

Abstract
Purpose
Because of its high expression on various types of tumors and its restricted distribution in normal tissues, chondroitin sulfate proteoglycan-4 (CSPG4) represents an attractive target for the antibody-based therapy of several solid tumors. We tested whether T cells transduced with a CSPG4-specific chimeric antigen receptor (CAR) inhibited the growth of CSPG4-expressing tumor cells both in vitro and in vivo.
Experimental Design
We first independently validated by immunohistochemistry (IHC) the expression of CSPG4 in an extensive panel of tumor arrays and normal tissues as well as queried public gene expression profiling datasets of human tumors. We constructed a second generation CSPG4-specific CAR also encoding the CD28 costimulatory endodomain (CAR.CSPG4). We then evaluated human T lymphocytes expressing this CAR for their ex vivo and in vivo anti-tumor activity against a broad panel of solid tumors.
Results
IHC showed that CSPG4 is highly expressed in melanoma, breast cancer, head and neck squamous cell carcinoma (HNSCC) and mesothelioma. In addition, in silico analysis of microarray expression data identified other important potential tumors expressing this target, including glioblastoma, clear cell renal carcinoma and sarcomas. T lymphocytes genetically modified with a CSPG4-CAR controlled tumor growth in vitro and in vivo in NSG mice engrafted with human melanoma, HNSCC and breast carcinoma cell lines.
Conclusions
CAR.CSPG4-redirected T cells should provide an effective treatment modality for a variety of solid tumors.
Introduction
Chondroitin sulfate proteoglycan-4 (CSPG4), also known as high molecular weight-melanoma associated antigen (HMW) and melanoma-associated chondroitin sulfate proteoglycan (MCSP), is a well characterized cell surface proteoglycan first identified on human melanoma cells (1). Subsequent studies showed it to be highly expressed on other solid tumors such as mesothelioma (2) and triple negative breast carcinoma (3) all of which often show an aggressive clinical course. In contrast, CSPG4 has a restricted distribution in normal tissues (4).
CSPG4 participates in tumor migration, invasion, angiogenesis, and metastasis (5). It interacts with α4β1 integrins to directly modulate cell adhesion, motility and metastasis as demonstrated by its ectopic expression in tumor cells (6). Given its restricted expression in normal tissues, high expression on various types of solid tumors and its role in the biology of tumor cells, CSPG4 is an attractive target for immunotherapy.
CSPG4 has been targeted with monoclonal antibodies (mAbs) in models of melanoma, mesothelioma, and breast carcinoma, resulting in the inhibition of tumor growth and survival in addition to thwarting the metastatic capability of tumor cells (7). Recent advances in potentiating the antitumor effects of a specific mAb rely on coupling its antigen-binding specificity with the effector function and long-term persistence of T lymphocytes to generate specific chimeric antigen receptors (CARs) (8–10). These molecules are obtained by fusing the extracellular antigen-binding domain of the mAb with the intracellular signaling domains derived from the CD3-ζ chain of the T-cell receptor, in tandem to costimulatory endodomains to support survival and proliferative signals (11–13). Since CAR-modified T cells function independently of a patient’s MHC and can readily be generated for clinical use (14–16), the value of targeting CSPG4 with a CAR based-approach is appealing.
We first validated the expression of CSPG4 in an extensive panel of tumor arrays and normal tissues as well as queried public gene expression profiling datasets of human tumors and confirmed its broad expression. We then generated a CSPG4-specific CAR (CAR.CSPG4) and showed that when expressed by T cells, not only was melanoma effectively targeted in vitro, as previously demonstrated (17), but antitumor activity was observed in vitro and in vivo against many solid tumors including breast carcinoma, HNSCC and mesothelioma. Redirecting T cells to CSPG4 using CARs may thus represent a robust platform to target multiple solid tumors.
Materials and Methods
Cell lines
The previously described SENMA, CLB and P1143 tumor cell lines were generated in our laboratory from melanoma biopsies (18). MDA-MB-231 was originally obtained from American Type Culture Collection (ATCC) and authenticated by the analysis of short tandem repeat sequences performed at MD Anderson Cancer Center, Texas, USA. UACC-812, PCI-30 and PHI cell lines were provided by Dr Ferrone and these cells, when maintained in culture for several passages, retained the same phenotypic expression of CSPG4 as the early cell passages. Previously described melanoma cell lines PLAODE, NE-18732, NE-18588, NE-8959, NE-4405 and NE-371952 were only used to confirm the expression of CSPG4 in a broad array of melanoma cell lines (18). All these cells, including SENMA, CLB, and P1143, when maintained in culture for several passages, retained the same phenotypic expression of CSPG4 as the early cell passages. SENMA, CLB, UACC-812, MDA-MB-231, and PCI-30 cell lines were cultured in DMEM (Invitrogen Grand Island, NY) or RPMI 1640 (P1143, UACC-812, and PHI) (Cambrex, East Rutherford, NJ) medium supplemented with 10% heat inactivated fetal calf serum (FCS) (HyClone, Thermo Fisher Scientific Inc., Wyman, MA), 200 IU/mL penicillin, 200 mg/mL streptomycin (Invitrogen), and 2 mmol/L GlutaMAX (Invitrogen) at 37°C in a 5% CO2 atmosphere. Tumor cell lines were transduced with a gamma retroviral vector encoding eGFP to obtain GFP+ tumor cells (>98% GFP+). Primary epithelial cells from normal small airway, kidney and prostate were purchased from ATCC and kept in culture according to ATCC recommendation.
