

Abstract
Background
While dezocine is a partial mu opioid receptor agonist, it is not a controlled substance. Thus, the characterization of the molecular targets of dezocine is critical for scientific and clinical implications. The goal of this study is to characterize molecular targets for dezocine and their implications.
Methods
A binding screen for dezocine was performed on 44 available receptors and transporter proteins. Functional assays for the novel targets were performed along with computation calculations to locate the binding site. A G protein activation study was performed for the human kappa opioid receptor to determine whether dezocine is a kappa antagonist. Data are presented as mean ± SE.
Results
The affinities for dezocine were 3.7±0.7 nM for the mu receptor, 527±70 nM for the delta receptor, and 31.9±1.9 nM for the kappa receptor. Dezocine failed to induce G protein activation with kappa opioid receptor and concentration dependently inhibited kappa agonist (salvinorin A and nalbuphine) induced receptor activation, indicating that dezocine is a kappa antagonist. Two novel molecular targets (norepinephrine transporter, NET; and serotonin transporter, SERT) were identified. Dezocine concentration-dependently inhibited norepinephrine and serotonin reuptake in vitro. The half maximal inhibitory concentrations (expressed as pIC50) were 5.68±0.11 for NET and 5.86 ± 0.17 for SERT. Dezocine occupied the binding site for known NET and SERT inhibitors.
Conclusions
The unique molecular pharmacological profile of dezocine as a partial mu receptor agonist, a kappa receptor antagonist and a norepinephrine and serotonin reuptake inhibitor (via NET and SERT) was revealed. These discoveries reveal potentially important novel clinical implications and drug interactions of dezocine.
Introduction
Dezocine is an opioid medication that is structurally similar to pentazocine, a mixed opioid receptor partial agonist/antagonist developed in 1970’s by the American Home Products Corporation.(1) Dezocine was approved by the Food and Drug Administration for perioperative care management but was discontinued with the closure of its parent company. PubMed lists only 74 papers related to dezocine*, with the first paper published in 1978 in Anesthesia & Analgesia demonstrating its use for postoperative pain management.(1) Although no longer used clinically in Western countries, dezocine is gaining popularity in China as an alternative medication for perioperative pain management.(2)
Dezocine is an opioid mu receptor partial agonist/antagonist. (1) Because of its partial mu agonism, it exhibits a “ceiling effect” for respiratory depression (a notorious and fatal side effect caused by commonly used clinical opiates). Though initially identified as a kappa receptor agonist, a later study suggests that dezocine is a kappa receptor antagonist. (3) Further study is needed to resolve this discrepancy. Since dezocine is a partial mu antagonist, in theory, a concerted use of dezocine together with a mu receptor agonist like morphine should decrease the analgesia effect of morphine significantly. However, it is reported that the combination of morphine and dezocine increases the analgesic effects significantly,(4) indicating that dezocine may induce analgesia through an additional mechanism(s).
Opiate receptors belong to the G protein coupled receptor (GPCR) family. It is highly possible that, in addition to opiate receptors, opioids could interact with other GPCRs. In this study, we hypothesized that dezocine might also act at other membrane receptors, and we therefore screened a large group of available recombinant GPCRs and transporter proteins in an attempt to identify novel pharmacological targets for dezocine. We further investigated whether dezocine was a kappa receptor agonist or antagonist. The molecular interactions with the target proteins were analyzed using available crystal structures and molecular models for docking calculations.
Materials and Methods
All chemicals (except those specified otherwise) were obtained from Sigma-Aldrich (St. Louis, MO) and were reagent grade or higher. Dezocine was obtained from Young's River pharmaceutical group (Taizhou, Jiangsu, China) with 99.9% purity. All chemicals were used without further purification. The chemical structures of the ligands that have the same targets as dezocine and were used for comparison purposes in this study are listed in figure 1.
Figure 1.

The structures of the ligands used in this study to probe the pharmacological properties of dezocine are listed. All structures were obtained from public domain without further modification or verification by a chemist except salvinorin A and JDTic. Salvinorin A (http://commons.wikimedia.org/wiki/File:Salvinorin-A_structure.png) and JDTic (http://commons.wikimedia.org/wiki/File:JDTic_cas_361444-66-8.svg) are obtained from Wikimedia Commons and are licensed under the Creative Commons Attribution-Share Alike 3.0 Unported license.
