

Abstract
Amoebiasis, a disease caused by Entamoeba histolytica, is usually diagnosed by microscopic examination, which does not differentiate the morphologically identical species of the E. histolytica/E. dispar complex. Furthermore, morphologically similar species such as Entamoeba hartmanni contribute to misidentification. Therefore, there is a need for more sensitive and specific methods. This study standardized a multiplex real-time PCR system for E. histolytica and E. dispar and a single real-time PCR for E. hartmanni. The multiplex protocol detected up to 0.0143 pg of E. histolytica DNA and 0.5156 pg of E. dispar DNA, and the average melting temperature (T m) was 73°C and 70°C, respectively. For E. hartmanni, the T m was 73°C and the amplification was successful down to 0.03 fg of plasmid DNA. Negative controls and other intestinal parasites presented no amplification. Among the 48 samples tested, E. dispar DNA was detected in 37; none exhibited E. histolytica DNA and 11 were negative in the multiplex protocol. In 4 of these 11 samples, however, E. hartmanni DNA was amplified. SYBR Green is demonstrated to be an interesting option and these combined PCR reactions can improve laboratory diagnosis of amoebiasis in developing countries.
1. Introduction
The genus Entamoeba contains many species, six of which are found in the human intestinal tract: Entamoeba histolytica, Entamoeba dispar, Entamoeba moshkovskii, Entamoeba coli, Entamoeba hartmanni, and Entamoeba polecki. Of these species, only E. histolytica is associated with pathological injuries; the others are considered to be nonpathogenic species [1]. E. histolytica is responsible for approximately 50 million cases of amoebiasis worldwide each year, resulting in up to 100,000 deaths [2, 3]. Because of these characteristics, E. histolytica is globally considered to be a leading parasitic cause of human mortality [1]. However, historical epidemiological data concerning E. histolytica infections can lead to overestimates because they were obtained before the formal separation of this parasite into two morphologically identical species: the potentially pathogenic E. histolytica and the nonpathogenic E. dispar [4]. More importantly, it is also estimated that 90% of diagnosed infections are caused by E. dispar [5].
Diagnosis of E. histolytica has historically relied on microscopic examination of protozoan morphology. Optical microscopy is a very useful tool for the diagnosis of amoebiasis because it is simple and cheap to execute. However, concentration techniques and stained smears are required to increase its low sensitivity [1, 6]. It is also influenced by the intermittent release of cysts, requiring the examination of multiple samples from different days [7]. Moreover, this method cannot differentiate between morphologically identical species such as E. dispar and E. moshkovskii, and the proficiency of the examiner can also be crucial for precise identification, as other Entamoeba species can present structures very similar to the features of E. histolytica [2, 8, 9].
Entamoeba hartmanni is one of these species with similar morphological forms because its cysts share characteristics with those of the E. histolytica/E. dispar/E. moshkovskii complex. The major difference between these species is the size of the cysts, which in E. hartmanni are generally smaller than in those from the complex [4, 10]. For this differentiation to be conducted by microscopy, an appropriate microscope capable of performing morphometric analysis is also required. Given that, this organism can still be difficult to identify depending on the circumstances and the condition of the sample. Contractile trophozoites or cysts of E. histolytica can be erroneously identified as E. hartmanni, and larger structures of E. hartmanni can also be misdiagnosed as the pathogenic species. Alternative molecular methods could be helpful to provide accurate and reliable identification of Entamoeba species. Polymerase Chain Reaction (PCR), including real-time PCR, has provided the means to identify E. histolytica, E. dispar, and E. moshkovskii in a variety of samples [11–17]. However, little attention has been devoted to the possibility of identifying E. hartmanni, whose structures confuse examiners at different proficiency levels [8]. Previously, data determined by microscopic examination have demonstrated a prevalence of E. hartmanni of 13.3% in HIV patients and 2.5% in children in Brazilian groups [18, 19]. Identification of Entamoeba species other than the complex using molecular techniques has been conducted in only a single study in Brazil, in which the evaluation of Brazilian clinical samples previously identified as positive for the complex by microscopic examination demonstrated the presence of E. hartmanni DNA in more than one sample, while E. moshkovskii DNA was not detected among the samples analyzed [20]. These results showed the presence of E. hartmanni in Brazil but yielded no data about E. moshkovskii.
Real-time PCR is a very sensitive methodology that could become an important tool to achieve these goals, and SYBR Green (N′,N′-dimethyl-N-[4-[(E)-(3-methyl-1,3-benzothiazol-2-ylidene)methyl]-1-phenylquinolin-1-ium-2-yl]-N-propylpropane-1,3-diamine) technology with its probe-free assays is a less expensive choice for establishing more efficient protocols in developing countries [12]. Because of these possibilities, the objectives of this study were to standardize SYBR Green real-time PCR protocols for the identification of E. histolytica, E. dispar, and E. hartmanni and to evaluate their efficiency on clinical stool samples.