Tissue microarrays and Immunohistochemistry (IHC)
Antigen retrieval was performed by placing the samples in 1X Dako Citrate Buffer followed by incubation at 90°C in a pressure cooker for 45 minutes. After blocking with normal goat serum diluted in Tris-Buffered Saline, samples were incubated with the CSPG4 mAb (Abcam, Anti-NG2 antibody [LHM 2], ref# ab104535) (1:300 dilution) either overnight at 4°C or at room temperature for 1 hour. Detection of CSPG4 was then assessed using the VECTASTAIN® ABC kit (Vector Laboratories, Inc. ref# PK-4001) following the manufacturer’s protocol. Tissue arrays were obtained from Cybrdi Inc (Rockville, MD) for breast cancer (CC08-10-001) and HNSCC (CC34-01-001), while melanoma (ME2082b), neuroblastoma (MC602), mesothelioma (T392), and normal tissue (FDA 808b) arrays were obtained from US Biomax, Inc. (Rockville, MD). Each array contained a range of 4 to 192 cores of tumor or 5 to 18 cores of normal tissue samples in duplicate, triplicate or quadruplicate in the case of mesothelioma. Expression of CSPG4 in tumor cells was scored in blind fashion by the pathologist Dr Michael Ittmann based on both intensity (0–3+) and extent of staining (1–3+). A multiplicative staining score was calculated by multiplying the intensity and extent scores to yield scores on a 10 point scale from 0–9 (19). In microarrays with multiple cores per patient, the individual scores were averaged to obtain a final score. In some cores, tumor was not identified due to artifacts. In the vast majority of cases, IHC showed uniform staining (3+) within a given core and in most cases cores from different patients were highly concordant. Areas of necrosis or acellular keratin were not included in the scoring. Cases were divided based on staining scores into three groups: negative/weak (0–3), moderate (4–6) or strong (7–9).
Generation of the CSPG4-specific CAR and transduction of T lymphocytes
The hybridoma 763.74 was generated from a BALB/c mouse immunized with cultured human melanoma cells (20). The scFv 763.74 was isolated from the hybridoma (21) and then cloned in frame with the human IgG1-CH2CH3 domains, the CD28 costimulatory endodomain and the CD3ζ chain into the SFG retroviral backbone (CAR.CSPG4), as previously described (22). The control CAR specific for the CD19 antigen (CAR.CD19) has been previously described (13). Transient retroviral supernatant was generated by co-transfection of 293T cells with the RD114 envelope (RDF plasmid), the MoMLV gag-pol (PegPam3-e plasmid) and the retroviral vector, as previously described (23). For the generation of CAR-T cells, peripheral blood mononuclear cells (PBMCs) were isolated from buffy coat preparations (Gulf Coast Regional Blood Center, Houston, TX) using Ficoll-Paque (Amersham Biosciences, Piscataway, N.J.). PBMCs were activated with OKT3 and CD28 (BD Biosciences PharMingen, San Diego, CA) mAbs, transduced with the retroviral supernatant by day 3 of culture and then expanded in complete medium containing 45% RPMI 1640 and 45% Click’s medium (Irvine Scientific, Santa Ana, CA, USA) supplemented with 10% FCS, 100 IU/mL penicillin, 100 mg/mL streptomycin, and 2 mmol/L GlutaMAX. Cells were fed with IL-2 (50 U/mL) (PeproTech; Rocky Hill, NJ) twice a week for 2 weeks (23).
Flow cytometry
Conjugated CD3, CD4, CD8, CD45RO, CD62L and CCR7 mAbs (BD Biosciences) were used to identify T lymphocytes, while the CSPG4 mAb (Miltenyi-Biotech Inc, Auburn, CA) was used to label tumor cells. CAR expression in T lymphocytes was assessed using an antibody recognizing the human IgG1-CH2CH3 fragment (Jackson ImmunoResearch, West Grove, PN) (24). Analyses were performed on a FACsCaliber flow cytometer using the BDFACs CellQuestPro software (BD Biosciences, San Jose, CA).
