many investigators. Notably, it was previously demonstrated that while intravenous rituximab induces significant C3 cleavage in serum, simultaneous activation of C3 in matched serial CSF specimens from a CNS lymphoma patient could barely be detected.44 Our results therefore identify one mechanism by which intraventricular rituximab may overcome the blood-brain barrier to mediate cytotoxicity against otherwise drug-resistant recurrent NHL. Nevertheless, while the time course of complement activation correlates with the onset of B-cell lysis in CSF, other mechanisms of rituximab-mediated efficacy such as antibody-dependent cellular cytotoxicity also likely contribute in this setting.
In addition, we demonstrate upregulation of C3 protein not only within the CSF but also within brain parenchyma. Given the pharmacokinetic evidence for brain penetration of rituximab, these results raise the possibility that rituximab-induced activation of the complement cascade within the brain may have the capacity to elicit anti-tumor responses throughout the neuroaxis, a result which has implications for the intravenous route of administration as well. In support of this hypothesis is our demonstration of the expression of the C5b-9 membrane attack complex not only within the CSF but also diffusely within 3 consecutive CNS lymphomas specimens analyzed (unpublished results). We also detected spontaneous complement activation within the CSF of patients with other types of brain tumors, including metastatic carcinoma and glioma (not shown). Somewhat surprisingly, high levels of the C3 cleavage product C3a were found to be associated with inferior prognosis in independent populations of CNS lymphoma patients. The activating signal for spontaneous complement activation within brain tumors is undefined. One possibility is that C3 convertase activity is mediated via the lectin pathway which may occur by presentation of mannose receptors expressed by alternatively-activated macrophages or by tumor cell apoptosis. Nevertheless, our results demonstrate for the first time that complement components in the CSF can be reproducibly quantified in the clinical setting in CNS lymphoma and other types of brain tumors and that C3a concentration in CSF may be a prognostic biomarker denoting high-risk disease. An emerging body of data implicates complement activation in the pathogenesis of multiple sclerosis and other neuro-inflammatory and/or neurodegenerative diseases.45,46, 47,48, 49 The fact that we demonstrated significantly higher levels of complement activation in CNS lymphoma compared to these and other non-malignant neurologic conditions underscores the potential physiologic significance of C3 activation in CNS lymphoma and other brain tumors.
An important question which emerges from these studies is the mechanism by which constitutive activation of complement C3 cleavage might promote CNS lymphoma progression. In addition to the generation of C3b which attenuates natural killer function,41 activation of the complement cascade at the level of C5a may potentiate intratumoral chemotaxis of myeloid suppressor cells, which may disrupt adaptive anti-lymphoma immunity.50 In addition, in experimental autoimmune encephalomyelitis, there is direct evidence that C3a itself promotes chronic inflammation in the CNS, as a chemoattractant.51 There is also evidence that complement C3 and C3a promote growth signals in cancer and potentiate CXCL12 signaling.52, 53 C3 and C3a may also regulate lymphoma-associated vascular permeability and/or angiogenesis, a possibility supported by recent observations regarding the anti-angiogenic effects of C3a and C5a on retinal vasculature.54 Notably, in support of this hypothesis, we detected a significant correlation between C3a concentration in CSF and the contrast-enhancing volume of CNS lymphomas (R2=0.65, data not shown).
The rapid decrease in rituximab concentrations in CSF indicates a rapid elimination and/or distribution in the cranial-spinal axis. At first, we modeled the CSF and brain tissue compartment as a single compartment. However, the necessity of treating the serum and brain tissue compartments separately became evident after unsuccessful attempts to fit the CSF and serum data, when V1 and V3 are combined. Such a model without a third compartment cannot explain the biphasic distribution of rituximab in CSF, as the distribution to serum was slow and serum levels were considerably lower than those measured in the CSF. The biphasic CSF rituximab indicates rapid distribution to a second biological space, consistent with penetration into brain tissue. Nevertheless, additional studies are required to directly quantify brain tissue penetration of rituximab.
