

Abstract
There is growing evidence that anti-angiogenic therapy stimulates cancer cell invasion and metastasis. However, the underlying molecular mechanisms responsible for these changes have not been fully defined. Here we report that anti-VEGF therapy promotes local invasion and metastasis by inducing collagen signaling in cancer cells. We show that chronic VEGF inhibition in a genetically engineered mouse model (GEMM) of pancreatic ductal adenocarcinoma (PDA) induces hypoxia, a less differentiated mesenchymal-like tumor cell phenotype, TGFβ expression, and collagen deposition and signaling. Additionally, we show that collagen signaling is critical for pro-tumorigenic activity of TGFβ in vitro. To further model the impact of collagen signaling in tumors, we evaluated PDA in mice lacking Sparc, a protein that reduces collagen binding to cell surface receptors. Importantly, we show that loss of Sparc increases collagen signaling and tumor progression. Together, these findings suggest that collagen actively promotes PDA spread and that enhanced disease progression associated with anti-VEGF therapy can arise from elevated ECM-mediated signaling.
Keywords: pancreatic cancer, VEGF, Sparc, collagen, Peak1, epithelial to mesenchymal transition
Introduction
Tumor growth beyond a few millimeters in diameter requires the establishment of a tumor-associated vasculature (1). This neovascularization is mediated by the activity of soluble growth factors, most notably vascular endothelial growth factor (VEGF), on existing endothelium (2). The reliance of tumors on a blood supply suggests that blocking angiogenesis would restrain tumor growth and dissemination (1, 3). Indeed transplantation, genetic, and pharmacologic approaches using animal models have demonstrated that blocking tumor angiogenesis represents a viable therapeutic avenue, and several anti-angiogenic agents are in various phases of clinical testing or are approved by the FDA (4, 5). The vast majority of these target VEGF or its receptors, vascular endothelial growth factor receptors 1 and 2 (VEGFR1 and VEGFR2).
Despite the progress from conception to FDA-approved therapy, preclinical success has not always translated to a robust clinical response for drugs that inhibit angiogenesis (5). Tumors that do respond to anti-angiogenic treatment often eventually relapse. Even more challenging, patients with glioblastoma multiforme (GBM) treated with the anti-VEGF monoclonal antibody bevacizumab had rapid tumor dissemination following a period of primary tumor response (6). A similar effect was observed in preclinical models of GBM and pancreatic neuroendocrine carcinoma (PNET) using the anti-VEGFR2 antibody DC101 and the small molecule tyrosine kinase inhibitor sunitinib (7). These observations suggest that anti-angiogenic therapy can, in some patients, promote tumor growth and spread.
The effect of anti-VEGF therapy on progression of pancreatic ductal adenocarcinoma (PDA) has not been studied in detail. PDA results in almost a quarter million deaths yearly worldwide. In the United States, approximately 90% of patients diagnosed with PDA present with metastatic disease and have a median survival of less than one year (8). Preclinical studies have shown that anti-angiogenic therapy can suppress the growth of PDA xenografts and prolong the survival of mice bearing PDA tumors (9, 10). Yet in two separate phase III trials, patients with metastatic PDA failed to show any increase in overall survival when bevacizumab was added to the standard of care agent gemcitabine (CALGB 80303), or gemcitabine plus erlotinib (AViTA) (11, 12).
Tumors develop and progress in the context of the extracellular matrix (ECM). The ECM of several different cancer types, especially PDA, are robust and rich in fibrillar collagens, which have been proposed to be major barrier to chemoresponse (13, 14). Collagen signaling facilitates TGFβ-mediated changes in tumor cell phenotype and can promote tumor cell survival and chemoresistance. This is particularly relevant to PDA, which is a desmoplastic disease (13, 15). Each cell in the PDA microenvironment interacts with fibrillar collagen, which has the potential to impact cell signaling events via crosstalk of its receptors with other signal cascades (15). Our goal was to study the biology of anti-VEGF therapy in a robust preclinical model of PDA and determine if therapy induced hypoxia drives changes in the ECM that contribute to the poor response of these tumors to therapy.
We assessed the effect of the anti-VEGF monoclonal antibody, mcr84 (16), in a clinically relevant genetically engineered mouse model (GEMM) of PDA. We found that mcr84 induced hypoxia, restrained tumor growth, and prolonged survival, yet tumors from mcr84-treated mice exhibited a less differentiated phenotype and had increased metastatic burden. Further, these tumors contained higher levels of fibrillar collagen and elevated collagen signaling which includes activation of discoidin domain receptor 1 (Ddr1), protein tyrosine kinase 2 (Pyk2) and pseudopodium-enriched atypical kinase 1 (Peak1). We also found that secreted protein acidic and rich in cysteine (Sparc) blocked collagen I from binding to Ddr1, and that PDA tumors grown in Sparc-/- mice (17, 18) exhibited increased collagen signaling and enhanced disease progression, similar to tumors from mice treated chronically with anti-VEGF therapy. Our findings support that collagen enhances PDA and that Sparc functions to limit collagen induced activation of Ddr1. Further these studies suggest that VEGF-blockade can promote tumorigenicity of PDA cells by promoting hypoxia-induced collagen production and stimulation of Ddr1.
Materials and Methods
Cell lines
mPLR cell line isolation
Isogenic cell lines were derived from 5 week old KIC transgenic (LSL-KrasG12D; Cdkn2alox/lox; p48Cre) and Sparc-/-; KIC (LSL-KrasG12D; Cdkn2alox/lox; p48Cre, Sparc-/-) mice. Mouse pancreata from 5-wk-old Sparc+/+ ; KIC and Sparc-/- ; KIC mice were minced and then subjected to digestion with 1% collagenase type 1, DME, 10 mM Hepes, 1% fetal bovine serum, and PBS at 37°C until a single-cell suspension was obtained. Cell suspensions were centrifuged at low speed to pellet large debris, resuspended in wash buffer, and passed through a 70 μm cell strainer. The resulting cell suspension was plated at low density to isolate tumor cell populations using cloning rings. Cells were confirmed to be tumor cells by immunocytochemistry and PCR. These cell lines were expanded and stained for tumor cell markers. Cell lines were confirmed to be pathogen-free before use. Clones mPLRB8 (Sparc+/+) and mPLR6C (Sparc-/-) were used in subsequent experiments. Cells were cultured in DMEM (Invitrogen) containing 5% fetal bovine serum and maintained at 37°C in a humidified incubator with 5% CO2 and 95% air.
Animal studies
All animals were housed in pathogen-free facility with access to food and water ad libitum. Experiments were approved and performed in accordance with the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. LSL-KrasG12D; Cdkn2alox/lox; p48Cre (KIC) mice were generated as previously described (19, 20). Mice 28-30 days old were randomized to receive treatment as indicated in Table 1. mcr84 and 1D11 were purified in our laboratory from tissue culture supernatant using Protein A or G affinity chromatography. The hybridoma producing 1D11 (21) was obtained from the Developmental Studies Hybridoma Bank (http://dshb.biology.uiowa.edu/). For endpoint studies experiments were stopped after 4 weeks of therapy. For survival studies, therapy was maintained until mice were moribund. At the time of sacrifice all mice were subjected to careful necropsy where visible metastases were noted and organs harvested for tissue analysis. Liver micrometastasis was assessed by hemotoxylin and eosin on the anterior lobes of the liver as well as qPCR for recombined Cdkn2a (Ink4a/Arf) allele.
Table 1.