Radioligand Binding Assays and Affinity Determination
A primary binding screen for dezocine was performed on 44 available receptors (mostly GPCRs, see Table 1). Evidence for interaction was based on the inhibition of the reference ligandbinding signal. Dezocine was diluted in standard binding buffer (50mM Tris-HCl, 10mM MgCl2, 0.1mM EDTA, pH 7.4) to a final concentration of 10 µM. Briefly, 50µL aliquots of radioactive ligand (5nM) were added to wells of a 96-well plate, which contained 25µL of the reference or test ligands. We employed transfected cell lines expressing mainly human (unless otherwise specified) recombinant receptors, monoamine transporters, or ion channels for crude membrane preparation. Detailed information about our membrane preparation can be obtained from the protocol online†. Crude membrane fractions containing the receptors were resuspended in standard binding buffer and 50µL aliquots added to each well. The reactions were incubated at room temperature for 1.5 hours to allow for radioligand binding equilibration. Bound radioactivity was harvested by rapid filtration through a 0.3% polyethyleneimine-treated, 96-well filter mats using a 96-wel Filtermate harvester (PerkinElmer, Waltham, MA). The dried filters were treated with melted scintillant and a Microbeta scintillation counter (PerkinElmer) was utilized to measure the radioactivity retained on the filter.
The secondary binding assay was performed only when the inhibition in the primary screen was over 50%. A secondary binding assay was utilized to determine the binding affinity for the identified receptor. Dezocine was prepared in standard binding buffer and serially diluted to the desired concentrations.
50µL aliquots of radioactive ligand (5nM) were added to wells of a 96-well plate, which contained 25µL aliquots of dezocine. 50µL of a crude membrane fraction of cells expressing the respective receptors were applied to each well. Reaction incubation, harvesting and radioactivity measurement procedures from the primary assay were repeated. Affinity is expressed as pKi (-logKi).
G protein activation by kappa receptor treated with agonists, partial agonists and antagonists
Membrane preparations of recombinant human kappa opioid receptor expressed in the mammalian cell line Chem-5 were obtained from Millipore (Billerica, MA). The effects of specific kappa opioid receptor ligands on the activation of the recombinant receptor were investigated by measuring G protein activation in vitro. Nalbuphine and salvinorin A (full agonist) and nor-binaltorphimine (antagonist) were utilized as controls.
The assay reports the initial rates of activation of heterotrimeric G proteins (Gαii1β1γ2) on an agonist-bound receptor by measuring the accumulation of [35S]-GTPγS (non-hydrolyzable analog of GTP) bound to the activated Gαi1 subunit. Myristoylated Gαi1 was expressed in E. coli and purified as previously described. (5) Recombinant human β1γ2 subunits of G protein were expressed in baculovirus-infected Sf9 cells and purified as previously described. (6) The G protein activation assay was conducted as follows (final concentrations in 50µL reaction mixture are given in parentheses): the membrane sample was diluted into ice-cold 10 mM 3-(N-morpholino) propanesulfonic acid (MOPS) buffer to reach a protein concentration of 40 ng/µL. 10 µL of the diluted dispersion were dispensed into pre-siliconized glass tubes and mixed with the ligand in MOPS buffer containing 0.1% (w/v) BSA. Upon addition of a mixture of Gαi1 (100 nM) and Gβ1γ2 (500 nM), the tubes were incubated on ice for 30 minutes. The reaction was started by addition of MOPS buffer pH=7.5 (50 mM), EDTA (1 mM), MgCl2 (3 mM), GDP (4 µM), BSA (0.3% w/v), NaCl (100 mM), DTT (1 mM), and [35S]-GTPγS (5 nM, 1250 Ci/mmol) followed by rapid transfer of the tubes to a water bath at 30 °C. The incubation continued for 45 minutes. The reaction was terminated by addition of 2 mL of ice-cold stop solution, TNMg (20 mM Tris-HCl pH=8.0, 100 mM NaCl, and 25 mM MgCl2). The reaction mixture was rapidly filtered through nitrocellulose filters (Millipore). Filters were washed four times with 2 mL each of cold TNMg buffer, dried, placed in scintillation vials filled with ScintiSafe Econo F scintillation liquid (Fisher, Waltham, MA), and the radioactivity counted. Duplicate samples corresponding to every ligand concentration point were counted.
To test whether dezocine could antagonize the full agonists, the kappa receptor was preactivated with either nalbuphine (250 nM) or salvinorin A (20 nM), a highly selective non-opioid kappa receptor agonists with strong affinity.(7) The kappa receptor was then treated with increasing concentrations of dezocine.