2. Material and Methods
2.1. DNA Samples
The positive controls used in the SYBR Green multiplex real-time PCR were genomic DNA from the standard strain of E. histolytica HM1-IMSS and E. dispar P2 (a strain isolated from patient samples in Piauí, Brazil). The DNA was measured in a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA), and 2-fold dilutions in water were prepared for testing using the SYBR Green real-time PCR protocol. For E. hartmanni, however, a cloning procedure was necessary to obtain a positive control because a protozoan culture of E. hartmanni was not available. For this process, positive DNA samples characterized in a previous study were used [20]. The specific E. hartmanni sequence was amplified with primers EhartF and EhartR2; this fragment was then cloned using the pCR 2.1-TOPO vector as described in the protocol with the pCR 2.1-TOPO TA Cloning Kit (Invitrogen). The extraction and purification of plasmid DNA containing the cloned fragment were performed using the Illustra PlasmidPrep Mini Spin Kit (GE Healthcare UK Limited, Buckinghamshire, UK), according to the manufacturer's instructions. This cloned plasmid DNA was measured in a Qubit fluorometer (Invitrogen) and 10-fold dilutions in water were prepared for testing with the SYBR Green real-time PCR protocol. In both protocols, DNA solutions from other intestinal parasites (two of each) were tested for a specificity assessment: E. moshkovskii, Cryptosporidium hominis, Giardia lamblia, and Schistosoma mansoni.
2.2. Clinical Samples
Stool samples from 2263 patients who were treated at Hospital Universitário Antônio Pedro (HUAP), Niterói, Rio de Janeiro, Brazil, between August 2008 and June 2009 were submitted to a microscopic examination in order to detect the presence of parasites. The procedure was conducted at the HUAP's parasitology laboratory. Moreover, a group of 292 stool samples from individual residents of Sumidouro, Rio de Janeiro, Brazil, a rural area, was also subjected to the same examination. Those samples identified as positive for the E. histolytica/E. dispar complex were selected to evaluate this SYBR Green real-time PCR system. Additional stool samples from 50 individuals with negative direct parasitological examinations were included as negative controls. This study was reviewed and approved by the Human Investigation Committee of the Universidade Federal Fluminense with protocol number 020/07.
2.3. Microscopic Examination
The microscopic examination was conducted by HUAP's parasitology laboratory staff. Prior to morphologic analysis, this service concentrated the stools using a spontaneous sedimentation method [21]. Then the sediments were analyzed in wet mounts stained with Lugol's iodine solution. For the microscopic examination, the laboratory staff used a Nikon Eclipse E20 optical microscope (Nikon). After the analysis, the sediments were stored at −20°C for DNA extraction.
2.4. DNA Extraction
DNA templates were extracted using the Fast Prep DNA Kit and the FastPrep FP120 Disrupter (MP Biomedicals, Solon, OH, USA). Further Purification was performed using the QIAquick PCR purification Kit (QIAGEN Inc., Valencia, CA, USA), and the purified DNA was stored at −20°C. Both methods were performed according to the manufacturer's instructions.
2.5. Conventional PCR Amplification
A multiplex PCR reaction was performed according to a protocol previously described by Santos et al. [22], with some modifications. The specific primers for E. histolytica and E. dispar were previously defined by Núñez et al. [23], E. histolytica (EHP1—5′CGATTTTCCCAGTAGAAATTA3′ and EHP2—5′CAAAATGGTCGTCTAGGC3′) and E. dispar (EDP1—5′ATGGTGAGGTTGTAGCAGAGA3′ and EDP2—5′CGATATTGACCTAGTACT3′). These primer sets were used to amplify specific sequences of 132 bp for E. histolytica and 96 bp for E. dispar. Each reaction was performed in a volume of 50 μL using PCR Supermix (Invitrogen), 20 pmol of each primer, 0.1% bovine serum albumin (BSA Sigma Chem. Co., USA), and 2 μL of the DNA sample. The PCR reaction was carried out using a Veriti 96 Well Thermal Cycler (Applied Biosystems, CA, USA), and the amplification parameters were as follows: 3 minutes at 94°C; 30 cycles of 30 seconds at 94°C, 30 seconds at 55°C, and 30 seconds at 72°C; and finally 7 minutes at 72°C. The amplified product was visualized with ethidium bromide staining after electrophoresis on a 2.0% agarose gel.
2.6. Multiplex SYBR Green Real-Time PCR Standardization
This PCR reaction was standardized for the simultaneous detection and identification of E. histolytica and E. dispar. The specific primers used for the detection of E. histolytica and E. dispar were the same oligonucleotide sequences employed for conventional multiplex PCR. Initially, a single format was designed in order to amplify E. histolytica and E. dispar separately. This format allowed us to know the specific melting temperature (T m) of the sequence amplified from each species and the feasibility of standardizing a multiplex reaction with these primers. The simplex PCR cycling parameters consisted of 2 minutes at 50°C, 2 minutes at 95°C and 35 cycles of 15 seconds at 95°C and 33 seconds at 55°C, using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) and the same PCR reagents and conditions. A dissociation stage was then performed; the parameters consisted of 15 seconds at 95°C, 1 minute at 60°C, 15 seconds at 95°C, and 15 seconds at 60°C. Finally, the multiplex format was standardized in a final volume of 25 μL. Four different amounts of primers were tested: 1.25, 2.5, 3.75, and 5 pmol. Two amounts of ROX Reference Dye (Invitrogen) were also tested in the standardization process: 0.25 pmol and 1.25 pmol. All reactions were carried out in an ABI 7500 System thermocycler (Applied Biosystems) and the PCR cycling parameters were the same as described for the single format, with an additional comparison between two annealing temperatures: 55°C and 60°C. The amplified products were visualized with ethidium bromide staining after electrophoresis on a 2.0% agarose gel for confirmation.