Animal experiment overview
| Endpoint: Anti-VEGF (mcr84) treatment | Age of experiment start | 4 weeks old |
|
| ||
| Experiment length | 4 weeks | |
|
| ||
| Treatment groups | Saline (n=8) | |
| mcr84 (n=12): 500 μg/week | ||
| mcr84 + Gem (n=9): mcr84 500 μg/week, Gem 250 μg 3x/week | ||
|
| ||
| Associated figures | Figures 2A-2C; Supplementary Figure 2A | |
|
| ||
| Survival: Anti-VEGF (mcr84) treatment | Age of experiment start | 4 weeks old |
|
| ||
| Experiment length | Until moribund | |
|
| ||
| Treatment groups | Saline (n=27) | |
| mcr84 (n=15): 500 μg/week | ||
| mcr84 + Gem (n=15): mcr84 500 μg/week, Gem 250 μg 3x/week | ||
|
| ||
| Associated figures | Figures 2D-2H, 3A-3C; Supplementary Figures 2B, 3A-3B | |
|
| ||
| Survival: Anti-TGFβ (1D11) treatment | Age of experiment start | 4 weeks old |
|
| ||
| Experiment length | Until moribund | |
|
| ||
| Treatment groups | Saline (n=4) | |
| mcr84 +Gem (n=5): mcr84 500 μg/week, Gem 250 μg 3x/week 1D11 (n=7): 150 μg 2x/week | ||
| mcr84 + 1D11 (n=7): mcr84 500 μg/week, 1D11 150 μg 2x/week, Triple (n=8): mcr84 500 μg/week, 1D11 150 μg 2x/week, Gem 250 μg 3x/week | ||
|
| ||
| Associated figures | Figure 4; Supplementary Figure 4 | |
Histology
Formalin-fixed tissues were embedded in paraffin, sectioned and stained with Masson’s Trichrome, Alcian Blue-PAS, or Picrosirius Red by the Molecular Pathology Core (UT Southwestern. Immunohistochemistry was performed with antibodies against: phospho-Ddr1 (Tyr792, Cell Signaling #11994), Ddr1 (D1G6, Cell Signaling #5583), phospho-Src (Tyr416, Cell Signaling #2101), phospho-Pyk2 (Tyr402, Cell Signaling #3291), phospho-p130 Cas (Tyr165, Cell Signaling #4015), α-Amylase (D55H10, Cell Signaling #3796), vimentin (Millipore AB5733), endomucin clone V.5C7 (Millipore MAB2624), NG2 clone 132.38 (Millipore 05-710), Peak1 (Millipore 09-274) and phospho-Peak1 (Tyr665, Millipore #ABT52). Fluorescent images were captured with Photometric Coolsnap HQ camera using NIS Elements AR 2.3 Software (Nikon). Color images were obtained with a Nikon Eclipse E600 microscope using a Nikon Digital Dx1200me camera and ACT1 software (Universal Imaging Corporation). Pictures were analyzed using NIS Elements (Nikon).
Immunocytochemistry
Cell lines were cultured in 4-well chamber slides. Cells were then fixed with methanol, washed with PBS, permeabilized with PBS+0.1% TX-100, and then blocked with TBST+20% Aquablock. Antibodies diluted TBST+5% BSA were added and allowed to incubate overnight at 4°C. Cells were then washed with PBS and incubated overnight with the appropriate secondary antibodies. Following another round of washes with PBS, coverslips were mounted with ProLong Gold with DAPI.
Hypoxia studies
Hypoxia studies with pimonidazole in vivo
Three mice per group were injected intravenously with 60 mg/kg of pimonidazole (30 mg/mL in 0.9% saline, Hypoxyprobe Plus;HPI Inc.) that was allowed to circulate for 90 minutes before sacrificing animals. Frozen tissue sections were interrogated with FITC-conjugated anti-pimonidazole primary antibody (Chemicon) and endothelial cell markers (CD31, Dianova; Meca-32, DSHB; or Endomucin, Santa Cruz Biotechnology). Eight images per tissue area were obtained and analyzed using NIS Elements.
Hypoxia studies with hypoxic chamber in vitro
Cell lines were grown in 4-well chamber slides in a humidified atmosphere containing 5% CO2 and 1% O2 in a modular incubator chamber (Billups-Rothenberger) for 48 hr. For control conditions cells were cultivated in an incubator with a humidified atmosphere containing 5% CO2 and 20% O2 (normoxic conditions). Immunocytochemical analysis was conducted for the validation of Hif1α induction.
Liquid colony forming assay
Cell lines were cultured in 6-well tissue culture plates at low density (250 cells per well) in 2 ml 5% DMEM. Cells were plated on respective culture conditions and allowed to sit for 72-96 hours, or until significant colony formation. Cells were then fixed with 10% formalin and stained with crystal violet. Images were analyzed with Image J or NIS Elements.
Sircol collagen assay
Cell lines were prepared as indicated by the Sircol manual (Sircol Collagen Assay Kit, Oubis Ltd). Sircol dye reagent is used to saturate the collagen in a sample. The principle of the Sircol collagen assay is the binding of a dye to collagen. A collagen-dye complex forms and precipitates out, is recovered by centrifugation, eluted with alkali, and measured using a spectrophotometer at 555 nm. The intensity of color measurement is proportional to the collagen concentration in a sample.
Western blot analysis
Sub-confluent monolayers of cells were lysed, supernatants were recovered by centrifugation, protein concentrations were measured and equal amounts of total protein were separated by SDSPAGE. Proteins were transferred to PVDF membranes (Bio-Rad, Hercules, CA) followed by blockade in 5% milk in TBS-T. The membranes were incubated overnight at 4°C with primary antibody. The membranes were then incubated with the corresponding HRP-conjugated secondary antibody (Pierce Biotechnologies, Santa Cruz, CA). Specific bands were detected using the enhanced chemiluminescence reagent (ECL, Perkin Elmer Life Sciences, Boston, MA) on autoradiographic film.
Competitive ELISA (Binding assay)
96-well ELISA plates were coated with 10 μg/ml rat tail collagen I (BD Biosciences) or 2% Casein Acid Hydrolysate (Sigma) in PBS (Casein-PBS) and blocked with 2% Casein. For competitive binding assays, human recombinant DDR1-Fc (R&D Systems) and human recombinant SPARC (R&D Systems) were added to the wells at respective concentrations in triplicate in ELISA Binding Buffer and incubated overnight at 4°C. Binding of DDR1 or SPARC was detected with primary and secondary antibodies for human DDR1 (R&D Systems) or human SPARC (R&D Systems) in 2% Casein-PBS. 50 μl TMB One Component HRP - Microwell Substrate (TMB) (SurModics-BioFx) was added per well to detect bound protein, along with addition of 50 μl 10% hydrochloric acid (HCl) to quench the TMB reaction.
Statistical analysis
Quantification of immunohistochemistry was conducted using NIS Elements 3.2 software (Nikon Instruments). All data were analyzed using GraphPad Prism 5.0 software (GraphPad Software Inc.). Datasets were analyzed by Student t test or ANOVA followed by Dunn posttest or Tukey’s MCT and results were considered as significant at p < 0.05. Results are shown as mean ± SEM.
