

Abstract
Androgen receptor (AR) signaling is critical in the development and progression of prostate cancer, leading to intensive efforts to elucidate all potential points of inflection for therapeutic intervention. These efforts have revealed new mechanisms of resistance and raise the possibility that known mechanisms may become even more relevant in the context of effective AR suppression. These mechanisms include tumoral appropriation of alternative androgen sources, alterations in AR expression, AR mutations, truncated AR variants, alterations and cross-talk in recruitment of co-factors to AR binding sites in the genome, and AR-driven oncogenic gene fusions. New agents such as enzalutamide, EPI-001, AR-specific peptidomimetics, novel HSP90 inhibitors and PARP inhibitors, as well as new approaches to co-targeting the AR pathway point to the potential for more complete and durable control of AR mediated growth.
Keywords: prostate cancer, androgens, androgen receptor
Background
Androgen Receptor Structure and Function in Prostate Cancer
Prostate cancer is the most common solid tumor and the second most common cause of cancer death in men in the United States, with over 29,000 men anticipated to die of metastatic disease in 2013(1). The androgen receptor (AR) is the critical driver of neoplastic prostate progression. Prostate cancer which has spread beyond the reach of definitive local therapy is treated with androgen deprivation therapy (ADT) to suppress AR activation.
The human AR, located on chromosome Xq11-12, is a nuclear receptor transcription factor structurally similar to other steroid hormone receptors. The AR is divided into distinct functional regions including the amino-terminal domain (NTD), DNA-binding domain (DBD), hinge-region (HR), and the carboxy-terminal ligand-binding domain (LBD). (Figure 1).
Figure 1.

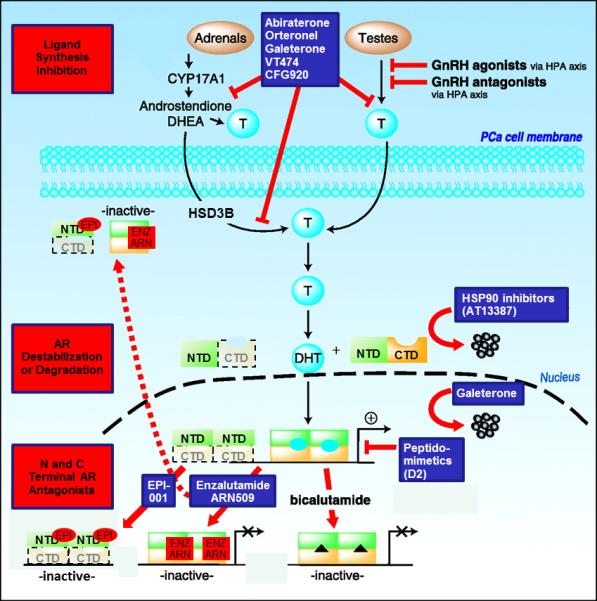
Figure A - Schematic of the full-length androgen receptor (a) and exon structure of major splice variants (ARV7 (b) and ARV567 (c)). Domains of AR include the amino (N) terminal domain, the DNA binding domain (DBD), the hinge region (HR), and the carboxy (C) terminal ligand-binding domain (LBD). Mutations which occur in specific regions are indicated. Adapted from Green and colleagues (68) with permission from Elsevier.
Figure 1B - ADVANCES: Inhibition of AR ligand production: Inhibition of CYP17A by Abiraterone, Orteronel, Galeterone, VT474, or CFG920 suppresses testicular, adrenal and tumoral androgen synthesis. Galeterone can suppress CYP17A, antagonize androgens and reduce AR levels. Abiraterone at high doses can inhibit AR and HSD3B. AR degradation: AR can be destabilized through inhibition of HSP90 binding. AR-cofactor association: Peptidomimetics (e.g. D2) can disrupt AR interaction with cofactors, decreasing AR driven gene transcription. AR antagonism: Enzalutamide and ARN509 block ligand/receptor interactions at the LBD, reduce AR-DNA association and AR nuclear accumulation. EPI-001 blocks AR NTD-coactivator association and nuclear accumulation, and is active against truncated AR variants lacking the C terminal domain (CTD). Adapted from Knudsen K, et al (69)
AR is activated by multiple steroid hormones, primarily testosterone (T) and dihydrotesterone (DHT) but also (at lower affinity) by adrenal androgens. Ligand binding releases receptor chaperones such as HSP90 and leads to nuclear translocation and receptor binding to androgen response elements (ARE). DNA binding induces formation of a signaling complex composed of coactivators and suppressors which then regulate cell type specific signaling
AR signaling normally promotes epithelial differentiation, but in prostate cancer AR modulates a broad array of genes regulating cell cycle, survival and proliferation driving tumor progression(2-5). Advanced prostate cancer is treated with androgen deprivation therapy (ADT), either as castration monotherapy or as combined therapy with AR antagonists. ADT induces nearly universal clinical responses; however, currently available agents do not achieve definitive tumor ablation and the majority of cancers become resistant to ADT. This phase of disease represents the lethal phenotype and carries significant morbidity and mortality within months to years. Despite anorchid testosterone blood levels, recapitulation of the intra-tumoral AR signaling pathway continues to drive progression and while previously considered ‘hormone refractory’, this phase is more appropriately considered “castration resistant” prostate cancer (CRPC).