Cytotoxicity and Co-culture Assays
The cytotoxic activity of control and CAR.CSPG4+ T lymphocytes was determined using a standard 51Cr release assay at different effector-to-target (E:T) (40:1, 20:1, 10:1 and 5:1) ratios using a gamma counter (Perkin-Elmer, Waltham, MA)(25). For the co-culture experiments, control and CAR.CSPG4+ T lymphocytes were plated at 1 × 106 cells/well in 24-well plates at different E:T ratios according to the kinetic growth of each tumor cell line. Tumor cell lines with a slow kinetic growth were plated at higher tumor ratio (T cells: tumor cells 3:1) compared to tumor cell lines with a fast kinetic growth (T cells: tumor cells 5:1). Supernatant was collected at 24 hours of culture to measure IFNγ and IL-2 release using specific ELISAs (R&D system, Minneapolis, MN). Following 72 hours of culture at 37° C, adherent tumor cells and T cells were collected and residual tumor cells and T cells assessed by FACs analysis based on GFP and CD3 expression, respectively.
Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) Assay
One week post transduction, control and CAR.CSPG4+ T lymphocytes were labeled with 1.5 μmol/L CFSE (Invitrogen) and plated with irradiated tumor target (SENMA) at an E:T ratio of 5:1. CFSE dilution was measured on CD4+ and CD8+ cells by flow cytometry by day 4 of co-culture.
Xenogenic mouse models
In vivo experiments were performed in accordance with Baylor College of Medicine’s Animal Husbandry guidelines. Antitumor activity of control and CAR.CSPG4+ T lymphocytes was evaluated using NOG/SCID/γc−/− mice (Jackson Lab, Bar Harbor, ME) engrafted with tumor cells. Eight to 9 week old mice were subcutaneously injected with 0.5 × 106 SENMA, 3 × 106 UACC-812 or 3 × 106 PCI-30 cells resuspended in Matrigel (BD Biosciences, San Jose, CA). On days 4, 6, and 8 following tumor cell injection, 1 × 107 control or CAR.CSPG4+ T lymphocytes were injected i.v. by tail vain. In summary, for the melanoma xenograft model 3 different preparations of CAR.CSPG4 T cells were generated from 3 different donors. Three doses, given two days apart, of 1 × 107 were infused i.v. into 5 mice per group. In total, 15 animals were treated for each group. The endpoint of the experiment was to examine differences in tumor volume up to day 30-post tumor injection. For the xenograft models of breast cancer and HNSCC 2 different preparations of T cells generated from 2 different donors were used. Two doses, given two days apart, of 1 × 107 were infused i.v. into 5 mice per group. In total, 10 animals were treated per group. In all tumor models, mice were sacrificed at 30 days or in accordance with our institution’s guidelines for the handling of sick animals. Weekly manual caliper measurements were performed post-treatment to evaluate tumor growth. Tumor volume was calculated using the modified ellipsoidal formula: .
Statistical Analysis
In vitro data are presented as mean ± standard deviation (SD) and a paired student’s t-test was used to determine statistical significance. The in vivo data are presented as mean ± standard error of the mean (SEM) and a paired student’s t-test was used to identify significant differences between CAR-treated and control-treated groups. Public gene expression profiling datasets of human tumors were queried for CSPG4, including data from The Cancer Genome Atlas (TCGA Data Portal; http://tcga-data.nci.nih.gov/tcga), Bittner multi-cancer dataset (unpublished, from www.oncomine.org)(26, 27) and GeneAtlas U133A data set (http://niogps.org).
Results
CSPG4 is expressed on a variety of solid tumors
As CSPG4 was originally identified as a melanoma associated antigen, we first independently validated its expression using IHC in a melanoma tissue array containing multiple primary cutaneous and visceral melanomas and metastatic lesions. Examples of either strong or negative/low staining are shown in Fig. 1A. Consistent expression of the antigen was documented in all types of lesions, regardless of their primary or metastatic origin, or their cutaneous and visceral source. We therefore analyzed melanomas as a whole group. Overall, 59% of melanomas showed strong staining and 25% displayed moderate staining (Fig. 1B). We then extended the analysis to include multiple samples of additional solid tumors including breast cancer, HNSCC, neuroblastoma, and mesothelioma. For the breast cancer array, staining was seen in invasive ductal and lobular carcinomas as well as in the small number of Paget’s disease and ductal in situ carcinoma cases present on the array. Staining for representative invasive ductal carcinomas (either strong or negative/low) are shown in Fig. 1A and summarized in Fig. 1B, while other lesions were not sufficient in number for a comparative analysis. Staining for CSPG4 in invasive ductal carcinoma was remarkable with 77% of cases showing moderate or strong staining. HNSCC most predominantly (50%) expressed moderate staining for CSPG4, with only 20% showing strong staining. Although neuroblastoma exhibited the weakest overall staining, there was still a fraction of cases with moderate to strong expression. Finally, despite the limited number of mesotheliomas, these lesions all consistently expressed CSPG4. We concluded that, at the protein level, all these malignancies exhibited variable but in most cases significant expression of CSPG4. To examine the expression of CSPG4 in a broader array of tumors we examined publically available databases for mRNA expression data. As shown in Fig. 1C, examination of TCGA datasets showed over expression of CSPG4 transcripts in melanoma and in glioma as anticipated based on the previously reported expression of the protein (28). Concordant with our protein expression data, CSPG4 mRNA expression was increased in HNSCC. We also found increased mRNA expression in clear cell renal carcinomas by in silico analysis, and overall, despite some intratumor variability, significant increased mRNA levels in all these tumor types relative to the corresponding normal tissues (Fig. 1D). Examination of the large Bittner multi-cancer dataset (www.oncomine.org) confirmed high CSPG4 mRNA expression in melanoma, clear cell renal carcinoma, HNSCC, multiple sarcoma types (chondrosarcoma, leiomyosarcoma, liposarcoma), gastrointestinal stromal tumors, skin, and vulvar squamous cell carcinomas (Supplementary Fig. 1). Of note several sarcoma cell lines have been previously reported to express CSPG4 protein (29). A number of other common malignancies such as colorectal, ovarian, and endometrial carcinoma did not show increased CSPG4 transcripts, consistent with the mRNA expression from the TCGA data sets. CSPG4 protein expression in an array of normal tissues was negative (Supplementary Fig. 2). In addition to the normal tissues represented in supplementary figure 2, we evaluated CSPG4 expression on a total of 33 different types of tissues, all of which were negative. Using the public Novartis GeneAtlas (http://biogps.org) and TCGA databases, CSPG4 mRNA expression was observed in a number of normal tissues. However, the levels of expression are remarkably lower than those of cancer tissues (Supplementary Fig. 3).