Results
Angiogenic stress and elevated VEGF in murine PDA model
Human PDAs frequently contain an activating mutation in the oncogene KRAS, and an inactivating mutation in the tumor suppressor CDKN2A (INK4A/ARF) (22, 23). Mice that harbor analogous genetic alterations specifically in the pancreas (LSL-KrasG12D; Cdkn2alox/lox; p48Cre, abbreviated here as KIC) develop aggressive, invasive pancreatic tumors (19, 24). Histological analysis of pancreatic tissue harvested from KIC animals revealed that these mice develop PanIN lesions that progress to invasive carcinoma by the time the mice are 4-8 weeks old (Supplementary Figure 1A). Alcian blue-PAS staining of mucin containing ducts showed the loss of normal ductal architecture and function during the progression of PDA in KIC mice (Supplementary Figure 1A) while picrosirius red and trichrome analysis revealed that KIC tumors have more collagen that WT pancreata (Supplementary Figure 1A-1B).
We evaluated vascular parameters in normal pancreatic and PDA tissue at various time points (Figure 1). NG2+ pericytes were localized to endomucin expressing endothelial cells in normal pancreatic tissue and in pancreatic tissue harvested from 4 and 7 week old KIC mice (Figure 1A). We found that microvessel density was significantly lower in PDA tissue than normal pancreas (Figure 1B). However, endothelial cells in the tumors were proliferative (Figure 1C) and displayed an angiogenic phenotype, typified by the presence of sprouts, which were not present in the vasculature of normal pancreas (Figure 1D). We also observed that the expression level of VEGF was higher in PDA tissue than normal pancreatic tissue (Figure 1E).
Figure 1. Activated endothelial cells are present in KIC PDA tumors.
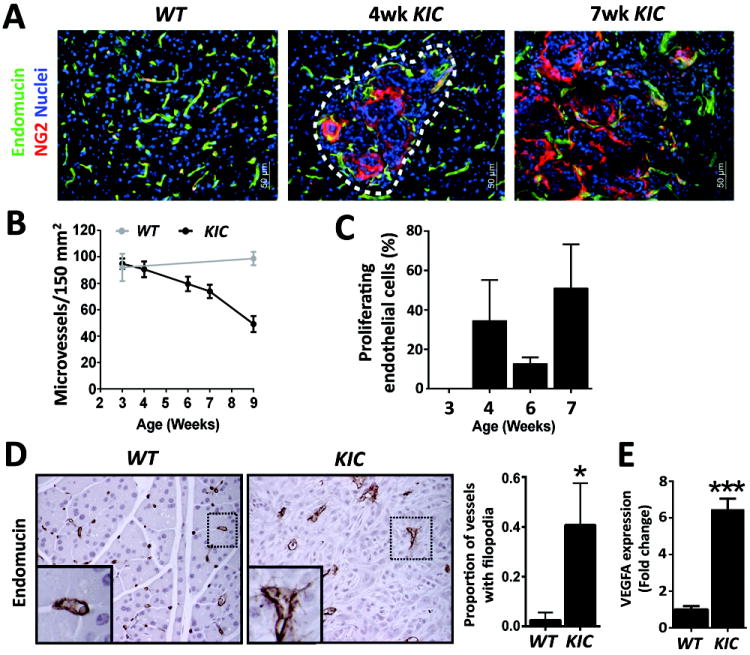
(A) KIC tumors exhibit abnormal vascular architecture. Wild-type (WT), and 4 and 7 week KIC samples were stained for a blood vessel marker (endomucin) and a pericyte marker (NG2). Quantification of microvessel density is displayed in (B). (C) Vessels undergo angiogenic stress as seen by increased vessel sprouting (Endomucin) in KIC tissues. (D) Endothelial cell activation in KIC PDA. WT pancreas and KIC PDA were stained for PCNA and endomucin. Inset in right panel highlights double-stained proliferating endothelial cells. Quantification of proliferating endothelial cells in KIC PDA is presented in (D). (E) Elevated expression of VEGF in PDA. VEGF expression in pancreata collected from 7 week old WT and KIC mice was assessed by qPCR. Error bars represent SEM (*, p < 0.05 vs WT; ***, p < 0.0005 vs WT). One way ANOVA with Tukey’s MCT.
VEGF-blockade prolongs PDA survival and increases hypoxia, TGFβ activity, and collagen deposition
To determine the contribution of VEGF to PDA progression, KIC mice (4 weeks old) were treated with saline, mouse chimeric r84 (mcr84), or mcr84 plus gemcitabine (Gem) (Figure 2A-2H, Table 1). mcr84 is a monoclonal antibody (mAb) that specifically binds VEGF and inhibits VEGF stimulation of VEGFR2 (16). Four weeks of therapy with mcr84 or mcr84 plus Gem reduced tumor burden as assessed by amylase expression (Figure 2A-2B) and total pancreas weight (Figure 2C). Anti-VEGF therapy alone reduced primary tumor weight by 36% (p<0.05 vs saline), while mcr84 plus Gem reduced tumor weight by 65% (p<0.0001 vs saline) (Figure 2C). To determine whether anti-VEGF +/- chemotherapy improved animal survival, KIC mice were treated with saline (n=27), mcr84 (n=15), or mcr84 + Gem (n=15) starting at the age of 4 weeks. Therapy was maintained until animals were moribund (Table 1). The median survival of each treatment cohort was determined. Mice that received saline had a median survival of 56 days, while treatment with mcr84 or the combination extended median survival to 56 and 82 days, respectively (Figure 2D). However, at the time of sacrifice liver metastatic burden was higher in in mice receiving mcr84. The extent of liver metastasis was determined by quantitative PCR for the recombined Cdkn2a allele (Figure 2E, Supplementary Figure 2A) and histological evaluation of macrometastases (Supplementary Figure 2B). The incidence of metastasis calculated by histology was 35, 63, and 46% in saline (n=14), mcr84 (n=11) and mcr84 + Gem (n=13) groups, respectively.
Figure 2. VEGF-blockade restricts tumor growth and promotes survival of KIC mice.
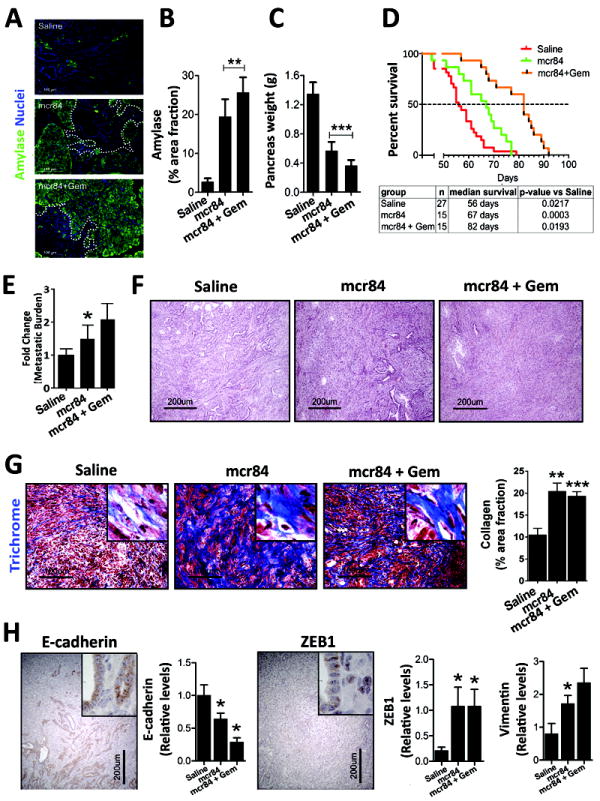
KIC mice were treated with saline (n=10), mcr84 (n=10), or mcr84 + Gem (n=11) from 4-8 weeks of age, after which pancreases were harvested (A-C). (A) mcr84 blocks the replacement of normal pancreatic parenchyma with PDA. Tissue sections from the indicated groups were stained for amylase, a marker of exocrine pancreas. Tumors in mcr84 and mcr84 plus Gem treated mice are circumscribed by a dotted line. Quantitation of amylase content is presented in (B). (C) mcr84 controls primary PDA growth. Mean pancreas weight was used a surrogate measurement of primary tumor burden. (D) Mice in a survival study were treated with saline, mcr84, or mcr84 + Gem from 4 weeks until moribund. (E) Metastasis to the liver was quantified by qPCR for recombined Cdkn2a allele (n=6-7 animals/group). (F) mcr84-treated KIC tumors exhibit a less differentiated phenotype as documented by H&E histology. (G) Collagen content is elevated after anti-VEGF therapy. Tumors from the survival study were stained with Masson’s trichrome and collagen was quantified and is presented in right panel. (H) VEGF-blockade enhances EMT. Tumors from the survival study were stained for E-cadherin, Zeb1, and vimentin. Insets demonstrate glandular E-cadherin staining, and nuclear Zeb1 staining in non-glandular tumor cells. Positive staining area was quantified and is presented in panels adjacent the micrographs. Error bars represent SEM. (*, p < 0.05 vs saline; **, p < 0.005 vs saline; ***, p < 0.0005 vs saline). One way ANOVA with Tukey’s MCT.