2.7. Standardization of SYBR Green Real-Time PCR for Identification of E. hartmanni
DNA sequence alignment was performed using CLC Sequence Viewer software (CLC bio). The sequences aligned were rRNA genes of the highly homologous species E. histolytica, E. dispar, E. moshkovskii, and E. hartmanni. Specific regions in the E. hartmanni sequence (GenBank access: AF149907) were identified. A forward primer EhartF (5′-CGTTCAAGACATGAGTGTGA-3′) and two reverse primers (EhartR1—5′-CCGTAGATCTCCTATTCACTTT-3′); EhartR2 (5′-ACAACACATTCATGTCGCA-3′) were designed.
To verify the identity of the target sequence of the primers, a conventional PCR was performed in a volume of 50 μL, using PCR Supermix (Invitrogen), 20 pmol of each primer, 0.1% bovine serum albumin (BSA Sigma Chem. Co), and 2 μL of DNA sample. The PCR reaction was carried out using a Veriti 96-Well Thermal Cycler (Applied Biosystems), and the amplification parameters were as follows: 3 minutes at 94°C; 40 cycles of 30 seconds at 94°C, 30 seconds at 55°C, and 30 seconds at 72°C; and finally 7 minutes at 72°C. The amplified product was visualized by ethidium bromide staining after electrophoresis on a 2.0% agarose gel and sent to the sequencing platform of the Genomic Unit at the Laboratory of Cell Physiology Darcy Fontoura (Federal University of Rio de Janeiro, Brazil). After confirming the identity of the amplified product on the GenBank database, the standardization process of the real-time PCR began. The PCR reaction using primers EhartF and EhartR2 was performed in a reaction volume of 25 μL, using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). Analyses concerning the amounts of primers and ROX Reference Dye were conducted just as described in the previous section for the multiplex protocol. Also, the reactions were carried out in the same thermocycler. The PCR cycling parameters consisted of 2 minutes at 50°C, 2 minutes at 95°C and 35 cycles of 15 seconds at 95°C and 33 seconds at 60°C. A dissociation stage was then performed; the parameters consisted of 15 seconds at 95°C, 1 minute at 60°C, 15 seconds at 95°C, and 15 seconds at 60°C. The amplified products were visualized with ethidium bromide staining after electrophoresis on a 2.0% agarose gel for confirmation.
2.8. Stool Sample Evaluation
For a preliminary evaluation of the real-time PCR protocols during the standardization process, a selection of Entamoeba positive samples was run. Thus, only samples exhibiting structures of the E. histolytica/E. dispar complex under microscopic examination were analyzed by multiplex conventional PCR and multiplex real-time PCR for the accurate identification of E. histolytica and E. dispar. Those samples with negative results obtained by both methods were submitted for further investigation with real-time PCR for the identification of E. hartmanni.
3. Results
3.1. Multiplex SYBR Green Real-Time PCR
The preliminary step represented by standardizing a single format of this reaction was essential to achieve the final protocol for the multiplex SYBR Green real-time PCR. This reaction determined that the mean melting temperatures (T m) of the amplified sequences of E. histolytica and E. dispar were 73.2°C (72.8 to 73.5°C) and 70.2°C (69.1°C to 70.9°C), respectively. These data demonstrated the possibility of standardizing a multiplex protocol to detect and identify these species using SYBR Green technology. Thus, multiplex real-time PCR using those primers was performed, and DNA amplification was exhibited as expected. The melting curve analysis demonstrated similar T m values for each species, and the concomitant presence of genetic material from both species did not alter these conditions (Figure 1(a)).
Figure 1.