The parameters which regulate distribution of rituximab into this second biological space are not defined but may relate to dynamics of vascular permeability (perhaps mediated by complement), dynamics of tumor cellularity and interstitial pressure, intraventricular CSF pressure as well as hypothetical mechanisms of facilitated uptake of therapeutic molecules such as IgG1 into the brain. Despite these variables, the utility of this three-compartment pharmacokinetic model is underscored by its accurate prediction of the pharmacokinetics of intraventricular rituximab in a second, independent multicenter phase I trial.22
The emerging body of data regarding the physiology of the complement system within the CSF in CNS lymphoma suggests several possible interventions to improve efficacy or to overcome resistance to intraventricular rituximab - for example, via the co-administration of pharmacologic inhibitors of complement antagonists such as clusterin, which is upregulated within the CSF in CNS lymphoma, as a means to potentiate rituximab-mediated cytotoxicty.55 In addition, we provide the first pharmacokinetic model of the distribution of a naked monoclonal antibody after intraventricular administration in humans. We believe that this model may provide a framework for the design of future studies of the intraventricular injection, as well as infusion, of therapeutic biological molecules within the CSF and neuroaxis.
Supplementary Material
Statement of Translational Relevance.
Among the many impediments to the development of successful strategies to treat brain tumors are problems of drug delivery and an incomplete understanding of the tumor microenvironment, in particular, the innate and adaptive immune response within the brain. Using the brain ventricle as a window to this microenvironment, we explored for the first time the pharmacodynamics of complement activation within the CSF upon intrathecal rituximab administration. In addition, we applied data generated from two recent phase I trials of intraventricular rituximab in recurrent central nervous system lymphoma to develop and validate a novel, compartmental pharmacokinetic model that describes monoclonal antibody distribution after intraventricular administration in humans. We demonstrated rapid complement activation in CSF that may contribute to the lymphocytoxic effects of intraventricular rituximab. Distribution of rituximab was measured via CSF and blood samples and supported a three-compartment pharmacokinetic model with rituximab penetration into the brain. We hypothesize that this information may provide insights into the basis for efficacy of intraventricular rituximab in the treatment of lymphoma within the neuroaxis, and be of general relevance to the development of novel therapeutic strategies for brain diseases that are based upon the intra-CSF administration of therapeutic antibodies and other macromolecules.
Acknowledgments
Grant Support. This research was supported by the Leukemia & Lymphoma Society, the UCSF Brain Tumor SPORE, Gabrielle’s Angel Foundation, and by NIH R01CA139-83-01A1 (JLR).
Literature Cited
- 1.Oldham RK, Dillman RO. Monoclonal antibodies in cancer therapy: 25 years of progress. J Clin Oncol. 2008;26(11):1774–7. doi: 10.1200/JCO.2007.15.7438. [DOI] [PubMed] [Google Scholar]
- 2.Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366(21):2008–16. doi: 10.1056/NEJMct1114348. [DOI] [PubMed] [Google Scholar]
- 3.Gerecitano J, Portlock C, Hamlin P, Moskowitz CH, Noy A, Straus D, et al. Phase I trial of weekly and twice-weekly bortezomib with rituximab, cyclophosphamide, and prednisone in relapsed or refractory non-Hodgkin lymphoma. Clin Cancer Res. 2011;17(8):2493–501. doi: 10.1158/1078-0432.CCR-10-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller C, Murawski N, Wiesen MH, Held G, Poeschel V, Zeynalova S, et al. The role of sex and weight on rituximab clearance and serum elimination half-life in elderly patients with DLBCL. Blood. 2012;119(14):3276–84. doi: 10.1182/blood-2011-09-380949. [DOI] [PubMed] [Google Scholar]