The observation that anti-VEGF therapy did not reduce metastatic burden is similar to prior studies (7, 25), which reported that anti-VEGF therapy drove phenotypic changes in tumor cells consistent with a loss of differentiation. To examine if these changes were present in KIC tumors after treatment with mcr84, we evaluated histology (H&E and trichrome) of the primary tumors and expression of epithelial and mesenchymal markers. Tumors harvested from mice in the survival study treated with mcr84 alone or in combination with Gem displayed a loss of differentiation by histology (Figure 2F) and a dramatic increase in Masson’s trichrome stain, an indicator of collagen deposition (Figure 2G, Supplementary Figure 3A). Consistent with the histological changes, expression of E-cadherin, a typical marker of epithelial cells, was reduced while expression of the mesenchymal markers, zeb1 and vimentin, were elevated in tumors from mice treated with mcr84 alone or in combination with Gem (Figure 2H).
Therapy-induced hypoxia is a predicted outcome of VEGF blockade and has been reported to drive an aggressive tumor phenotype (7, 25, 26). To validate the general mechanism of mcr84, microvessel density and the level of hypoxia were determined. mcr84 alone or in combination with Gem significantly reduced tumor microvessel density (Figure 3A). This was coupled with an elevation in hypoxia as measured by companion studies using pimonidazole (Figure 3B). Concordant with the induction of hypoxia, the expression of collagen I, Hif1α, and hexokinase II were elevated in tumors from mice treated with mcr84 or mcr84 plus Gem (Supplementary Figure 3B). Furthermore, Tgfβ2, a hypoxia-responsive cytokine (27) that stimulates collagen expression and deposition (28) was elevated significantly following mcr84 therapy (Figure 3C). These results suggest that anti-VEGF therapy induces hypoxia, which in turn drives Tgfβ-stimulated activity including collagen deposition and EMT. To model this in vitro, primary murine KIC PDA cells were plated on collagen and treated with Tgfβ under normoxic and hypoxic conditions. We found that hypoxia stimulated soluble collagen secretion by PDA cells and primary NG2+ stromal cells (Supplementary Figure 3C-3D). Similar to the in vivo observations, in vitro stimulation with Tgfβ drove morphological changes consistent with a loss of differentiation and promoted loss of E-cadherin, gain of vimentin expression and protection from hypoxia-induced apoptosis (Figure 3D-3E). To determine if collagen was critical to the effect of Tgfβ, cells were plated on plastic or collagen and stimulated with Tgfβ. Tgfβ treatment in the absence of collagen resulted in induction of apoptosis but limited change in expression of E-cadherin or vimentin. In contrast cells plated on collagen and treated with Tgfβ underwent EMT and were protected from Tgfβ-induced apoptosis (Figure 3F-3G).
Figure 3. VEGF-blockade induces hypoxia and elevates TGFβ expression.
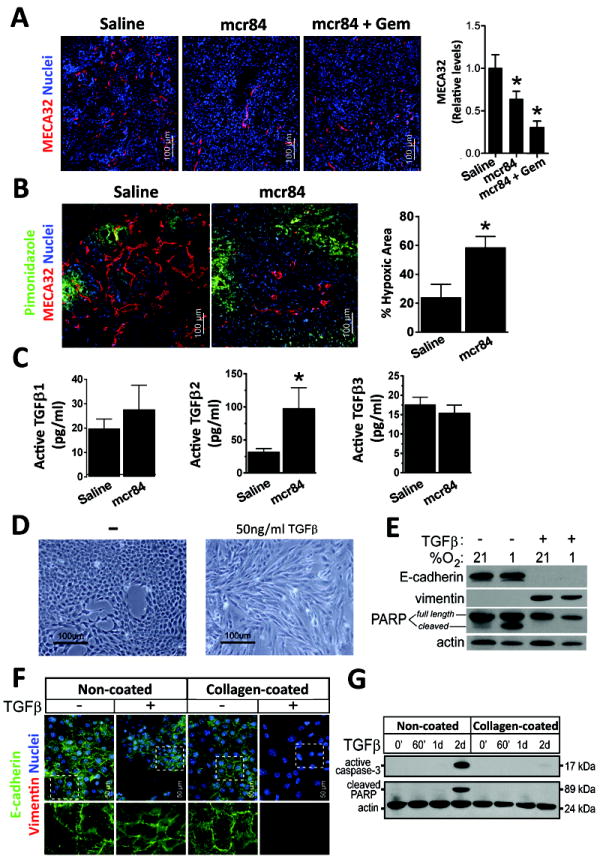
(A) Microvessel density in tumors from KIC mice treated with saline, mcr84, mcr84 + Gem was determined by immunohistochemistry with MECA32 (red). Quantification of MVD is shown. (B) 6 week old KIC mice were treated with saline or mcr84 for one week. Animals were sacrificed 1 hour after pimonidazole injection (iv). Tissue was stained for endothelial cells (MECA32, red) and pimonidazole adducts (Hypoxyprobe, green). Hypoxic area was quantified and is presented in right panel. (C) Lysates from tumors harvested from saline and mcr84 treated KIC animals were assessed for active TGFβ proteins by ELISA. The level of TGFβ2 was elevated significantly by blockade of VEGF with mcr84. (D) Stimulation of primary KIC cells with TGFβ for 48 hours resulted in morphological changes. (E) TGFβ stimulation promoted loss of E-cadherin, gain of vimentin expression and protection from hypoxia-induced apoptosis. (F, G) Collagen protects PDA cells from TGFβ-induced apoptosis. Murine KIC-derived PDA cells (clone 3B) were plated on plastic or collagen-coated surfaces and treated with 10 ng/ml TGFβ1 for 72 hrs or the indicated time. (F) Cells were examined by immunocytochemistry for E-cadherin (green) and vimentin (red). Dashed insets are enlarged and presented beneath each panel. (G) Cell lysates were probed for active caspase-3 and cleavage of the caspase-3 substrate PARP. Actin was used as a loading control. 1d = 24 hours; 2d = 48 hours. Error bars represent SEM (*, p < 0.05 vs saline)
To evaluate the contribution of Tgfβ to the tumor cell changes in vivo, we treated KIC mice with 1D11, a mAb that binds and inhibits each isoform of mouse Tgfβ (21). KIC mice were treated with saline (n=4), mcr84+Gem (n=5), 1D11 (n=7), 1D11+mcr84 (n=7), or the triple combination (n=8) for 4 weeks starting at 4 weeks of age (Table 1). 1D11 did not reduce primary tumor growth and abrogated the survival benefit of mcr84+Gem (Supplementary Figure 4A). However, blockade of Tgfβ with 1D11 reduced liver metastatic events more than treatment with mcr84 and dramatically curtailed mcr84-induced collagen deposition (Figure 4A, Supplementary Figure 4B-4C).