Norepinephrine transporter (NET) and serotonin transporter (SERT) reuptake assay
The potency of dezocine as an inhibitor of norepinephrine and serotonin uptake on human cloned NET and SERT, stably expressed in Human Embryonic Kidney 293 cells, was determined using the neurotransmitter assay kit from the Molecular Devices (Sunnyvale, CA) as described previously.(8) In brief, Human Embryonic Kidney 293 cells were plated in Poly-L-Lys (PLL) coated 384-well black clear bottom cell culture plates in DMEM + 1% dFBS, at a density of 15,000 cells per well in a total volume of 40 µl. The cells were incubated for a minimum of 6 hours before use in the assays. The medium was removed and 20 µl of assay buffer (20 mM HEPES, 1x HBSS, pH 7.40) was added, followed by 5 µl of 5x drug solutions. The plate was incubated at 37°C for 30 min. After the incubation, 25 µl of dye solution was added and fluorescence intensity was measured after 30 min at 37°C, using FlexStation II (bottom read mode, Excitation at 440 nm, Emission at 520 nm with 510 nm cut-off) from the Molecular Devices. Results (Relative Fluorescence Unit) were exported and plotted against drug concentrations in Prism 5.02 (GraphPad Software, Inc. La Jolla, CA) for nonlinear regression to obtain inhibitory potency. The half maximal inhibitory concentration were determined and expressed as pIC50 (pIC50= -log (IC50)).
Docking calculations
Docking calculations were carried out using DockingServer(9‡ as previously described to locate and visualize the binding site. (10) The coordinates of the crystal structure were obtained from the protein data bank (PDB§ with access code 4DKL for murine mu receptor (11) and 4DJH for kappa receptor. (12) The coordinates for the serotonin transporter were taken from the recently published model (13) based on the LeuT 3F3A crystal structure. (14) The coordinates for the norepinephrine transporter were obtained from the recently published model (15) based on the crystal structure (PDB ID code 2A65(16)) of LeuT from Aquifex aeolicus. Dezocine docking calculation on a LeuT crystal coupled with desipramine (PDB ID code 2QJU(17)) was also performed to identify the potential overlap of the binding sites. Semi-empirical charges calculated by MOPAC2009 were added to the ligand atoms**.(18) Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added to the receptor using AutoDock tools provided by the server. Grid maps of 30×30×30 Å grid points with 0.375 Å spacing centered at the known ligand binding site were generated using the Autogrid program. (19,20) All the ligand searches were performed using the Solis and Wets local search method with a Lamarckian genetic algorithm. (21) Initial position, orientation, and torsions of the ligand molecules were set randomly. The three-dimensional coordinates of the tested compound were obtained from the PubChem database††. PyMOL (Version 1.5.0.4, Schrodinger LLC, New York, NY) was used to render the graphics for presentation.
Data analysis
The data are presented as mean ± SE from three repeats. The results were analyzed using GraphPad Prism (version 5.02 Windows version). EC50s are determined using the following model as defined in GraphPad: Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)). Top and Bottom are plateaus in the units of the Y axis. EC50 is the concentration of a ligand that gives a response half way between Bottom and Top. HillSlope describes the steepness of the family of curves.
Results
Interaction with opioid receptors
While dezocine binds to all three major subtypes of opioid receptors (table 2), it only weakly interacts with the delta receptor. We determined affinities for dezocine as 3.7±0.7 nM for the human mu receptor, 527±70 nM for the human delta receptor, and 31.9±1.9 nM for the human kappa receptor (Table 3). As indicated in figure 2, dezocine docks to the known binding site for opioid ligands in both the mu and kappa receptor. Hydrogen bonding with ASP 147 (149 in human mu) contributes to the strong affinity of dezocine to the mu receptor. TYR326 also has polar interaction with dezocine in the mu receptor as demonstrated in figure 1A. In the case of the kappa receptor, dezocine hydrogen bonds with ASP 138 as predicted by docking calculations (figure 2B).
Figure 2.

A) Dezocine (magenta) overlaps with beta-Funaltrexamine (orange,) a mu receptor antagonist and the ligand found in the crystal structure of the mu opioid receptor (4DKL13, in the binding pocket. Polar interaction with ASP147 and TYR326 is predicted. B) Dezocine (magenta) overlaps with JDTic (orange), a kappa receptor antagonist and the ligand found in the crystal structure of the kappa opioid receptor (4DJH) 14, in the binding pocket. Some of interacting residues (ASP 138, TYR 139 and MET 142) are colored in yellow. A nitrogen in dezocine might hydrogen bond with oxygen atoms of ASP 138 (distance colored in magenta, 2.6 Å, or in green, 2.8 Å).
Kappa receptor antagonism
Consistent with published data, nalbuphine behaved as a full kappa receptor agonist and fully activated the G protein in the presence of membranes containing kappa receptor as indicated in figure 3A. There was no significant G protein activation with dezocine in the presence of kappa receptor, indicating that dezocine acted as an antagonist (figure 3A). To confirm this, the G protein was pre-activated with a full agonist (Nalbuphine or Salvinorin A), and then increasing amounts of dezocine were added. As indicated in figure 3B, dezocine inhibited the agonist effect concentration-dependently with a total blockage at high concentration. This finding correlated the lack of G protein activation observed in figure 3A. The IC50 (T=30 °C) values of inhibition were in a high nanomolar range (~ 350 nM for competition with nalbuphine or ~800 nM for competition with salvinorin A), suggesting that dezocine binds to the receptor at the same site as these full agonists. Interestingly, based on this G protein activation study, nor-binaltorphimine acted as an inverse kappa agonist.