Clinical-Translational Advances
Mechanisms of Resistance to AR Pathway Inhibition
Adaptive responses to ADT include tumoral appropriation of alternative androgen sources, alterations in AR expression, structural alterations in the AR including mutation and truncated AR variants, alterations in co-factor recruitment, and AR activation via cross-talk with signal transduction pathways(6). These ligand and AR-related alterations have been validated as important targets in CRPC based on the clinical efficacy of new agents designed to target them.
Tumor androgen levels in metastases from castrate patients exceed tissue androgen levels in primary prostate tumors from untreated patients(7). Potential non-gonadal sources of intra-tumor androgens include circulating adrenal androgens, as well as de novo or intracrine synthesis of androgens within prostate cancer cells(7-9). Abiraterone is a selective irreversible inhibitor of the steroidogenic enzyme CYP17 and suppresses serum and tissue androgen levels more effectively than standard ADT(10-12). Abiraterone in chemotherapy naïve and post-docetaxel treated CRPC provided survival and quality of life benefits, leading to FDA approval in both settings(13, 14), and supporting the importance of inhibiting non-gonadal androgen sources in CRPC.
CRPC tumors also respond to ADT by upregulating AR expression. While 20-30% of CRPC tumors demonstrate amplification of the AR locus, other means include increased transcription rates or stabilization of mRNA or protein(15). Increased AR expression contributes to prostate cancer growth by compensating for castrate androgen levels, and in PCa models was both necessary and sufficient to induce tumor growth(16). Novel AR antagonists have, accordingly, demonstrated encouraging clinical efficacy in CRPC. Enzalutamide (formerly MDV3100) is a competitive AR antagonist which binds AR with 5-8 fold greater affinity than other anti-androgens, decreases AR nuclear translocation, and reduces chromatin occupancy at canonical ARE’s(17). Unlike bicalutamide, enzalutamide did not demonstrate agonist potential in vitro, and retained activity against AR mutations associated with bicalutamide resistance (W741C). In a phase III randomized (AFFIRM) study enzalutamide improved overall survival by 37% compared to placebo in men with CRPC previously treated with docetaxel(18), confirming that therapy directly targeting AR provided clinical benefit and confirming the relevance of this mechanism of resistance in CRPC(19). A phase III study of enzalutamide (PREVAIL, NCT01212991) in Docetaxel-naive men has completed accrual with interim results pending.
Insights into mechanisms of resistance mediated by AR signaling led to rapid completion of phase III studies culminating with FDA approval of abiraterone and enzalutamide. These observations fundamentally changed the landscape of CRPC, credentialing the AR signaling axis as the most relevant target in CRPC, and emphasizing that novel approaches to co-targeting AR and the networks on which it depends is an important focus moving forward.
AR Adaptations in CRPC – Novel Insights and Biology
AR Mutations
Under the pressure of androgen deprivation and AR antagonism the AR is susceptible to both somatic mutation and aberrant transcription. Specific mechanisms of resistance have been associated with mutations in specific regions of the receptor, including broadening of ligand specificity and conversion of antagonists to agonists (Figure 1)(20, 21). In vitro selection with enzalutamide has revealed a new mutation (F876L) which mediates conversion of enzalutamide to an AR agonist(22-24), while maintaining sensitivity to first generation agents (e.g. bicalutamide). The frequency of AR mutation in CRPC tumors treated with LHRH agonist and first generation AR antagonists is low (8-25%)(25). However the frequency of these mutations may become a more significant mechanism of resistance in context of more complete suppression of AR signaling.