Figure 4. Collagen promotes PDA cell survival and EMT.
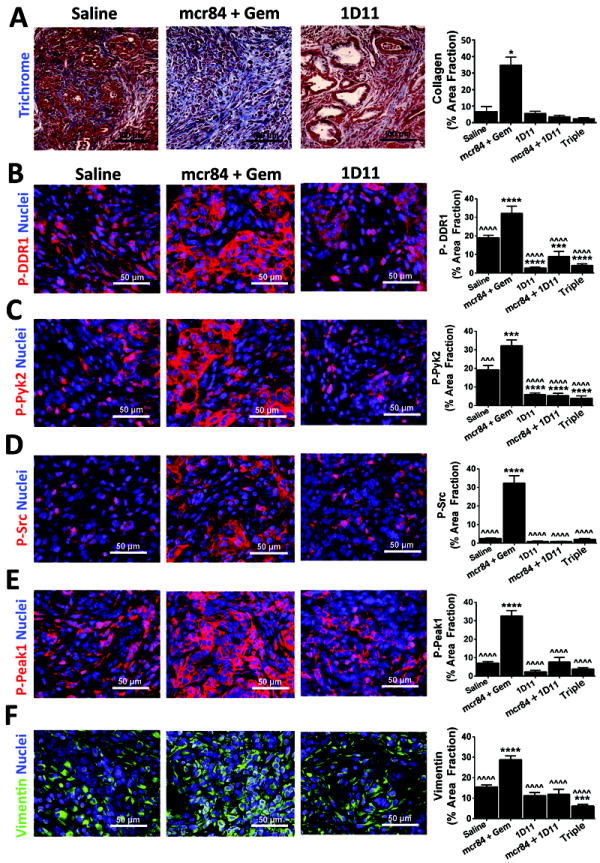
KIC mice were treated with saline (n=4), mcr84 + Gem (n=5), 1D11 (n=6), 1D11+mcr84 (n=6), or the triple combination (n=8) in the context of a survival study. Pancreatic tissue was harvested and subjected to Masson’s Trichrome (A) or immunohistochemistry for phosphorylated Ddr1 (B, p-DDR1), phosphorylated Pyk2 (C, P-Pyk2), phosphorylated Src (D, P-Src), phosphorylated Peak1 (E, P-Peak1) or vimentin (F). Quantification of signal intensity for each target in each treatment group is shown. Error bars represent SEM (*, p < 0.05 vs saline; ***, p < 0.0005 vs saline; ˆˆˆ, p < 0.0005 vs mcr84+gem; ****, p < 0.00005 vs saline; ˆˆˆˆ, p < 0.00005 vs mcr84+gem). One way ANOVA with Tukey’s MCT.
To determine if Tgfβ blockade affected collagen signaling we analyzed the activation status of the collagen receptor Ddr1 and signaling intermediates (Pyk2 and Src) thought to function downstream of this receptor (29). Tumors from mice treated with mcr84 plus Gem had elevated levels of phosphorylated Ddr1, Pyk2 and Src. Treatment with 1D11 significantly reduced the level of each phosphorylated target (Figure 4B-4D). We also evaluated the level of active pseudopodium-enriched atypical kinase I (Peak1), a non-receptor tyrosine kinase previously linked to Src activity and ECM signaling (30, 31). Phosphorylated Peak1 levels were also elevated in tumors from mice treated with mcr84 plus Gem but reduced to baseline levels by Tgfβ blockade (Figure 4E). In addition, the mesenchymal marker vimentin was elevated by mcr84 in combination with Gem and this was reduced by Tgfβ blockade (Figure 4F). These results suggest that collagen levels and collagen mediated signaling increase in PDA tumors treated with anti-VEGF therapy and that blockade of collagen deposition via Tgfβ inhibition decrease collagen mediated signaling.
Sparc inhibits collagen-Ddr1 interaction
Due to the challenges of interpreting the effect of loss of collagen signaling in the context of Tgfβ inhibition (32) we sought to model enhanced collagen signaling in PDA using mice deficient in secreted protein acidic and rich in cysteine (Sparc). Structural studies indicate that SPARC and the DDRs bind to the same epitope (GVMGFO) on fibrillar collagens (33, 34). To determine if SPARC interferes with collagen binding to DDR1 we evaluated the effect of recombinant human SPARC on the binding of human DDR1/Fc to immobilized collagen I in an ELISA-type assay. SPARC reduced DDR1 binding to collagen in a concentration-dependent manner (Figure 5A). To assess the effect of Sparc in vivo we crossed Sparc-/- animals with KIC mice. The absence of endogenous Sparc resulted in reduced survival and increased primary tumor size (Figure 5B-5C, Supplementary Figure 5A). Additionally, tumors in Sparc-/- mice were less differentiated as determined by reduced mucin expression by Alcian blue-PAS histology (Figure 5D). The increase in progression in the absence of Sparc correlated with a greater activation of Ddr1 and increased staining of mesenchymal marker (vimentin). This suggests the elevation of collagen signaling is due to increased collagen binding to the surface of tumor cells in Sparc-/- KIC model (Figure 5E-5J). Consistent with this, the activation of Ddr1, Src, Pyk2, and Peak1 was elevated in tumors from Sparc-/- animals (Figure 5E-5H), while the level of total Peak1 was not different between tumors in Sparc+/+ and Sparc-/- mice (Figure 5I). Additionally, vimentin levels were elevated in tumors from Sparc-/- mice (Figure 5J).
Figure 5. Sparc attenuates PDA progression by reducing collagen signaling.