Figure 3.

A) Nalbuphine and salvinorin A, full agonists of kappa opioid receptors, concentration dependently activate the G protein in the presence of kappa receptor. Dezocine fails to induce any G protein activation, indicating receptor antagonism. Based on the activity of the G protein in its presence, nor-binaltorphimine is an inverse agonist of kappa opioid receptor. B) G protein was pre-activated with a full agonist (Nalbuphine, 250 nM or Salvinorin A, 20 nM) and then increasing amounts of dezocine was added. Dezocine inhibited the agonist effects of nalbuphine and salvinorin A concentration-dependently with a total blockage at high concentration, confirming the kappa receptor antagonism effect of dezocine. The relationship is plotted using the following model: Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)).
Amine transporter proteins as novel targets of dezocine
As indicated in table 2, in addition to binding to the opioid receptor, dezocine also inhibits the NET with pKi of 6.00 ± 0.10 and the SERT with pKi of 6.96±0.08. These interactions were further confirmed by norepinephrine and serotonin reuptake studies. The pIC50s at NET were 7.57±0.23 for nisoxetine (positive control) and 5.68±0.11 for dezocine (figure 4A). The pIC50s of SERT were 5.99 ± 0.07 for nisoxetine and 5.86 ± 0.17 for dezocine (figure 4B).
Figure 4.
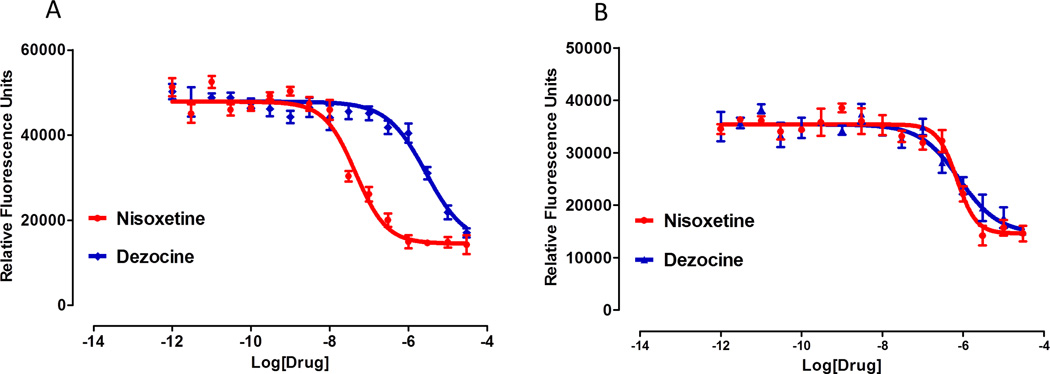
A) Norepinephrine reuptake is dose dependently inhibited by both dezocine and nisoxetine. However, inhibition is weaker in the case of dezocine. B) Serotonin reuptake is concentration dependently inhibited in the presence of dezocine and nisoxetine with comparable potency. The relationship is plotted using the following model: Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)).
Binding site location in NET and SERT
Consistent with the competitive binding assay, dezocine is predicted to share the same binding site with nisoxetine in the norepinephrine transporter as indicated in figure 5. Dezocine is located in close proximity to TRP103, TYR127, GLU281, and LEU368 and might form hydrogen bonds with these residues. Based on the docking prediction shown in figure 6A, dezocine binds to the preformed ligand-binding pocket in the model of human serotonin transporter. This pocket has been demonstrated to be the binding site for many selective serotonin reuptake inhibitors including fluoxetine, citalopram, sertraline, fluvoxamine and tricyclic antidepressants such as amitriptyline, desipramine, and imipramine. Mutation of the residues lining this pocket (Y95, D98,I172, Y176, F335, F341 and S438) changed the binding capability of these ligands significantly.(13) Dezocine shares the same binding site for desipramine found in the crystal structure of LeuT as indicated in figure 6B. Both findings indicate that dezocine may share the same site as selective serotonin reuptake inhibitors or tricyclic antidepressants.
Figure 5.

Docking result of dezocine and nisoxetine in the model of norepinephrine transporter (NET). Dezocine (magenta) shares the same binding site of nisoxetine (cyan), a NET inhibitor, as indicated by the close overlap. Dezocine is located in close proximity to TRP103, TYR127, GLU281, and LEU368 which are all colored in yellow.
Figure 6.