- 5.Duchnowska R, Dziadziuszko R, Czartoryska-Arlukowicz B, Radecka B, Szostakiewicz B, Sosinska-Mielcarek K, et al. Risk factors for brain relapse in HER2-positive metastatic breast cancer patients. Breast Cancer Res Treat. 2009;117(2):297–303. doi: 10.1007/s10549-008-0275-z. [DOI] [PubMed] [Google Scholar]
- 6.Musolino A, Ciccolallo L, Panebianco M, Fontana E, Zanoni D, Bozzetti C, et al. Multifactorial central nervous system recurrence susceptibility in patients with HER2-positive breast cancer: epidemiological and clinical data from a population-based cancer registry study. Cancer. 2011;117(9):1837–46. doi: 10.1002/cncr.25771. [DOI] [PubMed] [Google Scholar]
- 7.Feugier P, Virion JM, Tilly H, Haioun C, Marit G, Macro M, et al. Incidence and risk factors for central nervous system occurrence in elderly patients with diffuse large-B-cell lymphoma: influence of rituximab. Ann Oncol. 2004;15(1):129–33. doi: 10.1093/annonc/mdh013. [DOI] [PubMed] [Google Scholar]
- 8.Tai WM, Chung J, Tang PL, Koo YX, Hou X, Tay KW, et al. Central nervous system (CNS) relapse in diffuse large B cell lymphoma (DLBCL): pre- and post-rituximab. Ann Hematol. 2011;90(7):809–18. doi: 10.1007/s00277-010-1150-7. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto W, Tomita N, Watanabe R, Hattori Y, Nakajima Y, Hyo R, et al. Central nervous system involvement in diffuse large B-cell lymphoma. Eur J Haematol. 2010;85(1):6–10. doi: 10.1111/j.1600-0609.2010.01438.x. [DOI] [PubMed] [Google Scholar]
- 10.Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood. 2003;101(2):466–8. doi: 10.1182/blood-2002-06-1636. [DOI] [PubMed] [Google Scholar]
- 11.Schulz H, Pels H, Schmidt-Wolf I, Zeelen U, Germing U, Engert A. Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematologica. 2004;89(6):753–4. [PubMed] [Google Scholar]
- 12.Antonini G, Cox MC, Montefusco E, Ferrari A, Conte E, Morino S, et al. Intrathecal anti-CD20 antibody: an effective and safe treatment for leptomeningeal lymphoma. J Neurooncol. 2007;81(2):197–9. doi: 10.1007/s11060-006-9217-y. [DOI] [PubMed] [Google Scholar]
- 13.Rubenstein JL, Fridlyand J, Abrey L, Shen A, Karch J, Wang E, et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol. 2007;25(11):1350–6. doi: 10.1200/JCO.2006.09.7311. [DOI] [PubMed] [Google Scholar]
- 14.van de Glind G, de Graaf S, Klein C, Cornelissen M, Maecker B, Loeffen J. Intrathecal rituximab treatment for pediatric post-transplant lymphoproliferative disorder of the central nervous system. Pediatr Blood Cancer. 2008;50(4):886–8. doi: 10.1002/pbc.21297. [DOI] [PubMed] [Google Scholar]
- 15.Liu CY, Teng HW, Lirng JF, Chiou TJ, Chen PM, Hsiao LT. Sustained remission and long-term survival of secondary central nervous system involvement by aggressive B-cell lymphoma after combination treatment of systemic high-dose chemotherapy and intrathecal rituximab. Leuk Lymphoma. 2008;49(10):2018–21. doi: 10.1080/10428190802311375. [DOI] [PubMed] [Google Scholar]
- 16.Villela L, Garcia M, Caballero R, Borbolla-Escoboza JR, Bolanos-Meade J. Rapid complete response using intrathecal rituximab in a patient with leptomeningeal lymphomatosis due to mantle cell lymphoma. Anticancer Drugs. 2008;19(9):917–20. doi: 10.1097/CAD.0b013e32830cebc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaime-Perez JC, Rodriguez-Romo LN, Gonzalez-Llano O, Chapa-Rodriguez A, Gomez-Almaguer D. Effectiveness of intrathecal rituximab in patients with acute lymphoblastic leukaemia relapsed to the CNS and resistant to conventional therapy. Br J Haematol. 2009;144(5):794–5. doi: 10.1111/j.1365-2141.2008.07497.x. [DOI] [PubMed] [Google Scholar]
- 18.Perissinotti AJ, Reeves DJ. Role of intrathecal rituximab and trastuzumab in the management of leptomeningeal carcinomatosis. Ann Pharmacother. 2010;44 (10):1633–40. doi: 10.1345/aph.1P197. [DOI] [PubMed] [Google Scholar]