Figure 1. CSPG4 expression in primary solid tumors and tumor-derived cell lines.

Panel A. Representative immunohistochemistry (IHC) and scoring of analyzed solid tumor tissue arrays. Representative melanoma and breast carcinoma samples are shown at 200X magnification. Panel B. Scoring summary of a panel of solid tumors which includes melanoma, breast carcinoma (Breast CA), HNSCC, neuroblastoma (NeuroB) and mesothelioma (MesoT). Panel C. CSPG4 mRNA expression by TCGA in a variety of solid tumors. Box plots show median, 25%/75% range, 5%/95% range, and minimum/maximum. Panel D. CSPG4 mRNA expression analysis, comparing tumor versus corresponding normal tissues, for astrocytoma/glioblastoma (GBM), HNSCC, clear cell renal carcinoma, and melanoma. Indicated P-values were calculated by t-test. Panel E. CSPG4 expression in the indicated array of melanoma cell lines as assessed by flow cytometry (FACS). Panel F. FACS analysis of CSPG4 expression in the selected mesothelioma (MILL and PHI), HNSCC (PCI-30), and breast cancer- derived (MDA-MB-231 and UACC-812) cells lines. Dotted and bold lines indicate isotype and CSPG4 mAbs, respectively.
We next examined CSPG4 expression in a series of cell lines from a variety of tumor types analyzed above. Expression of CSPG4 was detected in 8 of the 9 melanoma cell lines screened (Fig. 1E). Importantly, CSPG4 was detected on tumor cell lines representative of the above identified solid tumors such as mesothelioma (MILL and PHI), HNSCC (PCI-30) and breast cancer [MDA-MB-231 (adenocarcinoma) and UACC-812 (ductal carcinoma)] (Fig. 1F) all consistent with our analysis of human tumor samples.
T lymphocytes expressing the CSPG4-specific CAR are cytotoxic against CSPG4+ tumor cell lines but not against primary normal tissues
To target CSPG4+ tumors we generated a CSPG4-specific CAR containing the CD28 costimulatory endodomain (CAR.CSPG4) (Fig. 2A). T lymphocytes from 4 healthy donors were engineered to express the CAR.CSPG4 using a gamma retroviral vector. Transduction efficiency was 80% ± 3%, and both CD4 and CD8 T cells stably expressed the CAR (26% ± 9% and 51% ± 16%, respectively), as assessed by phenotypic analysis by day 7 of culture (Fig. 2B). The majority of CAR.CSPG4+ T cells were CD45RO+ (76% ± 7%) and a fraction retained CD62L expression (51% ± 7%) and CCR7 (13% ± 2%), indicating that they were mainly composed of effector-memory T cells (Fig. 2C). The expression of CAR.CSPG4 by T cells was comparable to that obtained with a previously described CD19-specific CAR (CAR.CD19) (Supplementary Fig. 4) (30), which was used as an irrelevant-CAR control population.
Figure 2. Expression and function of CAR.CSPG4 in T cells.

Panel A. Schematic representation of the retroviral vector encoding the CSPG4-specific CAR. The CAR incorporates the CD28 costimulatory endodomain. Panel B. Representative FACS analysis showing the expression of the CAR in CD3, CD4 and CD8 T cells after retroviral transduction. Panel C. Representative expression of the CD62L, CD45RO, and CCR7 markers on control and CAR.CSPG4+ T cells by flow cytometry on day 14 of culture. Numbers represent percentages of cells per quadrant.