A) Dezocine (magenta) sits in the preformed ligand binding pocket for selective serotonin reuptake inhibitors in the model of human serotonin transporter. The key interacting residues lining the pocket (Y95, D98, I172, Y176, F335, F341, and S438) are colored in yellow. This binding pocket has been demonstrated to be the binding site for many important clinical drugs such as fluoxetine, sertraline, and amitriptyline. B) Dezocine (magenta) shares the same binding pocket and overlap well with desiprimine (orange), the ligand in the LeuT crystal structure (2QJU).
Discussion
This study details the discovery of the pharmacological interactions of dezocine with the human NET and SERT proteins as well as the molecular characterization of these interactions. This study also confirms the interaction of dezocine with three opioid receptors with different affinities and verifies that dezocine acts as an antagonist of the kappa receptor rather than as an agonist. Collectively, these findings have significant implications, as they help elucidate the mechanisms underlying dezocine’s pharmacological effects and present evidence supporting the compound’s potential novel clinical applications.
Interaction with opioid receptors
Consistent with published data, (22) dezocine exhibits strong affinity for both mu and kappa receptors, but relatively weak interactions with the delta receptor. The determined affinities for all three receptors are comparable with those published previously. (22) It is predicted that dezocine shares the same binding site with the known ligands in the crystal structures. The predicted hydrogen bonding and polar interactions of dezocine in the binding sites of mu and kappa receptors could be used to explain why the addition of sodium ion in the binding buffer can decrease the affinities significantly. (22)
Dezocine is a well-known partial mu agonist (23–25) that has been used for perioperative pain management because of its analgesic effects.23,25 Similar to other opioids, dezocine could decrease anesthetic requirement by up to 50%. (26) In a double- blinded study comparing the analgesic effects of dezocine and meperidine in patients for postoperative pain management (n=187), 10 mg of dezocine produced the same analgesic activity as 50 mg of meperidine.(1) Due to its partial agonism on the mu opioid receptor, common side effects observed in opioids with full agonism are significantly reduced. (27) Most importantly, dezocine is not a scheduled medication classified by the World Health Organization and no addiction related to its use has been reported. (27) Since addiction is one of the most significant side effects of opioids, it is critical to understand why dezocine is not addictive. This compound could serve as a key molecular probe to elucidate the mechanism(s) of addiction by comparing the molecular targets with other known addictive opioid molecules.
Dezocine is structurally related to the kappa agonist pentazocine. Due to this similarity, dezocine was initially considered a kappa agonist with the potential to generate the psychotomimetic effects observed with pentazocine. However, such effects were not observed in analgesic dosages for dezocine used in clinical trials. (1) In fact, a recent study suggested that dezocine is a kappa antagonist.(3) We confirmed this finding by demonstrating the compound’s inability to activate the receptor and its capacity to antagonize the agonist effects of salvinorin A, a potent and highly selective kappa receptor agonist, and nalbuphine, a non-selective kappa receptor agonist. Further study is needed to demonstrate whether this kappa receptor antagonism is connected to the compound’s lack of addictive properties. (28) Buprenorphine, a partial mu agonist and kappa antagonist, has been successfully used for addiction treatment for many years with the outcome equivalent to methadone therapy. (29) However, buprenorphine itself is an addictive Schedule III medication (30) and its chronic use creates significant difficulty for optimal perioperative pain management due to its high affinity to the receptor and long half-life. (31–33) Similar to buprenorphine, dezocine is also a partial mu agonist and a kappa antagonist based on our current findings. Further study is warranted to demonstrate whether dezocine, a non-addictive compound, could be used as a potential alternative medication for addiction management. Its shorter half-life allows for easier titration to an optimal effect as well as rapid removal when full agonism is required during the perioperative period.
While nor-binaltorphimine was traditionally considered a kappa antagonist, this study’s finding is consistent with the report that nor-binaltorphimine is an inverse kappa agonist, a property which may be related to its antidepressive effects.(34)
Interaction with NET and its related clinical implications
While dezocine is a mu opioid receptor partial antagonist, theoretically, it could antagonize the anti-nociceptive effects of morphine. However, when used in combination with morphine, dezocine concentration-dependently enhances the analgesic effects of morphine. (35) The combination of morphine and dezocine also reduces the anesthetic requirement. (26) These suggest the existence of alternative targets for dezocine (beside the opioid receptors) that could have additive effects for other opioids. Our findings support these earlier pharmacological observations. Dezocine interacts with NET and inhibits the norepinephrine reuptake. The competitive binding assay and the computational docking calculation suggest that dezocine interacts with NET directly at the binding site for the intrinsic NET ligand. Interestingly, a recent study indicates that pentazocine, a compound structurally similar to dezocine, inhibits NET function by reducing the amount of NET in cell surface membranes through an opioid receptor-independent pathway. (36) It is unclear whether dezocine could have similar properties as pentazocine in regards to indirect NET interaction. NET plays an important role in pain pathways and norepinephrine reuptake inhibition could be used for treating pain, (37) especially in neuropathic pain. (38) While its direct interaction with NET is relatively weak, it is still worthwhile to investigate whether dezocine could be used to target neuropathic pain in addition to its opioid receptor mediated analgesia.