- 19.Kim MG, Park SY, Kim EJ, Kim YM, Kim HY, Lee YK, et al. Hepatitis B virus reactivation in a primary central nervous system lymphoma patient following intrathecal rituximab treatment. Acta Haematol. 2011;125(3):121–4. doi: 10.1159/000321792. [DOI] [PubMed] [Google Scholar]
- 20.Bonney DK, Htwe EE, Turner A, Kelsey A, Shabani A, Hughes S, et al. Sustained response to intrathecal rituximab in EBV associated Post-transplant lymphoproliferative disease confined to the central nervous system following haematopoietic stem cell transplant. Pediatr Blood Cancer. 2012;58(3):459–61. doi: 10.1002/pbc.23134. [DOI] [PubMed] [Google Scholar]
- 21.Czyzewski K, Styczynski J, Krenska A, Debski R, Zajac-Spychala O, Wachowiak J, et al. Intrathecal therapy with rituximab in central nervous system involvement of post-transplant lymphoproliferative disorder. Leuk Lymphoma. 2012 doi: 10.3109/10428194.2012.718342. [DOI] [PubMed] [Google Scholar]
- 22.Rubenstein JL, Li J, Chen L, Advani R, Drappatz J, Gerstner E, et al. Multicenter phase I trial of intraventricular immuno-chemotherapy in recurrent CNS lymphoma. Blood. 2012 doi: 10.1182/blood-2012-07-440974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laufman LR, Forsthoefel KF. Use of intrathecal trastuzumab in a patient with carcinomatous meningitis. Clin Breast Cancer. 2001;2(3):235. doi: 10.1016/S1526-8209(11)70419-0. [DOI] [PubMed] [Google Scholar]
- 24.Stemmler HJ, Mengele K, Schmitt M, Harbeck N, Laessig D, Herrmann KA, et al. Intrathecal trastuzumab (Herceptin) and methotrexate for meningeal carcinomatosis in HER2-overexpressing metastatic breast cancer: a case report. Anticancer Drugs. 2008;19(8):832–6. doi: 10.1097/CAD.0b013e32830b58b0. [DOI] [PubMed] [Google Scholar]
- 25.Mir O, Ropert S, Alexandre J, Lemare F, Goldwasser F. High-dose intrathecal trastuzumab for leptomeningeal metastases secondary to HER-2 overexpressing breast cancer. Ann Oncol. 2008;19(11):1978–80. doi: 10.1093/annonc/mdn654. [DOI] [PubMed] [Google Scholar]
- 26.Colozza M, Minenza E, Gori S, Fenocchio D, Paolucci C, Aristei C, et al. Extended survival of a HER-2-positive metastatic breast cancer patient with brain metastases also treated with intrathecal trastuzumab. Cancer Chemother Pharmacol. 2009;63(6):1157–9. doi: 10.1007/s00280-008-0859-7. [DOI] [PubMed] [Google Scholar]
- 27.Ferrario C, Davidson A, Bouganim N, Aloyz R, Panasci LC. Intrathecal trastuzumab and thiotepa for leptomeningeal spread of breast cancer. Ann Oncol. 2009;20(4):792–5. doi: 10.1093/annonc/mdp019. [DOI] [PubMed] [Google Scholar]
- 28.Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47(2):115–23. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy S, Josephson SA, Fridlyand J, Karch J, Kadoch C, Karrim J, et al. Protein biomarker identification in the CSF of patients with CNS lymphoma. J Clin Oncol. 2008;26(1):96–105. doi: 10.1200/JCO.2007.12.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulido JS, Bakri SJ, Valyi-Nagy T, Shukla D. Rituximab penetrates full-thickness retina in contrast to tissue plasminogen activator control. Retina. 2007;27(8):1071–3. doi: 10.1097/IAE.0b013e31804ac032. [DOI] [PubMed] [Google Scholar]
- 31.Shahar J, Avery RL, Heilweil G, Barak A, Zemel E, Lewis GP, et al. Electrophysiologic and retinal penetration studies following intravitreal injection of bevacizumab (Avastin) Retina. 2006;26(3):262–9. doi: 10.1097/00006982-200603000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Zigova T, Pencea V, Wiegand SJ, Luskin MB. Intraventricular administration of BDNF increases the number of newly generated neurons in the adult olfactory bulb. Mol Cell Neurosci. 1998;11(4):234–45. doi: 10.1006/mcne.1998.0684. [DOI] [PubMed] [Google Scholar]