Cytotoxic activity of control and CAR.CSPG4+ T cells, after 1–2 weeks of culture, was assessed against K562, to measure natural killer cell-mediated activity, and against the melanoma derived cells lines P1143 (as CSPG4− target) and SENMA (as CSPG4+ target) (Fig. 1E) at various E:T ratios (Fig. 3A). CAR.CSPG4+ but not control T lymphocytes significantly lysed the CSPG4+ target (59% ± 5% vs. 11% ± 8% at 20:1 ratio)(p< 0.01), while both CAR.CSPG4+ and control T cells showed minimal activity against K562 (12% ± 9% vs. 13% ± 11%) and the CSPG4− target (<10% in both cases). The antitumor activity of CAR.CSPG4+ T lymphocytes was also evaluated in a 72 hour co-culture assay (Fig. 3B and C). CAR.CSPG4+ and control T lymphocytes were co-cultured with GFP-expressing tumor cell lines at an E:T ratio ranging from 5:1 to 3:1 according to the kinetic growth of each cell line. CAR.CSPG4+ T cells significantly controlled the growth of all CSPG4+ cell lines tested: SENMA (residual tumor cells = 0.1% ± 0.06%), CLB (0.1% ± 0.1%), UACC-812 (6% ± 6%), MILL (3% ± 5%), MDA-MB-231 (3% ± 3%), PHI (4% ± 3%), and PCI-30 (0.5% ± 0.5%), but not of the CSPG4− target P1143 (residual tumor cells 38% ± 10%). As expected, all tumor cell lines tested rapidly grow in the presence of control T lymphocytes (residual tumor cells for: SENMA = 62% ± 3%, CLB = 70% ± 6%, UACC-812 = 47% ± 15%, MILL = 50% ± 8%, MDA-MB-231 = 42% ± 11%, PHI = 29% ± 6%, PCI-30 = 17% ± 3%, and P1143 = 45% ± 10%). In all cases, the effects of CAR.CSPG4+ T cells were significantly greater than those of control T cells (from p<0.05 to p<0.001). T cells expressing the control CAR.CD19 showed cytotoxic activity neither against CSPG4+ nor CSPG4− targets (Supplementary Fig. 4). As illustrated in Fig. 3, commercially available primary normal epithelial cell lines (small airway, kidney and prostate) derived from tissuses found to express low levels of CSPG4 mRNA (Supplementary Fig. 3) did not express detectable levels of the protein by flow cytometry (Fig. 3D), and were not lysed by CAR.CSPG4+ T cells when tested in 51Cr release assays (Fig. 3E).
Figure 3. Cytotoxic function of CAR.CSPG4+ T cells against CSPG4+ tumors but not against epithelial cells from lung, kidney and prostate.

Panel A. Cytotoxic activity of control T cells and CAR.CSPG4+ T cells evaluated in a 6 hour 51Cr release assay. Target cells used were the CSPG4+ tumor cell line (SENMA), CSPG4− target cell line (P1143) and K562 to quantify natural killer activity. Data show averages and SD results of T cells from 4 donors. Panel B. Co-culture experiments of control and CAR.CSPG4+ T cells with GFP+ tumor cell lines, assessed by flow cytometry 72 hours later. The plots describe a representative experiment of T cells co-cultured with SENMA (CSPG4+ target) or P1143 (CSPG4− target). Numbers represent percentages of cells per quadrant. Panel C. Summary of co-culture experiments of control and CAR.CSPG4+ T cells against a panel of CSPG4+ tumor targets. Data represent averages ± SD of 4 donors. * = p<0.05, and *** = p<0.001. Panel D. FACS analysis of CSPG4 expression in primary epithelial cells derived from normal small airway, kidney and prostate. Dotted and bold lines indicate isotype and CSPG4 mAbs, respectively Panel E. Cytotoxic activity of control T cells and CAR.CSPG4+ T cells from a representative donor of two independent experiments evaluated in a 5 hour 51Cr release assay against these normal epithelial cells.
CAR.CSPG4+ T lymphocytes secrete Th1 cytokines and proliferate in response to CSPG4+ tumors
Since CAR.CSPG4 contains the CD28 costimulatory endodomain, we studied CAR.CSPG4+ T lymphocyte proliferation in response to CSPG4+ tumor cells using a CFSE dilution assay. When CFSE-labeled control and CAR.CSPG4+ T cells were cultured with irradiated SENMA tumor cells for 96 hours, a significant CFSE dilution occurred for CAR.CSPG4+ T cells, with both CD4 and CD8 T cells proliferating at a higher percentage (66% ± 12% and 68% ± 8%, respectively) compared to control CD4 and CD8 T cells (8% ± 7% and 14% ± 10%, respectively) (p<0.05 and p<0.01, respectively) (Fig. 4A and B). T cells transduced with the control CAR.CD19 also containing the CD28 endodomain did not show significant proliferation in response to CSPG4+ targets (Supplementary Fig. 4). We also evaluated whether the inclusion of a “late” co-stimulatory endodomain, such as 4-1BB, in addition to CD28 (third generation construct) provided these T cells with additional proliferative and cytotoxic activity, but found no further benefits (Supplementary Fig. 5).
Figure 4. T lymphocytes transduced with CAR.CSPG4 proliferate and release IL-2 and IFN γ upon specific antigen engagement.