AR Splice Variants
An alternative response to ADT is induction of AR variants (ARVs) with deletion of the LBD resulting in ligand-independent constitutive activity(26-31) (Figure 1). ARVs homo- and heterodimerize with full length AR, promoting nuclear localization and increasing AR signaling in absence of ligand(30). Variants most commonly identified in CRPC tumors, ARV7 and ARV567, have both unique and overlapping transcriptional programs compared to wild-type AR(32). While ARVs lacking the LBD clearly drive ligand-independent growth when evaluated in prostate cancer models, the specific role of ARVs in prostate cancer development and progression is still under debate. That ARV7 is found in normal prostate epithelium and associates with a shorter time to recurrence after prostatectomy, suggests variants play a role in prostate cancer pathogenesis(28, 29). Transgenic expression of ARV567 in prostate epithelium leads to adenocarcinoma by 50 weeks, also consistent with a potential role for ARVs in mediating prostate carcinogenesis (S. Plymate, in preparation). High levels of ARV7 and ARV567 were associated with shorter survival in patients with CRPC and bone metastases, consistent with a role in tumor progression(26, 28, 29). Notably, a subgroup of bone metastases demonstrated nearly equivalent protein levels of full-length and truncated AR variants by western blot. Rapid induction of ARVs following castration may itself facilitate prostate tumor survival, or may serve a bridging role until induction of additional tumor growth mechanisms(33). Alternatively, castration may promote outgrowth of ligand-independent tumor cell clones in which ARV expression is mediated by genomic rearrangement of AR(34).
AR Nuclear Transport
AR Nuclear localization occurs via binding to the microtubule-based dynein motor (via a binding site in exon 4 of the canonical NLS)(35). While the primary mechanism of taxane activity in CRPC is microtubule stabilization and disruption of cellular division, taxanes also inhibit microtubule-mediated nuclear AR transit, and cytoplasmic sequestration of AR in circulating tumor cells (CTC’s) correlated with clinical response to taxane therapy(36). Importantly, LBD-deleted ARVs may demonstrate differential sensitivity to taxane-mediated nuclear exclusion. While the nuclear transit of ARV567 (which retains exon 4) is taxane sensitive, the NLS of AR-V7 is located in the cryptic exon and is unique from that of full length AR(37). Accordingly, ARV7 is not susceptible to taxane-mediated nuclear exclusion(38). Thus, AR antagonists targeting the NTD may enhance taxane-efficacy in ARV expressing tumors. This may be of particular relevance in abiraterone and enzalutamide-resistant tumors, as pre-clinical data demonstrate that AR variants may contribute to resistance to these agents, and emerging clinical data suggests these patients have diminished sensitivity to docetaxel(39).
Nuclear Receptor Superfamily Participation in AR Signaling
Under selection pressure the AR can broaden ligand specificity to include ligands of the closely related steroid receptor superfamily (e.g. progesterone, cortisol). Conversely, members of the nuclear receptor super-family may be able to maintain AR signaling in androgen-deprived environments by inducing a cistrome (the genome-wide locations of a transcription factor’s binding-sites) which closely resembles the AR cistrome. The glucocorticoid receptor (GR), has been shown to share response elements with AR in multiple gene targets, and activates a transcriptional program which largely overlaps with that induced by AR(40). The pioneering transcription factor FOXA1 regulates differential binding of GR or AR to these targets, potentially functioning as a critical regulator of GR function in prostate cancer(40). Preliminary studies suggest GR may play an important role in resistance to second generation AR antagonists, as in vitro selection of enzalutamide-resistant cells revealed dramatically upregulated GR levels, and GR knockdown partially abrogated resistance to enzalutamide in these cells grown as xenografts(41). An analysis of AFFIRM, the phase III study of enzalutamide in post-docetaxel patients, suggested use of glucocorticoids was associated with inferior survival (independent of other known prognostic factors) and may be driving an adverse biology(42). A similar analysis in COU-301, a phase III study of abiraterone in the same patient population, suggested that glucocorticoids were used in patients with greater comorbidity and worse prognosis disease, potentially explaining the inferior outcomes(43). Whether GR biology drives progression in CRPC will require additional analysis of clinical samples and the impact of glucocorticoid use in patients with CRPC.