- 33.Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci. 2001;21(17):6706–17. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15(3):331–7. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubenstein JL, Fridlyand J, Shen A, Aldape K, Ginzinger D, Batchelor T, et al. Gene expression and angiotropism in primary CNS lymphoma. Blood. 2006;107(9):3716–23. doi: 10.1182/blood-2005-03-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubenstein JL, Li J, Chen L, Advani R, Drappatz J, Gerstner E, et al. Multicenter phase 1 trial of intraventricular immunochemotherapy in recurrent CNS lymphoma. Blood. 2013;121(5):745–51. doi: 10.1182/blood-2012-07-440974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsukamoto H, Ueda A, Nagasawa K, Tada Y, Niho Y. Increased production of the third component of complement (C3) by monocytes from patients with systemic lupus erythematosus. Clin Exp Immunol. 1990;82(2):257–61. doi: 10.1111/j.1365-2249.1990.tb05436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadoch C, Dinca EB, Voicu R, Chen L, Nguyen D, Parikh S, et al. Pathologic correlates of primary central nervous system lymphoma defined in an orthotopic xenograft model. Clin Cancer Res. 2009;15(6):1989–97. doi: 10.1158/1078-0432.CCR-08-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57(1):1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 40.Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem. 2004;88(4):844–56. doi: 10.1046/j.1471-4159.2003.02213.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111(3):1456–63. doi: 10.1182/blood-2007-02-074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petereit HF, Rubbert-Roth A. Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult Scler. 2009;15(2):189–92. doi: 10.1177/1352458508098268. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Zhi J, Wenger M, Valente N, Dmoszynska A, Robak T, et al. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52(12):1918–26. doi: 10.1177/0091270011430506. [DOI] [PubMed] [Google Scholar]
- 44.Harjunpaa A, Wiklund T, Collan J, Janes R, Rosenberg J, Lee D, et al. Complement activation in circulation and central nervous system after rituximab (anti-CD20) treatment of B-cell lymphoma. Leuk Lymphoma. 2001;42(4):731–8. doi: 10.3109/10428190109099335. [DOI] [PubMed] [Google Scholar]
- 45.Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173(7):4708–14. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- 46.van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- 47.Kulkarni AP, Kellaway LA, Lahiri DK, Kotwal GJ. Neuroprotection from complement-mediated inflammatory damage. Ann N Y Acad Sci. 2004;1035:147–64. doi: 10.1196/annals.1332.010. [DOI] [PubMed] [Google Scholar]
- 48.Aiyaz M, Lupton MK, Proitsi P, Powell JF, Lovestone S. Complement activation as a biomarker for Alzheimer’s disease. Immunobiology. 2012;217(2):204–15. doi: 10.1016/j.imbio.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 50.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–35. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173(7):4708–14. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- 52.Honczarenko M, Ratajczak MZ, Nicholson-Weller A, Silberstein LE. Complement C3a enhances CXCL12 (SDF-1)-mediated chemotaxis of bone marrow hematopoietic cells independently of C3a receptor. J Immunol. 2005;175(6):3698–706. doi: 10.4049/jimmunol.175.6.3698. [DOI] [PubMed] [Google Scholar]
- 53.Schraufstatter IU, Discipio RG, Zhao M, Khaldoyanidi SK. C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. J Immunol. 2009;182(6):3827–36. doi: 10.4049/jimmunol.0803055. [DOI] [PubMed] [Google Scholar]
- 54.Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116(22):4395–403. doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zoubeidi A, Chi K, Gleave M. Targeting the cytoprotective chaperone, clusterin, for treatment of advanced cancer. Clin Cancer Res. 2010;16(4):1088–93. doi: 10.1158/1078-0432.CCR-09-2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.