Panel A. Control and CAR.CSPG4+ T cells, labeled with CFSE, were stimulated with irradiated CSPG4+ (SENMA) tumor target. The panel illustrates the CFSE dilution in CD4 or CD8 gated T cells after 96 hours of culture for a representative donor. Panel B. Summary of 3 independent CFSE dilution assays. Data represents mean ± SD. Panel C. IL-2 cytokine-release assessment using specific ELISA by T lymphocytes transduced with CAR.CSPG4 and control T cells 24 hours post co-culture (E:T ratio 5:1) with either CSPG4+ tumors or CSPG4− target cells (P1143). Results of 5 experiments are presented with mean ± SD. Panel D illustrates the detection of IFNγ in the same culture supernatant. Results of 5 experiments with mean ± SD are shown. * = P< 0.05, ** = p<0.01, and *** = p<0.001.
We finally quantified the IL-2 and IFNγ cytokines released in response to the antigen, by co-culturing control and CAR.CSPG4+ T lymphocytes with CSPG4+ or CSPG4− tumor cells. As expected CAR.CSPG4+ T lymphocytes secreted significantly more IL-2 than control T cells only in the presence of CSPG4+ tumor cells (Fig. 4C). A positive trend, although not statistically significant, was observed when CAR.CSPG4+ T lymphocytes were cultured with the MSCP+ tumor cell lines PHI, MILL, UACC-812 and MDA-MB-231 (data not shown). Similarly IFNγ production was significantly higher than control T cells when CAR.CSPG4+ T lymphocytes were cultured with CSPG4+ tumor cells (Fig. 4D).
CAR.CSPG4+ T lymphocytes control the growth of human melanoma, HNSCC and breast carcinoma cells engrafted in immunodeficient mice
To assess the in vivo relevance of our in vitro results, we engrafted NSG mice subcutaneously with cell lines derived from representative melanoma tumor (SENMA), HNSCC (PCI-30) or breast carcinoma (UACC-812). Four to seven days later (depending on the kinetics of the tumor growth), mice were infused via tail vein injection with either control or CAR.CSPG4+ T lymphocytes, and tumor growth quantified by sequential tumor volume measurements. In all three models, CAR.CSPG4+ T lymphocytes inhibited tumor growth significantly better than control T lymphocytes (Fig. 5). By day 30, melanoma tumors reached a volume of 879 mm3 ± 124 mm3 in mice receiving CAR.CSPG4+ T lymphocytes versus 8359 mm3 ± 958 mm3 in mice receiving control T cells (p<0.001) (Fig. 5A), and this corresponded to improved overall survival (Supplementary Fig. 6). Although HNSCC and breast carcinoma tumors were not as aggressive as melanoma in vivo, we observed that in both models CAR.CSPG4+ T lymphocytes controlled tumor growth. By day 30 the size of HNSCC tumors was 19 mm3 ± 10 mm3 in treated mice versus 190 mm3 ± 75 mm3 in control mice (p<0.001) (Fig. 5B) and the size of breast carcinoma tumors was 28 mm3 ± 13 mm3 in treated mice versus 166 mm3 ± 64 mm3 in control mice (p< 0.001) (Fig. 5C).
Figure 5. CAR.CSPG4+ T lymphocytes control tumor growth in vivo.
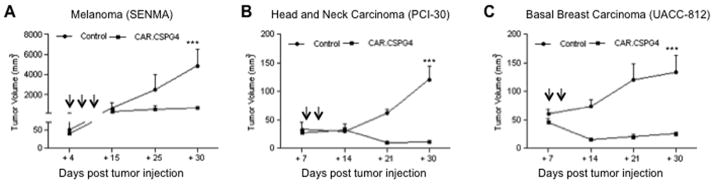
Panels show tumor growth, assessed by caliper measurement, of NSG mice engrafted subcutaneously with melanoma (SENMA) (Panel A), HNSCC (PCI-30), (Panel B) or breast carcinoma (UACC-812) (Panel C) cell lines and infused i.v. with either CAR.CSPG4+ (closed squares) or control (closed circles) T lymphocytes. Arrows indicate T-cell infusions. Shown are mean ± SD from 15 mice per group (3 independent experiments) for the melanoma model and 10 mice per group (2 independent experiments) for the HNSCC model and breast carcinoma models. *** = p< 0.001.
Discussion
The involvement in several signaling pathways associated with cell proliferation, survival, migration, and suggested high expression in various types of cancers highlight the critical role that CSPG4 has in promoting tumor growth and simultaneously make it an attractive target for immunotherapy. By IHC, we independently validated CSPG4 protein expression in several solid tumors with poor prognosis, such as melanoma, breast cancer, mesothelioma and HNSCC. In silico analysis of microarray expression data confirmed overexpression of CSPG4 in tumors that we validated by IHC as compared to normal tissues, and also disclosed CSPG4 overexpression in other important malignancies including glioblastoma, clear cell renal carcinoma and sarcomas suggesting that targeting this antigen may have a major impact on a broad array of solid tumors.