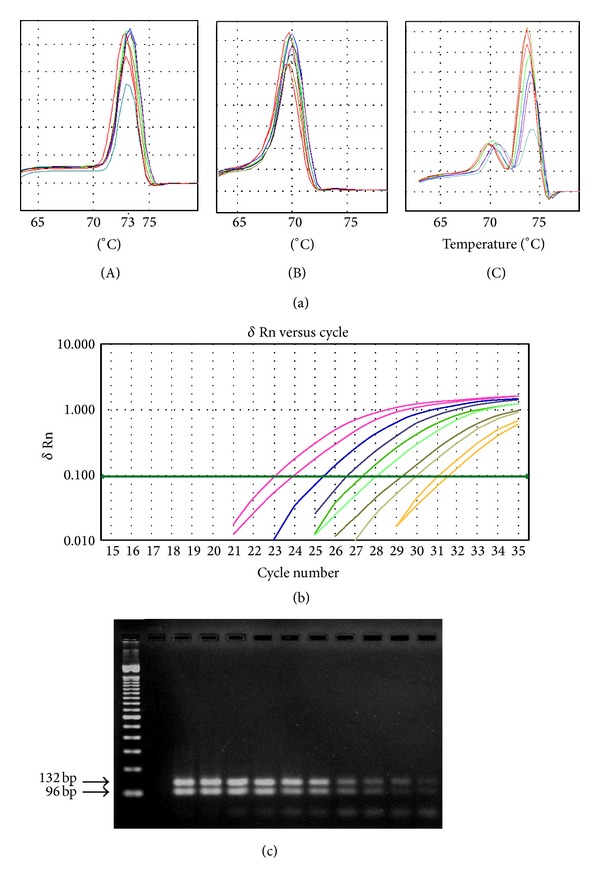
(a) Melting curve graph obtained through the SYBR Green real-time PCR for E. histolytica (A) and E. dispar (B) and the multiplex SYBR Green real-time PCR (C). (b) The software's graph demonstrating the DNA amplification of E. histolytica and E. dispar mixed DNAs in different amounts at the multiplex SYBR Green real-time PCR in descending order: 183.5 and 198.0 pg (E. histolytica and E. dispar, resp.); 91.5 and 99.0 pg; 45.9 and 49.5 pg; 22.9 and 24.7 pg; 11.5 and 12.4 pg; 8, 5.7 and 6.2 pg; 2.9 and 3.1 pg; 1.4 and 1.6 pg; 0.7 and 0.8 pg; 0.35 and 0.39 pg. (c) Agarose gel from multiplex real-time PCR products. Lane 1, 100 bp ladder standard; lane 2, no sample; lanes 3 to 12, PCR products in descending order of concentration.
Under the conditions for real-time PCR, the optimal concentration of primers was found to be 2.5 pmol. Other concentrations decreased the sensitivity of the assay or increased primer dimer formation. A concentration of ROX Reference Dye equivalent to 1.25 pmol gave the most reproducible and stable results. The best annealing temperature for amplification of both fragments was 55°C.
In the specificity assessment, no amplification was detected either in the presence of other intestinal parasite DNAs or in the negative control group. Specific DNA amplification was observed at all concentrations tested for both species, separately. For E. histolytica, amplification was tested successfully from 7.34 pg (C t: 21.2) to 0.0143 pg (C t: 30.2). For E. dispar, amplification was tested successfully from 264 pg (C t: 23.4) to 0.5156 pg (C t: 33.0). In order to model a mixed infection, genetic material from both species was tested together. A concomitant specific DNA amplification was observed at all concentrations tested, and the DNA of both species was amplified at levels as low as 0.3584 pg for E. histolytica and 0.3867 pg for E. dispar (Figures 1(b) and 1(c), Table 1). As expected, decreasing the DNA concentration also increased the C t value, revealing an inverse relationship between these values. In the latter situation, the specific T m value must be checked in the melting temperature graph to evaluate the presence of two peaks (Figure 1(a)).
Table 1.
Results of multiplex PCR obtained with different DNA concentration.
| DNA (pg) | C t value | |
|---|---|---|
| E. histolytica | E. dispar | |
| 183.5 | 198.0 | 23.1 |
| 91.5 | 99.0 | 23.9 |
| 45.9 | 49.5 | 25.4 |
| 22.9 | 24.7 | 26.5 |
| 11.5 | 12.4 | 27.3 |
| 5.7 | 6.2 | 28.1 |
| 2.9 | 3.1 | 29.3 |
| 1.4 | 1.6 | 30.0 |
| 0.7 | 0.8 | 31.1 |
| 0.36 | 0.39 | 31.5 |
3.2. SYBR Green Real-Time PCR for Identification of E. hartmanni
The alignment revealed highly specific regions in the E. hartmanni sequence that allowed the design of a forward primer (EhartF1) and a reverse primer (EhartR1). The fragment defined by these primers was 184 bp. However, GenBank database analysis showed a small region of homology with Cryptosporidium hominis. In an attempt to avoid homology with other intestinal parasites, a second reverse primer, EhartR2, was designed to exclude this homologous region. The new set of primers amplifies a sequence of 90 bp. In this real-time PCR protocol, the melting curve analysis revealed a mean melting temperature (T m) of 73°C for the amplified product (Figure 2). Agarose gel electrophoresis analysis confirmed the amplification of the specific 90 bp fragment. In the conditions for real-time PCR, the optimal concentration of primers was found to be 3.75 pmol. Other concentrations either decreased the sensitivity of the assay or increased primer dimer formation. For the ROX Reference Dye, 0.25 pmol was determined to be the concentration that gave the most reproducible and stable results.
Figure 2.