Interaction with SERT and its related clinical implications
One of the striking findings of this study is that dezocine interacts with SERT at its ligand binding site and that serotonin reuptake can be inhibited concentration-dependently by dezocine. It is important to note that since dezocine shares a binding site with some clinical antidepressant drugs such as fluoxetine, sertraline, and amitriptyline, cross interactions may occur. Thus precaution may be needed. The kappa opioid receptor, NET and SERT are all important targets for depression treatment.(39–42) Because dezocine interacts with all three targets, it is critical to investigate whether or not dezocine could be an alternative medication to treat depression and prevent opioid-induced depression. Opioids are the main class of medications used to manage moderate and severe pain; however, in addition to addiction, depression is another common side effect of chronic opioid usage. (43) Patients with chronic pain frequently report depression, a condition associated with high pain intensity and greater prevalence of chronic opioid therapy. (44) Identifying and treating depression symptoms in chronic pain patients are essential for reducing co-morbidity and disability. (45) 80% of patients with major depression without psychotic features have painful physical symptoms. (46) Consequently, there is a significant medical impetus for developing medications that simultaneously and effectively target pain and depression. In vivo studies are warranted to demonstrate whether dezocine is a medication of choice targeting both pain and depression due to its unique molecular targets of kappa receptor antagonism, NET and SERT inhibition. Potential drug interaction could occur when using similar drugs with shared binding site(s).
Limitation
The major limitation of this study is that the profiling was limited to 44 receptors available to us and it is possible that dezocine could interact with additional targets. No in vivo evidence is provided in this study.
In conclusion, this study explored the interaction of dezocine with three major opioid receptors, demonstrated that dezocine is a kappa antagonist, and suggested its potential use for addition treatment. Through molecular target profiling, we discovered two novel molecular targets of dezocine in vitro: NET and SERT. The binding sites were characterized using available structural models and docking experiments. Dezocine concentration-dependently inhibited norepinephrine and serotonin reuptake in vitro. These findings suggest the potential use of dezocine as a novel medication for the simultaneous treatment of pain and depression. Potential drug interactions should be considered when administering drugs with similar pharmacological targets. Further studies are warranted.
Supplementary Material
Acknowledgement
Dr. Liu thanks the homology model coordinates of serotonin transporter provided by Mari Gabrielsen, PhD, at the Medical Pharmacology and Toxicology, Department of Medical Biology, Faculty of Health Sciences, University of Tromsø, N-9037 Tromsø, Norway and the homology model coordinates of norepinephrine transporter provided by Avner Schlessinger, PhD, and Professor Andrej Sali at the Department of Bioengineering and Therapeutic Sciences, and California Institute for Quantitative Biosciences, University of California, San Francisco. Graphic assistance from Jose Manuel Perez-Aguilar, PhD, at Department of Physiology and Biophysics, Weill Medical College of Cornell University, New York, for figure 2 and Weiming Bu, PhD, at the Department of Anesthesiology and Critical Care at the University of Pennsylvania for figure 6 is much appreciated. The authors also appreciate the technical support from Felipe Matsunaga and Jingyuan Ma at the Department of Anesthesiology and Critical Care at the University of Pennsylvania.
Funding: This research was supported by National Institute of Health (NIH, K08-GM-093115) (PI:RL) and the Intramural Research Program of the National Institute on Alcohol Abuse and Alcoholism (NIAAA, Bethesda, MD) (PI, AY). This research is also supported by funding from the Department of Anesthesiology and Critical Care at the University of Pennsylvania, Philadelphia PA (PI, RL), and by the National Institute of Mental Health's Psychoactive Drug Screening Program, Chapel Hill, NC (NIMH PDSP; Contract # HHSN-271-2008-00025-C).
Footnotes
Conflict of Interest: The authors declare no competing interests.