Since CSPG4-specific mAbs can control tumor growth of CSPG4+ tumor cells, in both melanoma and breast cancer tumor models (7, 31), we proposed to improve the therapeutic benefits of this antibody-based approach by generating a CAR that targets the CSPG4 molecule. In contrast to mAb-based therapy, CAR-T cells should produce long-lasting effects, as engineered T cells can expand at the tumor site upon antigen stimulation if an appropriate costimulatory endodomain, derived from CD28, CD137 or CD134, is incorporated within the CAR (13, 31). In contrast to a previous report (21), we found that the CSPG4-specific CAR obtained from the same 763.74 single chain has potent antitumor activity. We traced these striking differences to two critical components we have introduced in our construct. First, the scFv in our CAR is coupled with the CD3-ζ endodomain of the TCR rather than the FcεRI-γ chain, which is known to promote a much weaker and less durable signaling (32, 33). Second, we incorporated the CD28 costimulatory endodomain within the CAR, to accomplish sustained IL-2 production and proliferation in response to CSPG4+ tumor cells, thus recapitulating previous observations for other CAR molecules (13). Of note, the inclusion of a second costimulatory endodomain derived from CD137 did not further improve the function of our CAR in vitro, supporting the concept that there is no single optimal configuration that is applicable to all CAR molecules, but that CAR receptor optimization remains largely empirical and required for each molecule.
The most critical improvement in the field by our work is the applicability of CAR.CSPG4+ T cells not only to target melanoma (17), but more broadly to other solid tumors generally characterized by poor prognosis with conventional treatments such as breast carcinoma, HNSCC and mesothelioma. We demonstrated that CAR.CSPG4+ T cells produce IFNγ and promote tumor elimination not only when challenged with tumor cells with high CSPG4 expression but also with tumor cell lines characterized by moderate/low CSPG4 expression, such as the breast carcinoma tumor cell lines UACC-812 and MBA-MB-231. This further supports the advantages of antibody specificity coupled with the T-cell effector function, as mastered by CAR-modified T cells, which can target tumors neglected by naked corresponding antibodies due to the suboptimal expression of the targeted antigen (34, 35).
Antitumor effects mediated by CAR.CSPG4+ T cells significantly limit tumor growth in xenograft mouse models of melanoma, HNSCC and breast carcinoma, strongly validating our in vitro findings. The lack of sustained and complete tumor eradication in these models was not caused by selection of CSPG4-negative tumor cells, as harvested tumors retained the expression of the antigen (Supplementary Fig. 6), but conversely are likely to be attributed to an intrinsic limitation of the models, as T cells do not persist long term in these immunodeficient mice (Supplementary Fig. 6).
To fully translate this approach, the differential expression of CSPG4 in tumor cells versus normal tissues needs to be ensured in order to limit potential toxicities (36, 37). We found the expression of CSPG4 absent or negligible in normal tissue arrays as assessed by IHC. The analysis of publically available data sets indicates that there is some level of CSPG4 mRNA expression in several normal tissues. However, when we compared normal tissues with cancer tissues, the cancers show consistent and dramatically higher expression of CSPG4 at mRNA levels, and our in vitro analyses illustrate that primary epithelial cells derived from some of these tissues do not express significant amount of the protein and are not targeted by CAR.CSPG4+ T cells. Even though the tissue screening, bioinformatics analysis and lack of toxicity by in vitro experiments support the relevance of CSGP4 as a targetable antigen in cancer patients, we cannot fully exclude that the low levels of mRNA in normal tissues, as reported in public data sets, may promote sufficient protein expression in specific physiological conditions to become a target for CSPG4-specific CAR-T cells. The clinical translation of this approach may thus benefit from the inclusion of a suicide gene within the vector cassette, to allow the rapid elimination of CAR-modified T cells in case of undesired toxicity (30, 38).
In summary, we provide ample data to support the use of CAR.CSPG4+ T cells to treat a broad range of solid CSPG4+ tumors for which the prognosis remains poor with conventional treatments. The combination of this approach with other biological agents may further increase their activity and thus clinical benefits.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Adoptive transfer of CAR-redirected T lymphocytes represents a promising therapy for patients with lymphoid malignancies. Here we extend the applicability of this strategy to a broad array of solid tumors by targeting the CSPG4 antigen; this antigen is over-expressed by numerous tumor types while having negligible expression in normal tissues. Our study provides preclinical evidence that CSPG4-redirected T cells can control the growth of human melanoma, HNSCC and breast cancer both ex vivo and in vivo in xenograft models.
Acknowledgments
Funding: This work was supported in part by RP110553 CPRIT Cellular Therapy of Cancer, P30 CA125123 NCI supporting the Human Tissue Acquisition and Pathology and Statistics and Bioinformatics Core Resources of the Dan L. Duncan Cancer and NIH CA 138188.
The authors would like to thank Dr Malcolm Brenner for the critical revision of the manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: Center for Cell and Gene Therapy has a collaborative research agreement with Celgene and Bluebird bio.