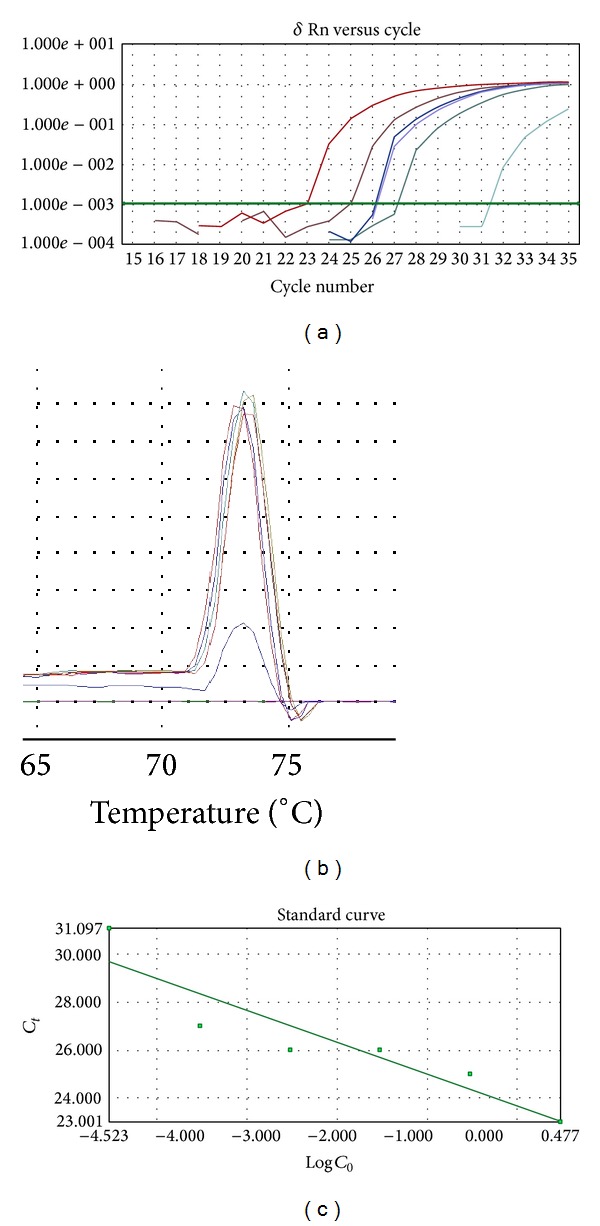
DNA amplification and the melting curve analysis using different DNA concentrations on SYBR Green real-time PCR for E. hartmanni. DNA concentrations: 3.0 pg, 0.3 pg, 0.03 pg, 0.003 pg, 0.0003 pg, and 0.00003 pg.
In the specificity assessment, no amplification was detected either in the presence of other intestinal parasite DNAs or in the negative control group. Specific DNA amplification was observed down to a concentration of 0.03 fg. As noted above, the decrease in DNA concentration corresponded to an increase in the C t value. The T m value was between 72.8 and 73.6°C (Figure 2, Table 2).
Table 2.
C t and T m values obtained with different DNA concentration from E. hartmanni.
| DNA (pg) | C t value | T m (°C) |
|---|---|---|
| 3.0 | 23.0 | 72.8 |
| 0.3 | 25.0 | 73.2 |
| 0.03 | 26.0 | 73.2 |
| 0.003 | 26.0 | 73.6 |
| 0.0003 | 27.0 | 73.2 |
| 0.00003 | 31.1 | 73.2 |
3.3. Stool Sample Evaluation
For this study, all samples were analyzed through microscopic examination. Of the 292 samples collected at Sumidouro, 24 specimens exhibited morphological structures characteristic of the E. histolytica/E. dispar complex, for a prevalence of 8.2%. In the HUAP's laboratory, these structures were detected in 24 specimens from the 2263 samples collected with a prevalence of 1.1%. These 48 samples were then selected and submitted to real-time multiplex PCR. The samples were also submitted to the corresponding multiplex conventional PCR format for comparison. Of these samples, 88% had polyparasitism with one or more parasites, mainly with Blastocystis hominis and Entamoeba coli (Figure 3(a)). The conventional multiplex PCR revealed amplification of the specific sequence of E. dispar in 34 samples, while real-time multiplex PCR was able to detect E. dispar in these 34 samples and in three other samples (77%). Amplification of the specific sequence of E. histolytica, however, was not observed in the samples tested by either methods (Figure 3(b)). The 11 samples negative for E. histolytica and E. dispar by both methods were then submitted to the SYBR Green real-time PCR protocol for E. hartmanni identification to investigate whether the morphological structure observed was misidentified. In four samples, the specific sequence of E. hartmanni was amplified while the remaining 7 samples remained unidentified (Figure 3(b)). To investigate the presence of inhibitory substances, these samples were contaminated with 11.7 fg of E. hartmanni DNA and subsequently submitted to E. hartmanni real-time PCR. DNA amplification was successful after this step, indicating that inhibitory substances did not influence the negative results.
Figure 3.