www.pubmed.com, last date accessed April 18, 2013
http://pdsp.med.unc.edu/PDSP%20Protocols%20II%202013-03-28.pdf, pages 18–24, last date accessed: October 15, 2013
http://www.dockingserver.com; last date accessed: October 15, 2013
http://www.rcsb.org/; last date accessed: October 15, 2013
http://openmopac.net/MOPAC2009.html; last date accessed: October 15, 2013
http://pubchem.ncbi.nlm.nih.gov/; last date accessed: October 15, 2013
References
- 1.Fragen RJ, Caldwell N. Comparison of dezocine (WY 16, 225) and meperidine as postoperative analgesics. Anesth Analg. 1978;57:563–566. doi: 10.1213/00000539-197857050-00010. [DOI] [PubMed] [Google Scholar]
- 2.Sun Q, Zhou W, Wu B, Ji MH, Peng YG. Dezocine: A novel drug to prevent fentanyl-induced cough during general anesthesia induction? J Anesth. 2012;26:470. doi: 10.1007/s00540-011-1318-x. [DOI] [PubMed] [Google Scholar]
- 3.Gharagozlou P, Hashemi E, DeLorey TM, Clark JD, Lameh J. Pharmacological profiles of opioid ligands at kappa opioid receptors. BMC Pharmacol. 2006;6:3. doi: 10.1186/1471-2210-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gal TJ, DiFazio CA. Ventilatory and analgesic effects of dezocine in humans. Anesthesiology. 1984;61:716–722. doi: 10.1097/00000542-198412000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Mumby SM, Linder ME. Myristoylation of G-protein alpha subunits. Methods Enzymol. 1994;237:254–268. doi: 10.1016/s0076-6879(94)37067-2. [DOI] [PubMed] [Google Scholar]
- 6.Wildman DE, Tamir H, Leberer E, Northup JK, Dennis M. Prenyl modification of guanine nucleotide regulatory protein gamma 2 subunits is not required for interaction with the transducin alpha subunit or rhodopsin. Proc Natl Acad Sci. 1993;90:794–798. doi: 10.1073/pnas.90.3.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iversen L, Gibbons S, Treble R, Setola V, Huang XP, Roth BL. Neurochemical profiles of some novel psychoactive substances. Eur J Pharmacol. 2013;700:147–151. doi: 10.1016/j.ejphar.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu R, Perez-Aguilar JM, Liang D, Saven JG. Binding site and affinity prediction of general anesthetics to protein targets using docking. Anesth Analg. 2012;114:947–955. doi: 10.1213/ANE.0b013e31824c4def. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrielsen M, Kurczab R, Ravna AW, Kufareva I, Abagyan R, Chilmonczyk Z, Bojarski AJ, Sylte I. Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol. Eur J Med Chem. 2012;47:24–37. doi: 10.1016/j.ejmech.2011.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc Natl Acad Sci. 2011;108:15810–15815. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stewart JJ. MOPAC: A semiempirical molecular orbital program. J Comput Aided Mol Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 19.Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 20.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solis FJ, Wets RJB. Minimization by Random Search Techniques. Math Oper Res. 1981;6:19–30. [Google Scholar]
- 22.Chen JC, Smith ER, Cahill M, Cohen R, Fishman JB. The opioid receptor binding of dezocine, morphine, fentanyl, butorphanol and nalbuphine. Life Sci. 1993;52:389–396. doi: 10.1016/0024-3205(93)90152-s. [DOI] [PubMed] [Google Scholar]
- 23.Malis JL, Rosenthale ME, Gluckman MI. Animal pharmacology of Wy-16,225: A new analgesic agent. J Pharmacol Exp Ther. 1975;194:488–498. [PubMed] [Google Scholar]
- 24.Downing JW, Brock-Utne JG, Barclay A, Schwegmann IL. WY 16225 (dezocine):A new synthetic opiate agonist-antagonist and potent analgesic: Comparison with morphine for relief of pain after lower abdominal surgery. Br J Anaesth. 1981;53:59–64. doi: 10.1093/bja/53.1.59. [DOI] [PubMed] [Google Scholar]
- 25.Gharagozlou P, Demirci H, David Clark J, Lameh J. Activity of opioid ligands in cells expressing cloned mu opioid receptors. BMC Pharmacol. 2003;3:1. doi: 10.1186/1471-2210-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowlingson JC, Moscicki JC, DiFazio CA. Anesthetic potency of dezocine and its interaction with morphine in rats. Anesth Analg. 1983;62:899–902. [PubMed] [Google Scholar]
- 27.Jacobs AM, Youngblood F. Opioid receptor affinity for agonist-antagonist analgesics. J Am Podiatr Med Assoc. 1992;82:520–524. doi: 10.7547/87507315-82-10-520. [DOI] [PubMed] [Google Scholar]
- 28.Chavkin C. The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacol. 2011;36:369–370. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraus ML, Alford DP, Kotz MM, Levounis P, Mandell TW, Meyer M, Salsitz EA, Wetterau N, Wyatt SA. Statement of the American Society Of Addiction Medicine Consensus Panel on the use of buprenorphine in office-based treatment of opioid addiction. J Addict Med. 2011;5:254–263. doi: 10.1097/ADM.0b013e3182312983. [DOI] [PubMed] [Google Scholar]