Reference List
- 1.Pluschke G, Vanek M, Evans A, Dittmar T, Schmid P, Itin P, et al. Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc Natl Acad Sci U S A. 1996;93:9710–5. doi: 10.1073/pnas.93.18.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivera Z, Ferrone S, Wang X, Jube S, Yang H, Pass HI, et al. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin Cancer Res. 2012;18:5352–63. doi: 10.1158/1078-0432.CCR-12-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, et al. CSPG4 in cancer: multiple roles. Curr Mol Med. 2010;10:419–29. doi: 10.2174/156652410791316977. [DOI] [PubMed] [Google Scholar]
- 4.Campoli MR, Chang CC, Kageshita T, Wang X, McCarthy JB, Ferrone S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol. 2004;24:267–96. doi: 10.1615/critrevimmunol.v24.i4.40. [DOI] [PubMed] [Google Scholar]
- 5.Iida J, Wilhelmson KL, Ng J, Lee P, Morrison C, Tam E, et al. Cell surface chondroitin sulfate glycosaminoglycan in melanoma: role in the activation of pro-MMP-2 (pro-gelatinase A) Biochem J. 2007;403:553–63. doi: 10.1042/BJ20061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang J, Price MA, Neudauer CL, Wilson C, Ferrone S, Xia H, et al. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J Cell Biol. 2004;165:881–91. doi: 10.1083/jcb.200403174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hafner C, Breiteneder H, Ferrone S, Thallinger C, Wagner S, Schmidt WM, et al. Suppression of human melanoma tumor growth in SCID mice by a human high molecular weight-melanoma associated antigen (HMW-MAA) specific monoclonal antibody. Int J Cancer. 2005;114:426–32. doi: 10.1002/ijc.20769. [DOI] [PubMed] [Google Scholar]
- 8.Dotti G, Savoldo B, Brenner M. Fifteen years of gene therapy based on chimeric antigen receptors: “are we nearly there yet?”. Hum Gene Ther. 2009;20:1229–39. doi: 10.1089/hum.2009.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 12.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–84. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 13.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns WR, Zhao Y, Frankel TL, Hinrichs CS, Zheng Z, Xu H, et al. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010;70:3027–33. doi: 10.1158/0008-5472.CAN-09-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yvon E, Del VM, Savoldo B, Hoyos V, Dutour A, Anichini A, et al. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15:5852–60. doi: 10.1158/1078-0432.CCR-08-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu W, Feng S, Dakhova O, Creighton CJ, Cai Y, Wang J, et al. FGFR-4 Arg(3)(8)(8) enhances prostate cancer progression via extracellular signal-related kinase and serum response factor signaling. Clin Cancer Res. 2011;17:4355–66. doi: 10.1158/1078-0432.CCR-10-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Temponi M, Gold AM, Ferrone S. Binding parameters and idiotypic profile of the whole immunoglobulin and Fab′ fragments of murine monoclonal antibody to distinct determinants of the human high molecular weight-melanoma associated antigen. Cancer Res. 1992;52:2497–503. [PubMed] [Google Scholar]
- 21.Reinhold U, Liu L, Ludtke-Handjery HC, Heuser C, Hombach A, Wang X, et al. Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol. 1999;112:744–50. doi: 10.1046/j.1523-1747.1999.00586.x. [DOI] [PubMed] [Google Scholar]
- 22.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–7. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu L, Qi CF, Schlom J, Kashmiri SV. Secretion of a single-gene-encoded immunoglobulin from myeloma cells. Proc Natl Acad Sci U S A. 1993;90:7995–9. doi: 10.1073/pnas.90.17.7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glouchkova L, Ackermann B, Zibert A, Meisel R, Siepermann M, Janka-Schaub GE, et al. The CD70/CD27 pathway is critical for stimulation of an effective cytotoxic T cell response against B cell precursor acute lymphoblastic leukemia. J Immunol. 2009;182:718–25. doi: 10.4049/jimmunol.182.1.718. [DOI] [PubMed] [Google Scholar]
- 26.Rickman DS, Bobek MP, Misek DE, Kuick R, Blaivas M, Kurnit DM, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–91. [PubMed] [Google Scholar]
- 27.Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–42. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- 28.Chekenya M, Krakstad C, Svendsen A, Netland IA, Staalesen V, Tysnes BB, et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene. 2008;27:5182–94. doi: 10.1038/onc.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, et al. CSPG4 in cancer: multiple roles. Curr Mol Med. 2010;10:419–29. doi: 10.2174/156652410791316977. [DOI] [PubMed] [Google Scholar]
- 30.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–70. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 32.Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, et al. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol. 2001;166:182–7. doi: 10.4049/jimmunol.166.1.182. [DOI] [PubMed] [Google Scholar]
- 33.Heuser C, Hombach A, Losch C, Manista K, Abken H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003;10:1408–19. doi: 10.1038/sj.gt.3302023. [DOI] [PubMed] [Google Scholar]
- 34.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res. 2013;19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 2009;17:1779–87. doi: 10.1038/mt.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 37.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.