AR-Driven ETS Fusion Genes
The most common mutation in prostate cancer is fusion of the AR regulated protease TMPRSS2 with members of ETS family of transcription factors(44). The most common AR-driven fusion, TMPRSS2:ERG, induces high level expression of ERG which associates endogenously with PARP1 (which ribosylates proteins involved in DNA mismatch repair)(45), and both ERG mediated transcriptional activity and cell invasion require PARP1 activity(46). Targeting of PARP in tumors deficient in other components of DNA repair such as BRCA1/BRCA2 induces significant clinical responses, a result of presumed “synthetic lethality” resulting from complete abrogation of DNA repair(47). Notably, ETS transcription factors including ERG drive DNA double-strand break formation, and concurrent inhibition of PARP in ERG overexpressing cells further increases DNA damage. This finding raises the potential that co-targeting the AR pathway with inhibitors of PARP in ERG fusion positive prostate tumors may reconstitute synthetic lethality, even in tumors which are BRCA1/2 mutation negative(46).
Clinical Implications
Clinically, resistance to first and second generation AR targeting agents is universally associated with reactivation of AR signaling, as manifested by a rising PSA. Induction of resistance to abiraterone and enzalutamide in preclinical models also remains largely dependent on AR signaling, either via induction of steroidogenesis, or upregulated expression of AR and truncated ARVs, and complete abrogation of AR function has yet to be realized(48-50). Leveraging previously unexplored biology, exploring rational combinations earlier in the course of disease, and improving efficacy of steroidogenesis inhibitors and AR antagonists all have potential to improve outcomes(51). One example of novel targeting includes inhibiting the AR NTD which has resisted drug targeting because of difficulty crystallizing its highly disordered structure. Targeting the NTD with novel agents carries the potential to address constitutively active AR (driven by mutation or splice variants affecting the LBD), as well as AR amplification. Screens of libraries for NTD binding agents identified EPI-001, a nonsteroidal compound which binds the NTD and inhibits the growth of castration sensitive cell lines(52, 53). Multiple analogs of EPI-001 have been developed with greater anti-tumor efficacy in both sensitive and castration resistant xenograft models, including those driven by ARV expression(52).
Alternative approaches include concurrent targeting of AR and steroidogenesis production. Drawing from experience in treating infectious diseases such as HIV and tuberculosis, combining effective agents with complementary mechanisms of action may provide more durable disease control through rapid diminution of the cellular pool available to undergo mutation in response to treatment stress(54). This provides rationale for testing combinations of abiraterone and second generation AR antagonists such as enzalutamide and ARN-509 in multiple stages of prostate cancer, from metastatic CRPC to treatment naïve, high risk localized disease (Table 1). ARN-509 is structurally similar to enzalutamide, with similar mechanism of action and activity in vitro, but superior activity in xenograft experiments(55). Phase I/II studies of ARN-509 as a single agent have been recently completed with results pending.
The same rationale underlies testing of single agents with activity against multiple sites of the AR signaling axis. Galeterone (TOK-001/VN-124-1) was identified through library screening for combination inhibitors of CYP17 and AR(56). In vitro it suppresses AR levels, providing a separate potential avenue for inhibiting ARV or mutant AR driven tumors(57, 58). A phase I study in chemotherapy-naïve CRPC demonstrated 50% PSA reduction in 22% of patients, with a 50% decline at the highest dose level. The maximally tolerated dose was not reached, with the most common side effects being fatigue, liver function test (LFT) abnormalities, pruritus, nausea, and diarrhea. A phase II study (ARMOR2) is currently ongoing in multiple cohorts of patients with CRPC (Table 1). Interestingly, abiraterone has also been found to exhibit weak antagonism against both wild type and various mutant androgen receptors, including those activated by exogenous glucocorticoids (T877A) and bicalutamide (W741C)(59), providing a potential rationale for dose escalation.
Table 1.
Actively recruiting clinical trials with AR targeting and co-targeting in CRPC (clinicaltrials.gov)
| Target | Drugs | Design | NCT# |
|---|---|---|---|
| AR and CYP17 | Enzalutamide and abiraterone | Phase I/II | NCT01650194 |
| AR and CYP17 | Galeterone | Phase II | NCT01709734 |
| AR and CYP17 | ARN−509 and Abiraterone | Phase Ib | NCT01792687 |
| HSP90 and CYP17 | AT13387 and Abiraterone | Phase I/II | NCT01685268 |
| AR and VEGFR | Enzalutamide and Tivozanib | Phase II single arm | NCT01885949 |
| AR and CYP17 | CFG920 | Phase I/II | NCT01647789 |
| AR and PARP | Velaparib and abiraterone | Phase II | NCT01576172 |
Another strategy is targeting AR-coregulator interactions. A recent study reports a peptidomimetic (small organic molecules without a peptide backbone) that mimics the LXXLL motif found in AR co-regulators. The compound, D2, disrupted binding of AR to proteins such as PELP1 (which plays a scaffolding role for assembly of the AR transcriptional complex), prevented androgen-induced nuclear translocation comparable to that obtained with enzalutamide, and inhibited growth of AR positive PCa cells in vitro and in vivo(60).