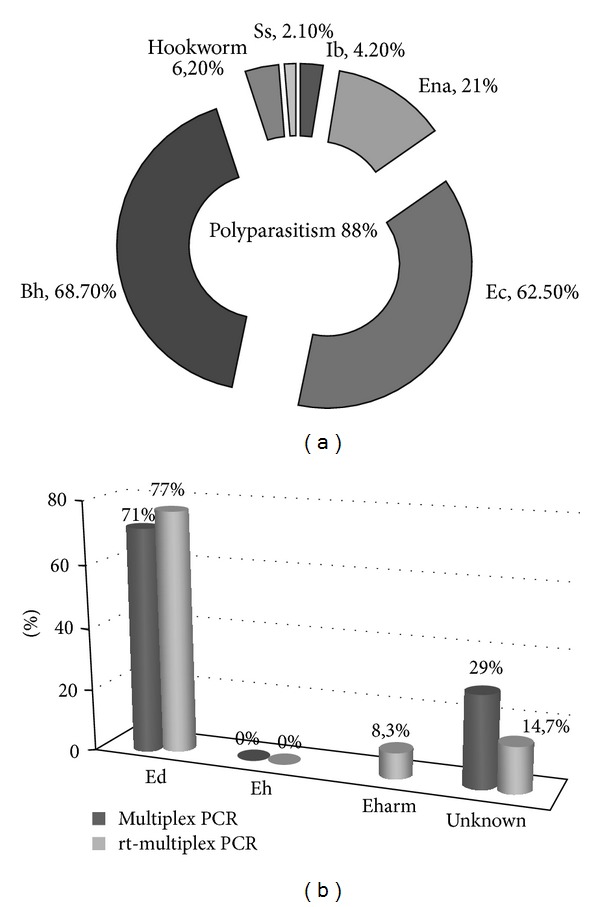
Results of the 48 samples with Entamoeba histolytica/Entamoeba dispar complex. (a) Frequency of polyparasitism and each detected parasite on microscopic examination. (b) Frequency of E. histolytica and E. dispar in conventional and multiplex real-time PCR and frequency of E. hartmanni by real-time PCR in negative samples from multiplex real-time PCR.
4. Discussion
Several authors have described the crucial importance of accurate differentiation of E. histolytica from E. dispar, E. moshkovskii, and other morphologically similar species of Entamoeba for proper clinical management of patients and for assessment of their actual prevalence in different geographical regions [13]. To date, several microscopy-based epidemiological surveys to study the prevalence of E. histolytica and E. dispar have been performed in different parts of Brazil, but these studies were carried out without using molecular methods to accurately identify the species. In Brazil, the standard clinical approach is to treat all asymptomatic individuals who present with cysts in their feces with an antiprotozoal agent. This approach to treatment results in the indiscriminate use of antiamoebic drugs and has led to increased minimum inhibitory concentrations of these therapeutic agents against E. histolytica, with the potential for resistant strains to appear [24, 25].
In this study, we described the development of a real-time PCR system for the differential diagnosis of three species of Entamoeba that share identical or similar morphology as both cysts and trophozoites. The standardization of these PCR reactions is an important step in developing an efficient system for the identification of Entamoeba species. Most molecular diagnostic tools detect only E. histolytica and do not detect other morphologically identical or similar species, giving rise to several questions. A previous study has already demonstrated a situation in which samples that were positive by microscopic examination for the E. histolytica/E. dispar complex were identified as E. hartmanni after analysis by two different molecular methods [20]. To avoid this potential confusion and provide complete epidemiologic data on parasitic infections, it is important to develop a reliable system for the identification and differentiation of pathogenic and nonpathogenic parasites. For this approach, real-time PCR methodology is an interesting possibility because of its high sensitivity and specificity as well as its speed in processing samples, while SYBR Green technology represents a less expensive option for developing real-time PCR protocols in developing countries. Additionally, the SYBR Green approach is especially attractive for those who already have the conventional PCR assay running and want to convert to the real-time format [12].
The presented assays successfully amplified positive controls from E. histolytica, E. dispar, and E. hartmanni, demonstrating no amplification of DNA from other intestinal parasites. Furthermore, the amplification was successfully tested on amounts of DNA ranging between 7.34 pg and 0.0143 pg for E. histolytica and between 264 pg and 0.5156 pg for E. dispar. These results demonstrated a high sensitivity. Other studies have presented detection limits at higher DNA concentrations, such as 0.2 pg for E. histolytica and 2.0 pg for E. dispar [16, 26]. For E. hartmanni, amplification was observed even with a lower DNA concentration. However, the lack of E. hartmanni parasites must be considered in analyzing the E. hartmanni detection protocol, a limitation that was circumvented by subjecting the specific fragment amplified to a cloning procedure. Certainly, the high number of copies of the target sequence in the cloned plasmids allowed the amplification of specific E. hartmanni DNA with lower quantities of DNA.
Among the 48 samples that were diagnosed as containing the complex by microscopic examination, 37 samples were identified as E. dispar. However, there was no DNA amplification of E. histolytica among these samples, neither with conventional PCR nor with real-time PCR. These results demonstrate the importance of accurate identification of Entamoeba species by alternative and sensitive molecular methods, considering the relevance of these results to ensuring adequate treatment. Two samples with E. dispar also presented Strongyloides stercoralis and hookworms, parasites that need specific treatment. Because E. histolytica was not detected by 2 different and sensitive protocols, there is a high chance no antiamoebic treatment was actually necessary. These sample results can be compared to the natural distribution of the parasite in Brazil, which has remained unknown since E. histolytica was separated into two species. It is possible that the pathogenic species has a low prevalence in Rio de Janeiro, in southeast Brazil, while E. dispar exists at a higher frequency in this area. This hypothesis corroborates other studies conducted over the past few years in different parts of Brazil, which have suggested that E. histolytica is more common in the north and extreme northeast of Brazil and is rare in other regions [7, 27]. If this is true, accurate diagnosis becomes even more important to clinical conduct because other Entamoeba species can lead to misidentification.