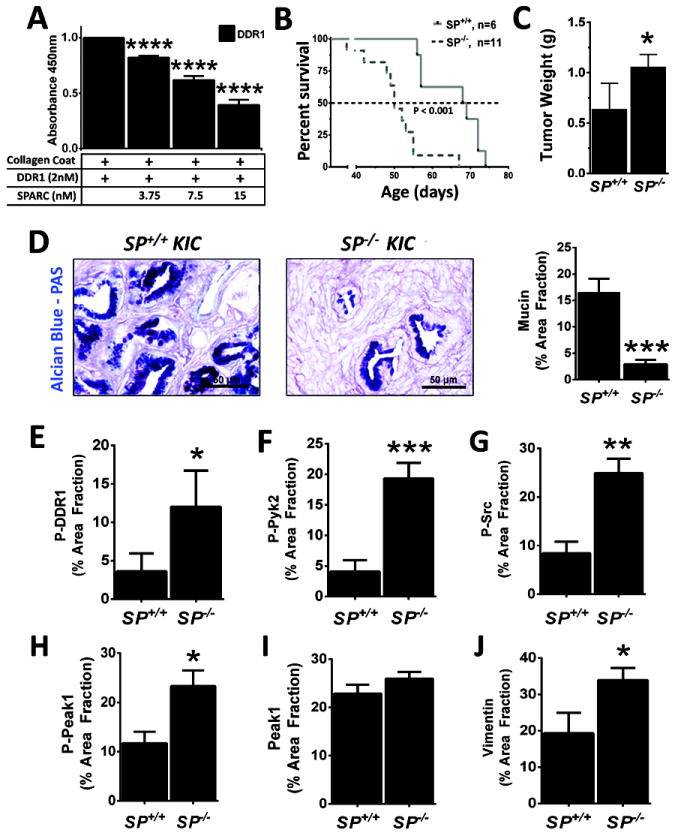
(A) SPARC blocks interaction between DDR1 and collagen. The binding of 2 nM DDR1/Fc to collagen I was determined by ELISA with collagen I being the solid phase. The binding to DDR1/Fc was detected via the Fc tag in triplicate. Increasing concentrations of recombinant human SPARC reduced the binding DDR1/Fc to collagen I. Error bars represent SEM (****, p < 0.00005 vs DDR1). One way ANOVA with Tukey’s MCT. (B) Decreased survival of Sparc-/-:KIC mice. Sparc-/- were crossed with KIC mice to generate Sparc+/-:KIC animals which were crossed with each other to generate KIC (SP+/+) and Sparc-/-:KIC (SP-/-) animals. Survival of these animals is shown. (C) Mean pancreas/tumor weight in Sparc+/+:KIC and Sparc-/-:KIC animals. (D) Alcian blue-PAS histology of tumors from Sparc+/+:KIC and Sparc-/-:KIC animals. (E-J) Expression of mediators of collagen signaling in murine PDA. Tumor was harvested from moribund KIC and Sparc-/-:KIC mice, sectioned and stained for phosphorylated Ddr1 (E, P-DDR1), phosphorylated Pyk2 (F, P-Pyk2), phosphorylated Src (G, P-Src), phosphorylated Peak1 (H, P-Peak1), total Peak1 (I), and Vimentin (J). Error bars represent SEM (*, p < 0.05 vs SP+/+; **, p < 0.005 vs SP+/+; ***, p < 0.0005 vs SP+/+; ****, p < 0.00005 vs SP+/+).
Increased collagen deposition and signaling drives DDR-mediated PDA progression
To investigate how Sparc interferes with collagen-stimulated Ddr1 signaling, primary cell lines developed from Sparc+/+ and Sparc-/- KIC tumors were plated on fibronectin and stimulated with soluble collagen (Figure 6). These PDA cells differentially express collagen receptors, Sparc, and collagen (Supplementary Figure 5B-5C). Maximal activation of Ddrs occurs after 4 hours (35). We found that a 4 hour stimulation with collagen resulted in robust activation of Ddr1, Pyk2, Src, and Peak1 (Figure 6A). Furthermore, Peak1 activation appears to be more potently induced by collagen I rather than collagen IV (35) (Figure 6B) and collagen induced Peak1 activation was elevated in cells that lack Sparc (Figure 6C; Supplementary Figure 5C, 6A). We also found that activation of Peak1 by collagen was sensitive to Ddr1 blockade by an anti-DDR1 antibody (36) or recombinant human SPARC (Figure 6D, Supplementary Figure 6B). Ddr1 blockade had functional consequences as it reduced collagen-supported colony forming capabilities of primary PDA cell lines (Figure 6E), as well as migration in the presence of Ddr1 inhibitors (Supplementary Figure 6B).
Figure 6. PDA cells express collagen receptors and Sparc.
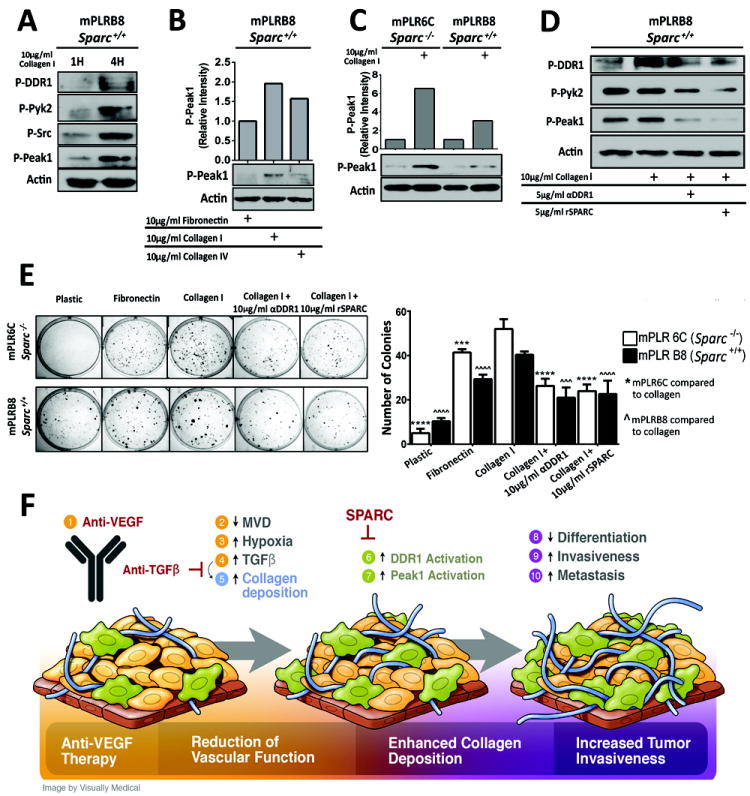
(A) mPLRB8 cells were plated on fibronectin and stimulated with soluble collagen I (10 μg/ml) for 1 or 4 hours. Lysates were probed for the indicated targets by western blotting. (B) mPLRB8 cells were stimulated for 4 hours with fibronectin, collagen I or collagen IV and the level of active Peak1 was determined by western blotting. (C) Activation of Peak1 was determined in Sparc-/- (mPLR6C) and Sparc+/+ (mPLRB8) KIC cells after 4 hours of collagen I stimulation. (D) Blockade of DDR1 reduces Peak1 activation. Sparc+/+:KIC cell line (B8) plated on 5 μg/ml fibronectin. Cells were stimulated with no treatment, collagen I, collagen I + anti-DDR1 (5 μg/ml) or collagen I + rSPARC (5 μg/ml). Peak1 activation was measured after 24 hours of stimulation. (E) Functional consequences of Ddr1 blockade. Sparc-/-:KIC clone (mPLR6C) and Sparc+/+:KIC clone (mPLRB8) were plated on plastic, 10 μg/ml fibronectin, or 10 μg/ml collagen I in the presence or absence of Ddr1 inhibitors (DDr1 blocking antibody or recombinant SPARC) and colony formation was determined. (F) Schematic displaying the proposed consequences of anti-VEGF therapy (1), which include reduced vascular function (2-4), enhanced collagen deposition (5-7) and increased tumor invasion (8-10). Epithelial like tumor cells (gold color) adopt a mesenchymal-like phenotype (olive color), a process that is increased by therapy-induced hypoxia and elevated collagen signaling. Error bars represent SEM (***, p < 0.0005 vs mPLR6C; ˆˆˆ, p < 0.0005 vs mPLRB8; ****, p < 0.00005 vs mPLR6C; ˆˆˆˆ, p < 0.00005 vs mPLRB8). One way ANOVA with Tukey’s MCT.
Discussion
The goal of this project was to assess the effect of anti-VEGF therapy on PDA and determine if changes in ECM contribute to tumor response to therapy. We found that the anti-VEGF mAb mcr84 reduced microvessel density, induced hypoxia, restrained tumor growth and prolonged survival, yet tumors from mcr84-treated mice exhibited epithelial plasticity and were more aggressive. We observed that VEGF inhibition increased Tgfβ-dependent collagen deposition and collagen I mediated signaling through Ddr1. Furthermore, we provide evidence that the recently identified kinase Peak1 is a signaling intermediate downstream of Ddr1 and that the matricellular protein Sparc reduces collagen I signaling through Ddr1. These findings highlight that collagen signaling through Ddr1 is a critical feature in the response of PDA to therapy and are summarized in Figure 6F.