- 30.Chowdhury S, Chowdhury AN. Emergence of buprenorphine addiction. J Indian Med Assoc. 1990;88:294. [PubMed] [Google Scholar]
- 31.Roberts DM, Meyer-Witting M. High-dose buprenorphine: Perioperative precautions and management strategies. Anaesth Intensive Care. 2005;33:17–25. doi: 10.1177/0310057X0503300104. [DOI] [PubMed] [Google Scholar]
- 32.Bryson EO, Lipson S, Gevirtz C. Anesthesia for patients on buprenorphine. Anesthesiol Clin. 2010;28:611–617. doi: 10.1016/j.anclin.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Chern SYS, Isserman R, Chen L, Ashburn M, Liu R. Perioperative Pain Management for Patients on Chronic Buprenorphine: A Case Report. J Anesth Clin Res. 2012;3:10. doi: 10.4172/2155-6148.1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Shi YG, Woods JH, Watson SJ, Ko MC. Central kappa-opioid receptor-mediated antidepressant-like effects of nor-Binaltorphimine: Behavioral and BDNF mRNA expression studies. Eur J Pharmacol. 2007;570:89–96. doi: 10.1016/j.ejphar.2007.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan D, Cook CD, Smith MA, Picker MJ. An examination of the interactions between the antinociceptive effects of morphine and various mu-opioids: The role of intrinsic efficacy and stimulus intensity. Anesth Analg. 1999;88:407–413. doi: 10.1097/00000539-199902000-00035. [DOI] [PubMed] [Google Scholar]
- 36.Obara G, Toyohira Y, Inagaki H, Takahashi K, Horishita T, Kawasaki T, Ueno S, Tsutsui M, Sata T, Yanagihara N. Pentazocine inhibits norepinephrine transporter function by reducing its surface expression in bovine adrenal medullary cells. J Pharmacol Sci. 2013;121:138–147. doi: 10.1254/jphs.12164fp. [DOI] [PubMed] [Google Scholar]
- 37.Hartrick CT. Noradrenergic reuptake inhibition in the treatment of pain. Expert Opin Investig Drugs. 2012;21:1827–1834. doi: 10.1517/13543784.2012.731393. [DOI] [PubMed] [Google Scholar]
- 38.Rojo ML, Rodriguez-Gaztelumendi A, Pazos A, Diaz A. Differential adaptive changes on serotonin and noradrenaline transporters in a rat model of peripheral neuropathic pain. Neurosci Lett. 2012;515:181–186. doi: 10.1016/j.neulet.2012.03.050. [DOI] [PubMed] [Google Scholar]
- 39.Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, Lucki I. Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology. 2010;35:752–763. doi: 10.1038/npp.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA. Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology. 2012;62:167–176. doi: 10.1016/j.neuropharm.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillman KW, Parker MF, Silva M, Degnan AP, Tora GO, Lodge NJ, Li YW, Lelas S, Taber M, Krause RG, Bertekap RL, Newton AE, Pieschl RL, Lengyel KD, Johnson KA, Taylor SJ, Bronson JJ, Macor JE. Design, optimization, and in vivo evaluation of a series of pyridine derivatives with dual NK1 antagonism and SERT inhibition for the treatment of depression. Bioorg Med Chem Lett. 2013;23:407–411. doi: 10.1016/j.bmcl.2012.11.094. [DOI] [PubMed] [Google Scholar]
- 42.Joensuu M, Tolmunen T, Saarinen PI, Tiihonen J, Kuikka J, Ahola P, Vanninen R, Lehtonen J. Reduced midbrain serotonin transporter availability in drug-naive patients with depression measured by SERT-specific [(123)I] nor-beta-CIT SPECT imaging. Psychiatry Res. 2007;154:125–131. doi: 10.1016/j.pscychresns.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Merrill JO, Von Korff M, Banta-Green CJ, Sullivan MD, Saunders KW, Campbell CI, Weisner C. Prescribed opioid difficulties, depression and opioid dose among chronic opioid therapy patients. Gen Hosp Psychiatry. 2012;34:581–587. doi: 10.1016/j.genhosppsych.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jamison RN, Edwards RR, Liu X, Ross EL, Michna E, Warnick M, Wasan AD. Relationship of negative affect and outcome of an opioid therapy trial among low back pain patients. Pain Pract. 2013;13:173–181. doi: 10.1111/j.1533-2500.2012.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valkanoff TA, Kline-Simon AH, Sterling S, Campbell C, Von Korff M. Functional disability among chronic pain patients receiving long-term opioid treatment. J Soc Work Disabil Rehabil. 2012;11:128–142. doi: 10.1080/1536710X.2012.677653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Husain MM, Rush AJ, Trivedi MH, McClintock SM, Wisniewski SR, Davis L, Luther JF, Zisook S, Fava M. Pain in depression: STAR*D study findings. J Psychosom Res. 2007;63:113–122. doi: 10.1016/j.jpsychores.2007.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.