Agents targeting suppression of the AR protein, such as inhibitors of the AR chaperone HSP90, carry potential to completely abrogate AR signaling, independent of tissue ligand levels, AR mutation or ARV structure(61, 62). Next generation HSP90 inhibitors show activity in preclinical models and have significant advantages over agents such as geldenamycin, including improved potency and lack of requirement for activation by enzymes such as diaphorase, which are not highly expressed in prostate tissue(63). Phase I studies with newer agents show promising activity against heavily pretreated CPRC, and phase II studies as monotherapy and in combination with abiraterone in patients with abiraterone-refractory CRPC are ongoing (Table 1).
Conclusions
Perhaps the most significant challenge to defining optimal sequencing and combinations of next generation of agents is the heterogeneity of CRPC. At inception of CRPC, molecular profiling of the AR-axis reveals specific patterns of AR pathway components. Some tumors are positive for steroidogenic machinery in conjunction with upregulated AR suggesting modulation by ligand, others show no evidence for steroidogenic potential but only upregulation of AR, suggesting modulation by AR itself, and a smaller number lack any signature for upregulation of ligand or receptor(64, 65). Efforts to characterize molecular phenotype of metastatic prostate cancers, such as the SU2C/AACR/PCF sequencing project (66)will be critical to defining which pathways are relevant for targeting in abiraterone and enzalutamide refractory cancers. In addition, correlating tissue biopsy with a common platform for analyzing circulating tumor cells and cell free DNA, as means of noninvasively sampling relevant biology, will await adoption of an assay which simultaneously detects and efficiently collects samples for analysis(67). Using tumor biopsy to enrich patient populations, as is being utilized in the study of PARP inhibition in TMPRSS2:ERG positive and negative tumors (NCT01576172 – Table 1), will be critical to moving novel combination therapies to the patients who need them as rapidly as possible(54).
Acknowledgments
Research Support: National Institutes of Health (Pacific Northwest Prostate Cancer SPORE) (EAM, SP, RBM), Prostate Cancer Foundation (EAM and RBM), Damon Runyon Cancer Research Foundation (EAM), and Department of Defense Congressionally Directed Medical Research Program (EAM, SP, RBM).
Footnotes
Disclosure Statement: R.B.M. has received research funding from Janssen, Medivation, Tokai, and Novartis. S.R.P.served as a compensated consultant to ESSA.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen KE, Arden KC, Cavenee WK. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J Biol Chem. 1998;273:20213–22. doi: 10.1074/jbc.273.32.20213. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–92. doi: 10.1158/0008-5472.CAN-05-4472. [DOI] [PubMed] [Google Scholar]
- 5.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–71. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013 doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 9.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 11.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–8. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostaghel EA, Marck B, Plymate S, Vessella RL, Balk SP, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logothetis CJ, Basch E, Molina A, Fizazi K, North SA, Chi KN, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. The lancet oncology. 2012;13:1210–7. doi: 10.1016/S1470-2045(12)70473-4. [DOI] [PubMed] [Google Scholar]
- 15.Shiota M, Yokomizo A, Naito S. Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. Journal of molecular endocrinology. 2011;47:R25–41. doi: 10.1530/JME-11-0018. [DOI] [PubMed] [Google Scholar]
- 16.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 17.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 19.Hoffman-Censits J, Kelly WK. Enzalutamide: a novel antiandrogen for patients with castrate-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1335–9. doi: 10.1158/1078-0432.CCR-12-2910. [DOI] [PubMed] [Google Scholar]
- 20.Taplin ME. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol. 2007;4:236–44. doi: 10.1038/ncponc0765. [DOI] [PubMed] [Google Scholar]
- 21.Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Curr Genomics. 2009;10:18–25. doi: 10.2174/138920209787581307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, et al. An F876L Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to MDV3100 (Enzalutamide) Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 23.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to 2nd generation anti-androgens enzalutamide and ARN-509. Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 24.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taplin ME. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol. 2007;4:236–44. doi: 10.1038/ncponc0765. [DOI] [PubMed] [Google Scholar]