Numerous reports (7, 26, 37-40) have identified increased tumor aggressiveness in murine models of cancer after anti-angiogenic therapy. In each of these studies, therapy induced hypoxia is a central mechanistic feature of the resulting tumor phenotype. Hypoxia-driven expression and/or activation of specific signaling cascades (e.g., c-Met, Axl, CTGF), recruitment of tumor promoting immune cells or induction of cell survival pathways including autophagy are supported by these and other reports (41-43). Our study is the first to show that hypoxia-induced driven changes in the ECM contribute to the aggressiveness of tumors post anti-VEGF therapy. Importantly, therapies that drive hypoxia do not always increase epithelial plasticity and tumor aggressiveness. For example, we have found that some anti-angiogenic strategies, including PG545, a heparanase inhibitor and BIBF1120, a multi-angiokinase inhibitor, potently increase hypoxia but do not increase epithelial plasticity or metastasis in pancreatic tumors in mice (20, 44). Interestingly, neither strategy increased collagen deposition.
Our studies relied on the use of the KIC model of PDA. One of the notable features of the KIC model is that the tumors often exhibit histologic features (sarcomatoid change) that are associated with aggressive clinical behavior in human cancers. Concordantly, PDAs arise in the KIC model with short latency, are desmoplastic and lethal (19, 24, 32). Tumors are first detected at 3-4 weeks of age and deaths from cancer occur by 8-10 weeks. These features make the KIC model particularly well-suited for endpoint and survival studies, which can be performed more rapidly and efficiently in this model than in other PDA GEMMs. Furthermore, human PDA displays a spectrum of histologies and response characteristics that mirror those of the KIC GEMM. Additionally, this model incorporates the two most common genetic lesions present in human PDA tumors (e.g., KRAS activation and INK4a loss).
We attempted to evaluate the contribution of Tgfβ to PDA progression in the context of anti-VEGF therapy. Our studies show that Tgfβ is a major driver of collagen deposition; however, Tgfβ has pleiotropic effects which include tumor suppression even in established KIC tumors. Thus blockade of Tgfβ increased tumor progression and reduced animal survival, results that are consistent with Hezel et al (32). Importantly, we found that Tgfβ blockade resulted in a reduction in metastatic burden in the liver as assessed by qPCR. We propose that inhibition of Ddr1 might be a functional alternative to direct Tgfβ inhibition. In support of this, we identified that collagen I signaling enhanced the tumorigenic properties of Tgfβ in vitro. Additionally, collagen signaling was reduced when Tgfβ was neutralized but was increased when Sparc was removed from the tumor microenvironment.
Sparc participates in the orchestration of collagen deposition but a unifying principle of Sparc function in the tumor microenvironment is lacking (45). Sparc is produced by multiple cell types (45), including stromal and tumor cells, though tumor cells often lose SPARC expression via hypermethylation (46, 47) and this correlates directly with poor chemoresponse (48). We found that Sparc blocked collagen I from binding to Ddr1, and that PDA tumors grown in Sparc-/- mice exhibited increased collagen signaling and enhanced disease progression. This is supported by the following observations: a) the amino acids that comprise the Sparc binding region on fibrillar collagens is the same sequence recognized by Ddr1 (34, 49); b) collagen is associated preferentially with cell surfaces instead of being deposited into the ECM in the absence of Sparc (50); c) Tgfβ-induced EMT is potentiated by collagen interaction with the cell surface (29); d) loss of Sparc expression by tumor cells correlates with the switch of Tgfβ from a suppressor to a promoter of PDA (46, 47); and e) pancreatic tumor cells are more aggressive in preclinical models of PDA in the absence of Sparc (51, 52). Further, these results are consistent with reports that collagen is associated with cellular processes that are prominent in PDA, including EMT and chemoresistance (13, 29). Activation of Ddr signaling is likely context dependent and may preferentially affect the cell that produces the collagen. Multiple cell types, including tumor cells, secrete collagen I in the tumor microenvironment. It is plausible that this collagen provides autocrine stimulation, especially in the absence of Sparc (50). In aggregate these studies suggest that inhibition of collagen signaling through the DDRs will enhance therapeutic response in PDA. Furthermore, SPARC inhibition of DDR activation by collagen provides a potential explanation for the observations that SPARC expression correlates with chemoresponse in PDA patients (48).
How Ddr1 promotes tumor progression is unclear; although we identified that collagen stimulated Ddr1 activation induces signaling through Pyk2 and Peak1. Peak1, a recently described kinase, has been implicated in invasion and tumor progression in colon and PDA xenografts (30). Inhibition of Peak1 or Ddr1 activity is an attractive strategy to potentially reduce collagen signaling in tumors. No Peak1 selective inhibitors have been described however the number of DDR selective small molecular inhibitors is increasing (53, 54). Some of these inhibitors including 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)-ethynyl) benzamide 7rh (7-4104) (53) have shown potent inhibition of colony formation and anchorage independent growth in tumor cells in vitro and favorable in vivo pharmacokinetic characteristics (53).
In summary, our findings demonstrate that collagen signaling enhances PDA, and suggest that VEGF-blockade can promote tumorigenicity of a subset of PDA cells by promoting hypoxia-induced collagen production and stimulation of Ddr1.
Supplementary Material
Acknowledgments
We gratefully acknowledge Jordan Braunfeld for technical assistance and members of the Brekken laboratory for helpful comments on the manuscript, as well as Richard Howdy at Visually Medical for preparation of the pathway figure. This work is supported in part by a sponsored research agreement from Affitech AS (RAB), the NIH (R01CA118240 to RAB; F31 CA168350 to KYA; U01CA141576 and R01CA137181 to DHC), and the Effie Marie Cain Scholarship for Angiogenesis Research (RAB).