The presence of other species identified as part of the complex is a problem that has been previously reported in the literature [22]. For this reason, this study also proposes the standardization of real-time PCR protocols for the identification of Entamoeba species other than the complex. Of the samples analyzed, 11 samples remained unidentified after the multiplex real-time PCR protocol and were submitted to screening for E. hartmanni DNA; this species' DNA was successfully amplified in four samples. Seven further samples remained negative after this analysis. However, further microscopic examination of these samples revealed another possible explanation for this negative result: the presence of Entamoeba coli cysts in the majority of these samples. It is possible that the presence of immature cysts in those samples resulted in a false positive diagnosis of the E. histolytica/E. dispar complex on microscopic examination.
Our results highlight the importance of accurate identification of Entamoeba species. In the diagnosis of amoebiasis, a simple detection at the level of the E. histolytica/E. dispar complex has been considered sufficient cause for clinical treatment. It is understandable for hospitals operating with limited funds to proceed in this manner. However, reference centers such as university hospitals or public health centers should use techniques such as PCR or antigen detection, described in the literature, for complete identification at the species level, of bacteria, viruses, fungi, and parasites [22, 28]. Accurate identification is extremely important for exact diagnosis and for providing correct epidemiological data.
Accurate diagnosis of amoebiasis is crucial in order for physicians to prescribe proper treatment. These laboratory results present important information that will help clinicians decide whether to apply an antiamoebic treatment or search for a different etiology for the presented symptoms. Only the specific detection of E. histolytica confirms a diagnosis of amoebiasis, while the identification of other nonpathogenic amoebas can lead clinical investigators to search for different pathologies with similar symptoms that would not be considered without this information.
5. Conclusion
The specificity and sensitivity demonstrated by the SYBR Green real-time PCR protocols indicate an interesting approach to the identification and differentiation of these Entamoeba species. The application of those protocols in different regions can be a helpful tool to provide more reliable parasitological diagnosis and more detailed epidemiological data on the prevalence of Entamoeba species around the world. The combination of both real-time PCR protocols may allow the development of a simple and useful Entamoeba identification system to improve the laboratory diagnosis of amoebiasis in developing countries.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Fotedar R, Stark D, Beebe N, Marriott D, Ellis J, Harkness J. Laboratory diagnostic techniques for Entamoeba species. Clinical Microbiology Reviews. 2007;20(3):511–532. doi: 10.1128/CMR.00004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Amoebiasis. Weekly Epidemiological Record. 1997;72:97–99. [Google Scholar]
- 3.Tanyuksel M, Petri WA., Jr. Laboratory diagnosis of amebiasis. Clinical Microbiology Reviews. 2003;16(4):713–729. doi: 10.1128/CMR.16.4.713-729.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali IKM, Clark CG, Petri WA., Jr. Molecular epidemiology of amebiasis. Infection, Genetics and Evolution. 2008;8(5):698–707. doi: 10.1016/j.meegid.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delialioglu N, Aslan G, Ozturk C, Ozturhan H, Şen S, Emekdas G. Detection of Entamoeba histolytica antigen in stool samples in Mersin, Turkey. Journal of Parasitology. 2008;94(2):530–532. doi: 10.1645/GE-1355.1. [DOI] [PubMed] [Google Scholar]
- 6.Tengku SA, Norhayati M. Public health and clinical importance of amoebiasis in Malaysia: a review. Tropical Biomedicine. 2011;28(2):194–222. [PubMed] [Google Scholar]
- 7.Santos FLN, Gonçalves MS, Soares NM. Validation and utilization of PCR for differential diagnosis and prevalence determination of Entamoeba histolytica/Entamoeba dispar in Salvador City, Brazil. Brazilian Journal of Infectious Diseases. 2011;15(2):119–125. [PubMed] [Google Scholar]
- 8.Garcia LS. Intestinal protozoa: amebae. In: Garcia LS, editor. Diagnostic Medical Parasitology. 5th edition. Washington, DC, USA: ASM Press; 2007. pp. 6–25. [Google Scholar]
- 9.Sard BG, Navarro RT, Sanchis JGE. Amebas intestinales no patógenas: una visión clinicoanalítica. Enfermedades Infecciosas y Microbiología Clínica. 2011;29(supplement 3):20–28. doi: 10.1016/S0213-005X(11)70023-4. [DOI] [PubMed] [Google Scholar]
- 10.Burrows RB. Morphological differentiation of Entamoeba hartmanni and E. polecki from E. Histolytica . The American Journal of Tropical Medicine and Hygiene. 1959;8(5):583–589. doi: 10.4269/ajtmh.1959.8.583. [DOI] [PubMed] [Google Scholar]