- 26.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011 doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. 2013 doi: 10.1038/onc.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–67. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71:6019–29. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thadani-Mulero M, Nanus DM, Giannakakou P. Androgen receptor on the move: boarding the microtubule expressway to the nucleus. Cancer Res. 2012;72:4611–5. doi: 10.1158/0008-5472.CAN-12-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–49. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matov A, Nanus DM, Plymate SR, P G. Portella L, Thadani-Mulero M, Matov A, Nanus DM, Plymate SR, Giannakakou P. AACR Annual Meeting; 2013. L P, M. T-M. 2013. [Google Scholar]
- 39.Fitzpatrick JM, de Wit R. Taxane Mechanisms of Action: Potential Implications for Treatment Sequencing in Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2013 doi: 10.1016/j.eururo.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 40.Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. The EMBO journal. 2011;30:3962–76. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawyers CL. Overcoming Resistance to Cancer Drug Therapy; ASCO Annual Meeting; 2013. 2013. [Google Scholar]
- 42.Scher HI, Fizazi K, Saad F, Chi K, Taplin ME, Sternberg CN, et al. Association Of Baseline Corticosteroid With Outcomes In A Multivariate Analysis Of The Phase 3 Affirm Study Of Enzalutamide (ENZA), An Androgen Receptor Signaling Inhibitor (ARSI) ESMO. 2012 2012. [Google Scholar]
- 43.Montgomery B, Kheoh S, Molina A, Li J, Bellmunt J, Ryan CJ, et al. Effect of corticosteroid (CS) use at baseline (CUB) on overall survival (OS) in patients (pts) receiving abiraterone acetate (AA): Results from a randomized study (COU-AA-301) in metastatic castration-resistant prostate cancer (mCRPC) post-docetaxel (D); 2013 ASCO Annual Meeting 2013. [Google Scholar]
- 44.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 45.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer discovery. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer cell. 2011;19:664–78. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 48.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferraldeschi R, Attard G, de Bono JS. Novel strategies to test biological hypotheses in early drug development for advanced prostate cancer. Clin Chem. 2013;59:75–84. doi: 10.1373/clinchem.2012.185157. [DOI] [PubMed] [Google Scholar]
- 52.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013 doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 54.Glickman MS, Sawyers CL. Converting cancer therapies into cures: lessons from infectious diseases. Cell. 2012;148:1089–98. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48:2972–84. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- 57.Soifer HS, Souleimanian N, Wu S, Voskresenskiy AM, Collak FK, Cinar B, et al. Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. The Journal of biological chemistry. 2012;287:3777–87. doi: 10.1074/jbc.M111.261933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, et al. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Molecular cancer therapeutics. 2008;7:2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K, et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72:2176–82. doi: 10.1158/0008-5472.CAN-11-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley WD, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nature communications. 2013;4 doi: 10.1038/ncomms2912. 1923. [DOI] [PubMed] [Google Scholar]
- 61.Centenera MM, Fitzpatrick AK, Tilley WD, Butler LM. Hsp90: still a viable target in prostate cancer. Biochim Biophys Acta. 2013;1835:211–8. doi: 10.1016/j.bbcan.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Gillis JL, Selth LA, Centenera MM, Townley SL, Sun S, Plymate SR, et al. Constitutively-active androgen receptor variants function independently of the HSP90 chaperone but do not confer resistance to HSP90 inhibitors. Oncotarget. 2013;4:691–704. doi: 10.18632/oncotarget.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Centenera MM, Gillis JL, Hanson AR, Jindal S, Taylor RA, Risbridger GP, et al. Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin Cancer Res. 2012;18:3562–70. doi: 10.1158/1078-0432.CCR-12-0782. [DOI] [PubMed] [Google Scholar]
- 64.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142–52. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett NC, Hooper JD, Lambie D, Lee CS, Yang T, Vesey DA, et al. Evidence for steroidogenic potential in human prostate cell lines and tissues. Am J Pathol. 2012;181:1078–87. doi: 10.1016/j.ajpath.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 66.Stand Up To Cancer Dream Team 2013 [cited 2013 September 17]; Available from: http://www.standup2cancer.org/dream_teams/view/precision_therapy_for_advanced_prostate_cancer)
- 67.Danila DC, Pantel K, Fleisher M, Scher HI. Circulating tumors cells as biomarkers: progress toward biomarker qualification. Cancer J. 2011;17:438–50. doi: 10.1097/PPO.0b013e31823e69ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Green SM, Mostaghel EA, Nelson PS. Androgen action and metabolism in prostate cancer. Mol Cell Endocrinol. 2012;360:3–13. doi: 10.1016/j.mce.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–8. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]