Footnotes
Conflict of Interest Statement: R. A. Brekken is a consultant for, has equity interest in, and is a recipient of a sponsored research grant from Peregrine Pharmaceuticals and Affitech AS. R. A. Brekken is also an author of a patent on technology that was used to develop the antibody r84 by Peregrine Pharmaceuticals and Affitech. Peregrine and Affitech did not participate in the planning, execution, or interpretation of the experiments.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Roskoski R., Jr Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62:179–213. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320:130–7. doi: 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–87. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 7.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghaneh P, Costello E, Neoptolemos JP. Biology and management of pancreatic cancer. Gut. 2007;56:1134–52. doi: 10.1136/gut.2006.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korpanty G, Carbon JG, Grayburn PA, Fleming JB, Brekken RA. Monitoring response to anticancer therapy by targeting microbubbles to tumor vasculature. Clin Cancer Res. 2007;13:323–30. doi: 10.1158/1078-0432.CCR-06-1313. [DOI] [PubMed] [Google Scholar]
- 10.Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–6. doi: 10.1158/0008-5472.CAN-07-6705. [DOI] [PubMed] [Google Scholar]
- 11.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28:3617–22. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, Van Laethem JL, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27:2231–7. doi: 10.1200/JCO.2008.20.0238. [DOI] [PubMed] [Google Scholar]
- 13.Grzesiak JJ, Ho JC, Moossa AR, Bouvet M. The integrin-extracellular matrix axis in pancreatic cancer. Pancreas. 2007;35:293–301. doi: 10.1097/mpa.0b013e31811f4526. [DOI] [PubMed] [Google Scholar]
- 14.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shields MA, Dangi-Garimella S, Redig AJ, Munshi HG. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem J. 2012;441:541–52. doi: 10.1042/BJ20111240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan LA, Carbon JG, Roland CL, Toombs JE, Nyquist-Andersen M, Kavlie A, et al. r84, a novel therapeutic antibody against mouse and human VEGF with potent anti-tumor activity and limited toxicity induction. PLoS One. 2010;5:e12031. doi: 10.1371/journal.pone.0012031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradshaw AD, Puolakkainen P, Dasgupta J, Davidson JM, Wight TN, Helene Sage E. SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. The Journal of investigative dermatology. 2003;120:949–55. doi: 10.1046/j.1523-1747.2003.12241.x. [DOI] [PubMed] [Google Scholar]
- 18.Norose K, Clark JI, Syed NA, Basu A, Heber-Katz E, Sage EH, et al. SPARC deficiency leads to early-onset cataractogenesis. Investigative ophthalmology & visual science. 1998;39:2674–80. [PubMed] [Google Scholar]
- 19.Dineen SP, Roland CL, Greer R, Carbon JG, Toombs JE, Gupta P, et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 2010;70:2852–61. doi: 10.1158/0008-5472.CAN-09-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostapoff KT, Awasthi N, Cenik BK, Hinz S, Dredge K, Schwarz RE, et al. PG545, an angiogenesis and heparanase inhibitor, reduces primary tumor growth and metastasis in experimental pancreatic cancer. Mol Cancer Ther. 2013;12:1190–201. doi: 10.1158/1535-7163.MCT-12-1123. [DOI] [PubMed] [Google Scholar]
- 21.Dasch JR, Pace DR, Waegell W, Inenaga D, Ellingsworth L. Monoclonal antibodies recognizing transforming growth factor-beta. Bioactivity neutralization and transforming growth factor beta 2 affinity purification. J Immunol. 1989;142:1536–41. [PubMed] [Google Scholar]
- 22.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 23.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 24.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2:270–87. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22:21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA. Cellular response to hypoxia involves signaling via Smad proteins. Blood. 2003;101:2253–60. doi: 10.1182/blood-2002-02-0629. [DOI] [PubMed] [Google Scholar]
- 28.Lee YC, Lane KB, Zoia O, Thompson PJ, Light RW, Blackwell TS. Transforming growth factor-beta induces collagen synthesis without inducing IL-8 production in mesothelial cells. The European respiratory journal. 2003;22:197–202. doi: 10.1183/09031936.03.00086202. [DOI] [PubMed] [Google Scholar]
- 29.Shintani Y, Fukumoto Y, Chaika N, Svoboda R, Wheelock MJ, Johnson KR. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J Cell Biol. 2008;180:1277–89. doi: 10.1083/jcb.200708137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelber JA, Klemke RL. PEAK1, a novel kinase target in the fight against cancer. Oncotarget. 2010;1:219–23. doi: 10.18632/oncotarget.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croucher DR, Hochgrafe F, Zhang L, Liu L, Lyons RJ, Rickwood D, et al. Involvement of Lyn and the atypical kinase SgK269/PEAK1 in a basal breast cancer signaling pathway. Cancer Res. 2013;73:1969–80. doi: 10.1158/0008-5472.CAN-12-1472. [DOI] [PubMed] [Google Scholar]
- 32.Hezel AF, Deshpande V, Zimmerman SM, Contino G, Alagesan B, O’Dell MR, et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012;72:4840–5. doi: 10.1158/0008-5472.CAN-12-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carafoli F, Hohenester E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbapap.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carafoli F, Bihan D, Stathopoulos S, Konitsiotis AD, Kvansakul M, Farndale RW, et al. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure. 2009;17:1573–81. doi: 10.1016/j.str.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 36.Castro-Sanchez L, Soto-Guzman A, Navarro-Tito N, Martinez-Orozco R, Salazar EP. Native type IV collagen induces cell migration through a CD9 and DDR1-dependent pathway in MDA-MB-231 breast cancer cells. European journal of cell biology. 2010;89:843–52. doi: 10.1016/j.ejcb.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Carbone C, Moccia T, Zhu C, Paradiso G, Budillon A, Chiao PJ, et al. Anti-VEGF treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin Cancer Res. 2011;17:5822–32. doi: 10.1158/1078-0432.CCR-11-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011;108:3749–54. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamagishi N, Teshima-Kondo S, Masuda K, Nishida K, Kuwano Y, Dang DT, et al. Chronic inhibition of tumor cell-derived VEGF enhances the malignant phenotype of colorectal cancer cells. BMC Cancer. 2013;13:229. doi: 10.1186/1471-2407-13-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phan VT, Wu X, Cheng JH, Sheng RX, Chung AS, Zhuang G, et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc Natl Acad Sci U S A. 2013;110:6079–84. doi: 10.1073/pnas.1303302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu YL, Jahangiri A, Delay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–9. doi: 10.1158/0008-5472.CAN-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, et al. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29:5254–64. doi: 10.1038/onc.2010.268. [DOI] [PubMed] [Google Scholar]
- 44.Kutluk Cenik B, Ostapoff KT, Gerber DE, Brekken RA. BIBF 1120 (nintedanib), a triple angiokinase inhibitor, induces hypoxia but not EMT and blocks progression of preclinical models of lung and pancreatic cancer. Mol Cancer Ther. 2013;12:992–1001. doi: 10.1158/1535-7163.MCT-12-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnold SA, Brekken RA. SPARC: a matricellular regulator of tumorigenesis. J Cell Commun Signal. 2009;3:255–73. doi: 10.1007/s12079-009-0072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao J, Song J, Huang H, Li Z, Du Y, Cao J, et al. Methylation of the SPARC gene promoter and its clinical implication in pancreatic cancer. J Exp Clin Cancer Res. 2010;29:28. doi: 10.1186/1756-9966-29-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, Su GH, et al. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22:5021–30. doi: 10.1038/sj.onc.1206807. [DOI] [PubMed] [Google Scholar]
- 48.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hohenester E, Sasaki T, Giudici C, Farndale RW, Bachinger HP. Structural basis of sequence-specific collagen recognition by SPARC. Proc Natl Acad Sci U S A. 2008;105:18273–7. doi: 10.1073/pnas.0808452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris BS, Zhang Y, Card L, Rivera LB, Brekken RA, Bradshaw AD. SPARC regulates collagen interaction with cardiac fibroblast cell surfaces. Am J Physiol Heart Circ Physiol. 2011;301:H841–7. doi: 10.1152/ajpheart.01247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold SA, Rivera LB, Carbon JG, Toombs JE, Chang CL, Bradshaw AD, et al. Losartan Slows Pancreatic Tumor Progression and Extends Survival of SPARC-Null Mice by Abrogating Aberrant TGFbeta Activation. PLoS One. 2012;7:e31384. doi: 10.1371/journal.pone.0031384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech. 2010;3:57–72. doi: 10.1242/dmm.003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao M, Duan L, Luo J, Zhang L, Lu X, Zhang Y, et al. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J Med Chem. 2013;56:3281–95. doi: 10.1021/jm301824k. [DOI] [PubMed] [Google Scholar]
- 54.Kim HG, Tan L, Weisberg EL, Liu F, Canning P, Choi HG, et al. Discovery of a Potent and Selective DDR1 Receptor Tyrosine Kinase Inhibitor. ACS Chem Biol. 2013 doi: 10.1021/cb400430t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.