- 11.Blessmann J, Buss H, Nu PAT, et al. Real-time PCR for detection and differentiation of Entamoeba histolytica and Entamoeba dispar in fecal samples. Journal of Clinical Microbiology. 2002;40(12):4413–4417. doi: 10.1128/JCM.40.12.4413-4417.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qvarnstrom Y, James C, Xayavong M, et al. Comparison of real-time PCR protocols for differential laboratory diagnosis of amebiasis. Journal of Clinical Microbiology. 2005;43(11):5491–5497. doi: 10.1128/JCM.43.11.5491-5497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fotedar R, Stark D, Beebe N, Marriott D, Ellis J, Harkness J. PCR detection of Entamoeba histolytica, Entamoeba dispar, and Entamoeba moshkovskii in stool samples from Sydney, Australia. Journal of Clinical Microbiology. 2007;45(3):1035–1037. doi: 10.1128/JCM.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khairnar K, Parija SC. A novel nested multiplex polymerase chain reaction (PCR) assay for differential detection of Entamoeba histolytica, E. moshkovskii and E. dispar DNA in stool samples. BMC Microbiology. 2007;7, article 47 doi: 10.1186/1471-2180-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck DL, Doğan N, Maro V, Sam NE, Shao J, Houpt ER. High prevalence of Entamoeba moshkovskii in a Tanzanian HIV population. Acta Tropica. 2008;107(1):48–49. doi: 10.1016/j.actatropica.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamzah Z, Petmitr S, Mungthin M, Leelayoova S, Chavalitshewinkoon-Petmitr P. Development of multiplex real-time polymerase chain reaction for detection of Entamoeba histolytica, Entamoeba dispar, and Entamoeba moshkovskii in clinical specimens. The American Journal of Tropical Medicine and Hygiene. 2010;83(4):909–913. doi: 10.4269/ajtmh.2010.10-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haque R, Kabir M, Noor Z, et al. Diagnosis of amebic liver abscess and amebic colitis by detection of Entamoeba histolytica DNA in blood, urine, and saliva by a real-time PCR assay. Journal of Clinical Microbiology. 2010;48(8):2798–2801. doi: 10.1128/JCM.00152-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moura H, Fernandes O, Viola JPB, Silva SP, Passos RH, Lima DB. Enteric parasites and HIV infection: occurrence in AIDS patients in Rio de Janeiro, Brazil. Memórias do Instituto Oswaldo Cruz. 1989;84(4):527–533. doi: 10.1590/s0074-02761989000400010. [DOI] [PubMed] [Google Scholar]
- 19.Machado ER, Santos DS, Costa-Cruz JM. Enteroparasites and commensals among children in four peripheral districts of Uberlândia, state of Minas Gerais. Revista da Sociedade Brasileira de Medicina Tropical. 2008;41(6):581–585. doi: 10.1590/s0037-86822008000600007. [DOI] [PubMed] [Google Scholar]
- 20.Santos HLC, Bandea R, Martins LAF, et al. Differential identification of Entamoeba spp. based on the analysis of 18S rRNA. Parasitology Research. 2010;106(4):883–888. doi: 10.1007/s00436-010-1728-y. [DOI] [PubMed] [Google Scholar]
- 21.Lutz AV. Shistosoma mansoni e schistosomose, segundo observações feitas no Brasil. Memórias do Instituto Oswaldo Cruz. 1919;11:121–125. [Google Scholar]
- 22.Santos HLC, Peralta RHS, de Macedo HW, Barreto MGM, Peralta JM. Comparison of multiplex-PCR and antigen detection for differential diagnosis of Entamoeba histolytica . Brazilian Journal of Infectious Diseases. 2007;11(3):365–370. doi: 10.1590/s1413-86702007000300013. [DOI] [PubMed] [Google Scholar]
- 23.Núñez YO, Fernández MA, Torres-Núñez D, et al. Multiplex polymerase chain reaction amplification and differentiation of Entamoeba histolytica and Entamoeba dispar DNA from stool samples. The American Journal of Tropical Medicine and Hygiene. 2001;64(5):293–297. doi: 10.4269/ajtmh.2001.64.293. [DOI] [PubMed] [Google Scholar]
- 24.Martínez-Palomo A, Martínez-Báez M. Selective primary health care: strategies for control of disease in the developing world. X. Amebiasis. Reviews of Infectious Diseases. 1983;5(6):1093–1102. doi: 10.1093/clinids/5.6.1093. [DOI] [PubMed] [Google Scholar]
- 25.Bansal D, Sehgal R, Chawla Y, Mahajan RC, Malla N. In vitro activity of antiamoebic drugs against clinical isolates of Entamoeba histolytica and Entamoeba dispar . Annals of Clinical Microbiology and Antimicrobials. 2004;3, article 27 doi: 10.1186/1476-0711-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamzah Z, Petmitr S, Mungthin M, Leelayoova S, Chavalitshewinkoon-Petmitr P. Differential detection of Entamoeba histolytica, Entamoeba dispar, and Entamoeba moshkovskii by a single-round PCR assay. Journal of Clinical Microbiology. 2006;44(9):3196–3200. doi: 10.1128/JCM.00778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dourado A, Maciel A, Aca IDS. Occurrence of Entamoeba histolytica/Entamoeba dispar in ambulatory patients of Recife, PE. Revista da Sociedade Brasileira de Medicina Tropical. 2006;39(4):388–389. doi: 10.1590/s0037-86822006000400015. [DOI] [PubMed] [Google Scholar]
- 28.Furrows SJ, Moody AH, Chiodini PL. Comparison of PCR and antigen detection methods for diagnosis of Entamoeba histolytica infection. Journal of Clinical Pathology. 2004;57(12):1264–1266. doi: 10.1136/jcp.2004.017822. [DOI] [PMC free article] [PubMed] [Google Scholar]